Introduction
Malouf syndrome (1), also known as dilated
cardiomyopathy-hypergonadotropic hypogonadism (DCM-HH) syndrome, is
a congenital disorder. The clinical features include congestive or
dilated cardiomyopathy, ovarian dysgenesis in females or primary
testicular failure in males, mental retardation, broad nasal base,
blepharoptosis, skin lesions, bone abnormalities and occasionally
marfanoid habitus. Not all features are present in every individual
case (1–5).
DCM-HH may be caused by heterozygous mutations in
the lamin A/C (LMNA) gene. Chen et al(6) identified three heterozygous missense
mutations A57P, R133L and L140R in the LMNA gene in four patients
reported to exhibit atypical Werner syndrome (adult progeria)
(7). The mutations altered
relatively conserved residues in LMNA.
The heterozygous missense mutation L59R, in the LMNA
gene, was identified by McPherson et al(8), who noted the phenotypic similarities
between their case, with cardiomyopathy and hypergonadotropic
hypogonadism, and female cases previously studied by Nguyen et
al(9) and Chen et
al(6), described as exhibiting
atypical Werner syndrome. Although these patients had a progeroid
appearance, none had severe growth failure, alopecia or rapidly
progressive atherosclerosis and McPherson et al(8) suggested that the phenotype represents
a distinct laminopathy involving DCM-HH.
In this study, we present the case of a young female
patient with DCM-HH, including the genetic analysis of the LMNA
gene. The study was approved by theNational Medical Ethics
Committee of the Republic of Slovenia (No.39/02/05) and written
informed consent was obtained from the patient’s family.
Case report
Patient history
The Caucasian female patient was the only child born
to unrelated parents. The height of the patient was 165 cm with a
weight of 50 kg. The patient had finished upper secondary school
and had no history of mumps, diabetes, surgery or radiation to the
pelvic region or autoimmune disorders. The first-degree relatives
of the patient had experienced normal puberties and were of normal
statures. There was no family history of menstrual disorders or
dysmorphic features. The patient had primary amenorrhea without the
somatic stigmata of Turner’s syndrome. The secondary sexual
characteristics of the patient were undeveloped, including breast,
pubic and axillary hair stages (Tanner stage B1, P1 and A1). The
features of the patient included a small chin, bilateral
blepharoptosis and marfanoid elongated fingers.
Since the age of 28, the patient had exhibited with
dyspnea, leg edemas and paroxysmal supraventricular tachycardia
inducing syncope. Initial ultrasound evaluation indicated dilated
cardiomyopathy with systolic dysfunction. X-rays revealed no
evidence of bone dysplasia.
Hormonal evaluation included follicle-stimulating
hormone (112.2 IU/l; menopausal range 21.7–153 IU/l), luteinizing
hormone (72.5 IU/l; menopausal range 11.3–40 IU/l), estradiol
(<0.03 nmol/l; menopausal range 0–0.11 nmol/l) and prolactin
(9.1 ng/ml; normal range 4.5–40 μg/l). Thyroid function tests were
consistent with subclinical hypothyroidism (thyroid stimulating
hormone 7.7 mIU/l, normal range 0.27–4.2 mIU/l). However, a normal
sized thyroid gland was detected by the ultrasound. The patient
refused to receive hormone replacement therapy.
When the patient was 29 years old, a
cardioverter-defibrillator was implanted due to symptomatic
paroxysms of ventricular tachycardia. The patient’s condition was
worsening every year due to recurrent cardiac decompensation.
Admission
At the age of 33 years, the patient was admitted to
the Division of Intensive Internal Medicine due to congestive heart
failure. At the time of admission the patient exhibited dyspnea,
orthopnea and lethargy. Major diastolic and systolic dysfunction
was observed upon ultrasound examination [end-diastolic dimension
(EDD) 7.7 cm, end-systolic dimension (ESD) 6.6 cm and the left
ventricular diastolic posterior wall dimension (LVPWd) 0.7 cm] with
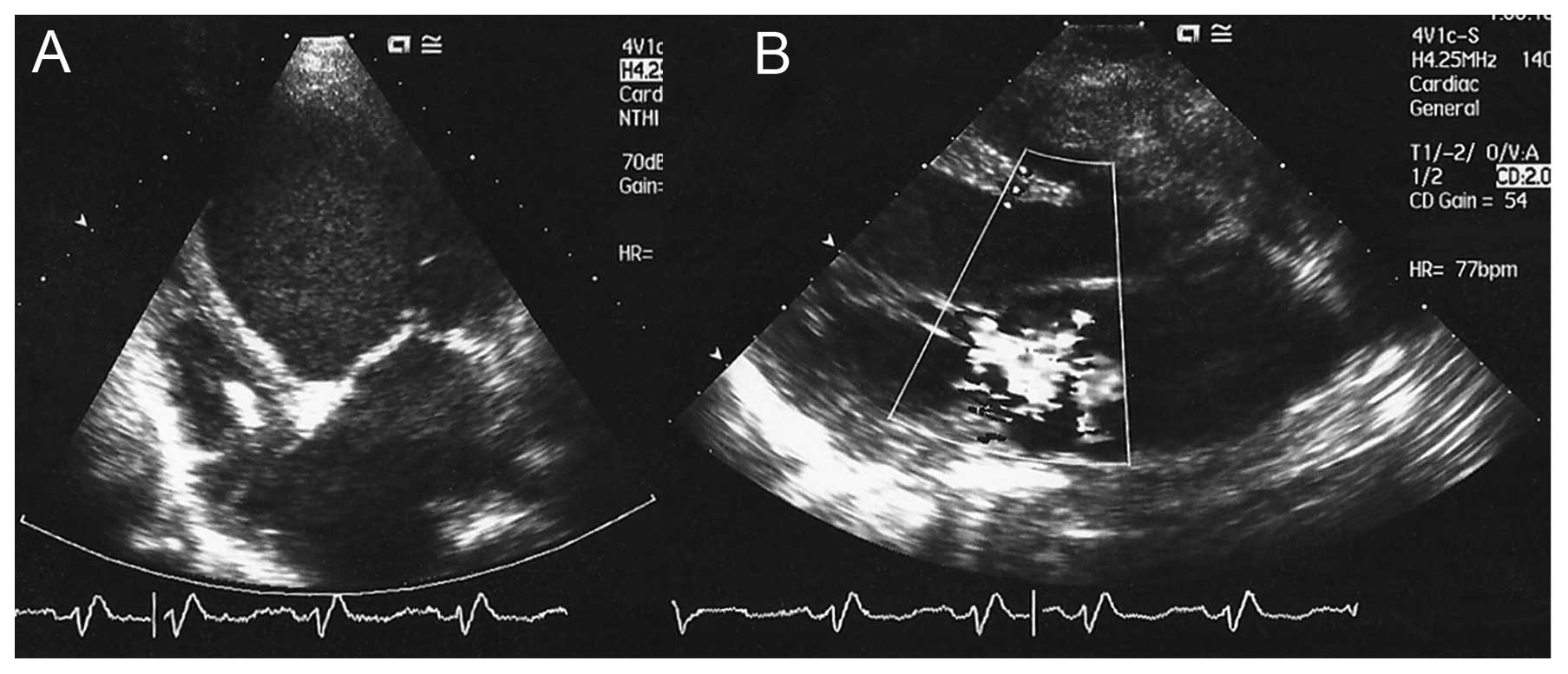
moderate mitral valve regurgitation (Figs. 1 and 2). The left ventricular ejection fraction
was 10–15%. On the inner wall of the left ventricle, a thrombus was
observed. The patient required an intra-aortic balloon pump, in
addition to inotropic and vasoactive support with noradrenalin and
dobutamine. Urgent transplantation of the heart was scheduled, but
the patient died on the fifth day of hospitalization due to
irreversible cardiogenic shock accompanied by multiple organ
failure.
Autopsy
An autopsy confirmed the diagnosis of asthenic
constitution without any secondary sexual characteristics. The
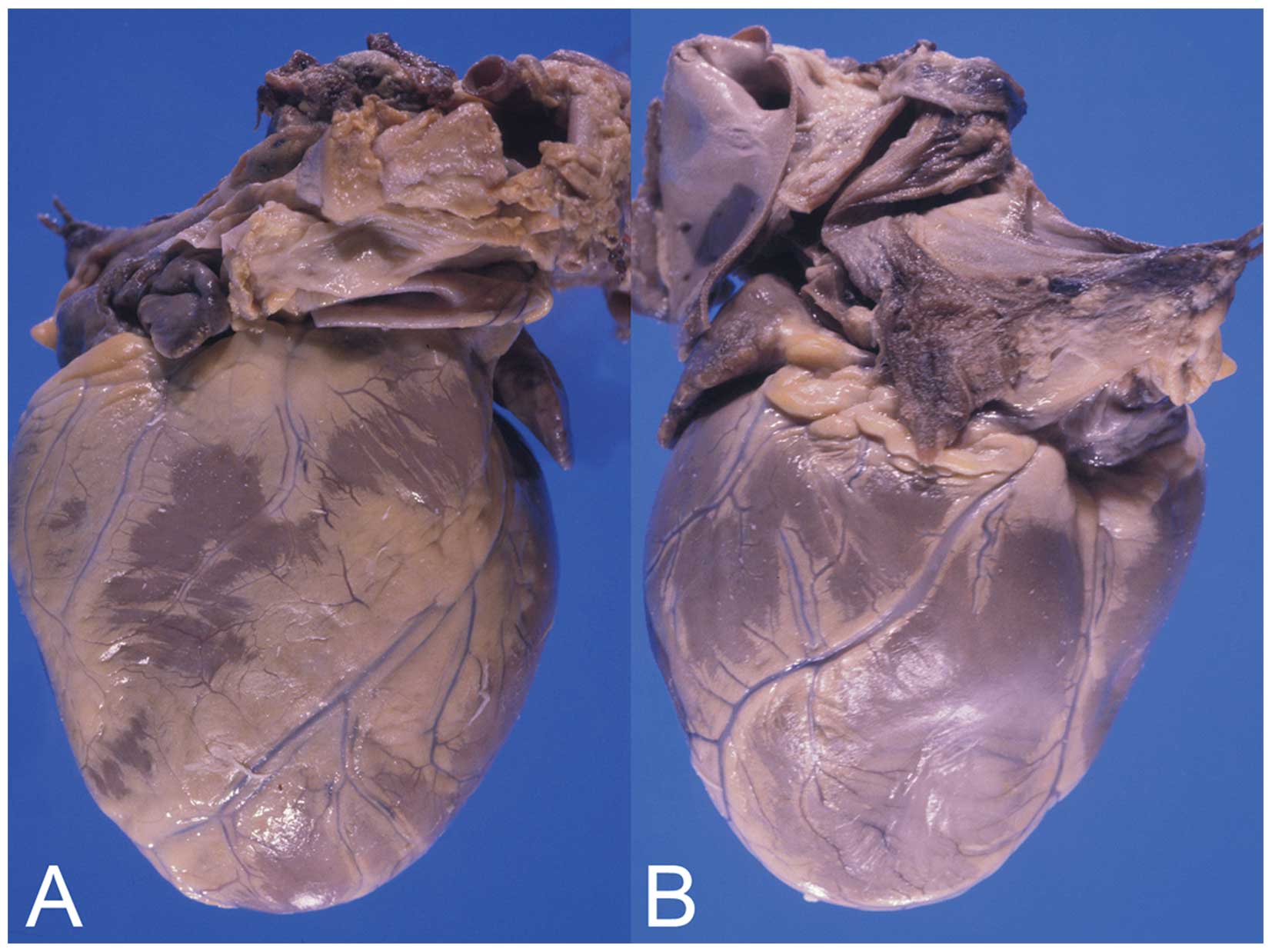
heart was enlarged, dilated and weighed 680 g (Fig. 3). The left ventricular wall and
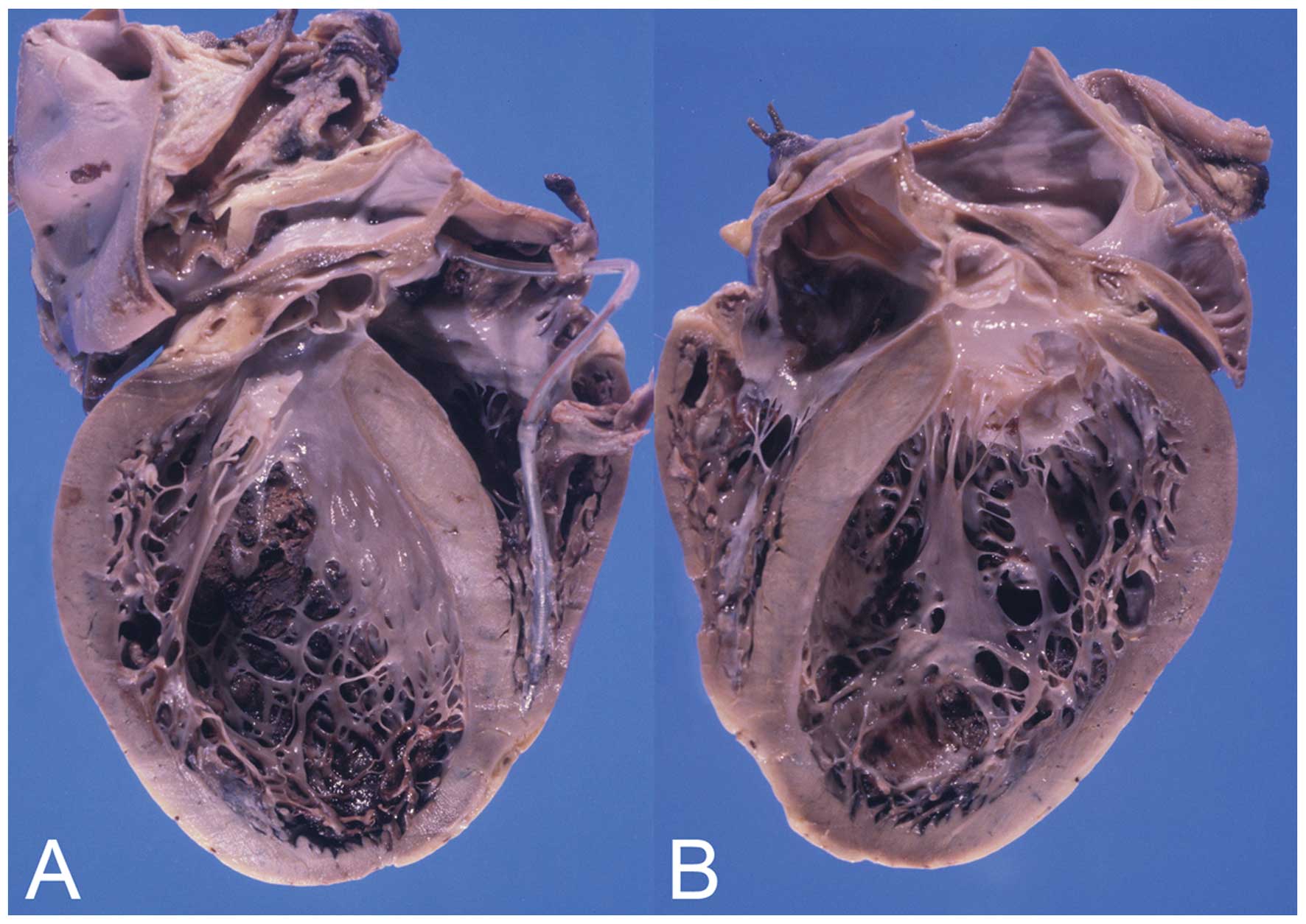
interventricular septum measured 11 and 12 mm, respectively; and
the right ventricular wall measured 3 mm (Fig. 4). Multiple fibrous scars were
observed in the inner section of the left ventricular wall and
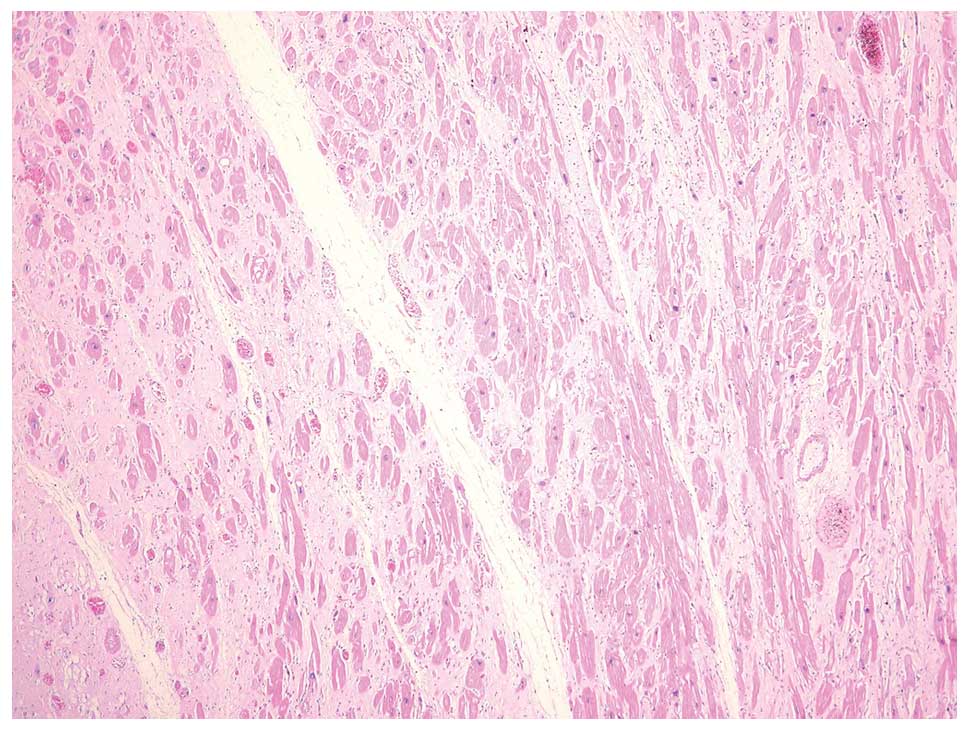
interventricular septum. Histological examination of the left
ventricular myocardium revealed hypertrophied myocytes with
polymorphic nuclei and multiple nucleoli (Fig. 5). Diffuse interstitial fibrosis
with rare, small foci of mononuclear infiltration was observed. The
endocardium was focally thickened, with parietal thrombi, the heart
valves were normal and the coronary arteries were slightly
atherosclerotic.
The uterus of the patient was small (3×1.5 cm) and
its cavity was covered with an atrophic endometrium and few glands.
The Fallopian tubes were almost obliterated with abnormal fimbriae
and there were hypoplastic streak gonads.
The thyroid gland was located on the anterior side
of the neck and comprised of two symmetrical lobes and isthmus. At
the microscopic level, the structure was normal.
Genetic analysis
Genomic DNA was used for polymerase chain reaction
(PCR) amplification of the 12 exons contained in the coding region
of the LMNA gene. The resulting PCR products were screened for
mutations by bidirectional sequencing using the automated
fluorescence dideoxy sequencing method. Sequence analysis
identified the benign polymorphism: homozygous 1698C>T (H566),
which is not associated with the disease.
Discussion
In the present study, we report on the sporadic case
of a young female with DCM-HH, facial dysmorphisms and
hypothyroidism. The majority of cases with DCM-HH are born in
consanguineous families. Najjar et al(3,10)
reported five boys in two families with primary testicular failure,
mental retardation and cardiomyopathy. Brothers with cardiomyopathy
and primary testicular failure were also described by Sacks et
al(2) and Thomas et
al(11). Malouf et al
reported two sisters who presented with congestive cardiomyopathy
associated with ovarian dysgenesis, bilateral ptosis and prominent
nasal bones (12). The girls also
had small chins and marfanoid long fingers, in a similar way to the
patient in this case report.
To the best of our knowledge, this is the second
reported case of DCM-HH associated with thyroid dysfunction.
Familial DCM-HH with thyroid hemiagenesis was first reported by
Gursoy et al(5).
The clinical heterogeneity of the syndrome is
evident. Certain patients have a more complex phenotype and it is
likely that several different conditions may be identified among
patients previously reported to have Malouf syndrome (8).
Molecular genetic analysis was performed for all 12
exons of the LMNA gene. The polymorphism 1698C>T (H566) was
detected, which had previously been registered in the National
Center for Biotechnology Information Single Nucleotide Polymorphism
database (http://www.ncbi.nlm.nih.gov/snp/). It is a benign
polymorphism with no known association with DCM-HH. No other
changes to the LMNA coding region were identified in our patient.
There is the possibility that the sequence analysis may have not
detected intronic mutations or mutations in portions of the 5′- and
3′-untranslated regions. Thus, this result neither confirms nor
rules out a mutation in the LMNA gene.
In spite of the features of our patient, the
sequence analysis of exons of the LMNA gene did not confirm the
clinical diagnosis of Malouf syndrome.
References
|
1
|
Online Mendelian Inheritance in Man.
Cardiomyopathy, dilated, with hypergonadotropic hypogonadism (ID
212112). http://www.ncbi.nlm.nih.gov/omim/212112.
Accessed April 11, 2012
|
|
2
|
Sacks HN, Crawley IS, Ward JA and Fine RM:
Familial cardiomyopathy, hypogonadism, and collagenoma. Ann Intern
Med. 93:813–817. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Najjar SS, Der Kaloustian VM and Ardati
KO: Genital anomaly and cardiomyopathy: a new syndrome. Clin Genet.
26:371–373. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Narahara K, Kamada M, Takahashi Y, et al:
Case of ovarian dysgenesis and dilated cardiomyopathy supports
existence of Malouf syndrome. Am J Med Genet. 44:369–373. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gursoy A, Sahin M, Ertugrul DT, et al:
Familial dilated cardiomyopathy hypergonadotrophic hypogonadism
associated with thyroid hemiagenesis. Am J Med Genet A.
140:895–896. 2006. View Article : Google Scholar
|
|
6
|
Chen L, Lee L, Kudlow BA, et al: LMNA
mutations in atypical Werner’s syndrome. Lancet. 362:440–445.
2003.
|
|
7
|
Online Mendelian Inheritance in Man.
Werner syndrome; WRN (ID #277700). http://www.ncbi.nlm.nih.gov/omim/277700.
Accessed January 11, 2013
|
|
8
|
McPherson E, Turner L, Zador I, et al:
Ovarian failure and dilated cardiomyopathy due to a novel lamin
mutation. Am J Med Genet A. 149A:567–572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nguyen D, Leistritz DF, Turner L, et al:
Collagen expression in fibroblasts with a novel LMNA mutation.
Biochem Biophys Res Commun. 352:603–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Najjar SS, der Kaloustian VM and Nassif
SI: Genital anomaly, mental retardation, and cardiomyopathy: a new
syndrome? J Pediatr. 83:286–288. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thomas IT, Jewett T, Lantz P, et al:
Najjar syndrome revisited. Am J Med Genet. 47:1151–1152. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Malouf J, Alam S, Kanj H, Mufarrij A and
Der Kaloustian V: Hypergonadotropic hypogonadism with congestive
cardiomyopathy: an autosomal-recessive disorder? Am J Med Genet.
20:483–489. 1985. View Article : Google Scholar : PubMed/NCBI
|