Introduction
It has been established that the osteoprotegerin
(OPG)/RANKL/RANK system is important in bone metabolism and
remodeling. The receptor activator of nuclear factor (NF)-κB
ligand, RANKL, is localized or expressed by osteoblasts and binds
to its receptor RANK, which is localized or expressed by
osteoclasts, leading to the enhancement of osteoclastogenesis. OPG
suppresses osteoclastogenesis via binding to RANKL. In addition to
its involvement in bone metabolism, OPG has been implicated in the
vascular system and in cartilage metabolism (1–5). It
has been suggested that OPG is involved in cartilage, as OPG almost
completely inhibited cartilage loss in an adjuvant-induced
arthritis (AIA) rat model (6). In
addition, Shimizu et al(5)
demonstrated that OPG-deficient mice exhibited thinning of the
articular cartilage layers, with superficial fibrillation and
proteoglycan defects. Intraarticular or systemic OPG treatment has
been demonstrated to protect articular cartilage from proteoglycan
depletion and destruction of the cartilage surface (5,7).
However, the precise mechanism of OPG protection of cartilage
remains largely unknown. Shimizu et al(5) hypothesized that OPG protected
cartilage by inhibiting the apoptosis of chondrocytes, as a direct
effect. By contrast, findings of another study (2) suggested that the protection effect of
OPG was obtained via subchondral bone protection. In in
vitro studies, which added exogenous RANKl to the supernatant
of cultured chondrocytes, it was observed that NF-κB was not
activated, the expression profile of genes encoding proinflammatory
mediators in chondrocytes was not altered and this effect was also
observed for the production of collagenase and nitric oxide
(3). These results suggest that
OPG exerts no effect on chondrocytes as it is currently understood
that OPG exerts its functions by counteracting RANKL. Kwan Tat
et al(8) demonstrated that
exogenous OPG-Fc (with an Fc domain) increased the production of
two catabolic factors, matrix metalloproteinase (MMP)-13 and
protease-activated receptor 2 (PAR-2). This finding suggested that
OPG exerts a direct effect on cartilage. In studies concerning the
involvement of OPG in other systems, Yongchaitrakul et
al(9) observed that OPG
increased osteopontin production via sydecan-1. Kobayashi-Sakamoto
et al(1) demonstrated that
OPG promoted the proliferation of human dermal microvascular
endothelial cells, and was protective from cell death induced by
Porphyromonas gingivalis, a pathogen which causes adult
periodontitis. Thus this study aimed to determine whether OPG
exerts a direct effect on chondrocytes.
Cartilage matrices are predominantly composed of a
fibrillar collagen network and aggregated proteoglycan (10). Chondrocytes are the only cell
elements that are responsible for the metabolism of cartilage. In
normal conditions, chondrocytes produce and maintain the
cartilaginous matrix, self-secrete several factors and create a
balanced local environment regulating the metabolism of cartilage.
Briefly, catabolic factors such as MMP-1, -3, -9 and -13, or
aggrecanase-1 and -2 (ADAMTS-4, -5) produced by chondrocytes
degrade the matrix of cartilage. Conversely these factors are also
able to increase the secretion of tissue inhibitor of
metalloproteinase (TIMP)-1, -2, -3, -4, insulin-like growth factor
(IGF)-I, transforming growth factor (TGF)-β, bone morphogenetic
protein (BMP)-2 and basic fibroblast growth factor (bFGF) to
inhibit the effect of MMPs, and reach a balance to maintain the
self-renewal of cartilage. Moreover, IGF-I, TGF-β, BMP-2 and bFGF
are predominant factors in the stimulation of the proliferation of
chondrocytes. Overloaded pressure on cartilage and inflammatory
factors such as interleukin (IL)-1, tumor necrosis factor (TNF)-α
strike this balance (11). During
the process of the development and progression of osteoarthritis
(OA) or rheumatoid arthritis (RA), there is often initial
destruction of superficial aggrecan by two predominant factors,
ADAMTS-4 and -5 (12).
Although an increasing number of studies have
indicated that OPG protects cartilage in vivo, whether OPG
affects chondrocytes directly and the underlying cellular and
molecular mechanisms of this effect remain to be established. The
aim of the present study was to investigate the direct effect of
OPG on chondrocyte viability and proliferation and the functional
consequences of OPG treatment.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and type
II collagenase were purchased from Gibco-BRL (Carlsbad, CA, USA),
and fetal bovine serum (FBS) and trypsin-EDTA were purchased from
HyClone (Logan, UT, USA). Cell Counting kit-8 (CCK-8) was purchased
from Dojindo Laboratories (Kumamoto, Japan), and 96-well culture
plates and 25-cm2 flasks were purchased from Axygen (San
Diego, CA, USA). Primer synthesis was performed by Shanghai Sangon
Biological Engineering Technology Services, Ltd. (Shanghai, China).
The SYBR-Green Real-Time PCR Master mix and TRIzol reagent were
purchased from Takara (Shiga, Japan). RevertAid First Strand cDNA
Synthesis kit was purchased from Fermentas (Amherst, NY, USA).
Immunodetection of cell signal proteins for western blot analysis
was achieved using the antibodies:
anti-mitogen-activated/extracellular signal-regulated kinase kinase
(MEK), anti-extracellular signal-regulated kinase (ERK)1/2,
anti-P38 mitogen-activated protein kinase (P38MAPK), anti-c-Jun
N-terminal protein kinase (JNK), anti-NF-κBp65, anti-phospho (p)
MEK, anti-p-ERK1/2, anti-p-P38MAPK, anti-p-JNK anti-p-NF-κBp65 and
anti-glyceraldehyde-3-phosphate dehydrogenate (GAPDH) from (Cell
Signaling Technology, Inc., Beverly, MA, USA). In addition, rabbit
anti-rat TIMP-4 and rabbit anti-rat ADAMTS-5 were purchased from
Abcam (Cambridge, UK) and mouse anti-rat BMP-2 was purchased from
Pierce (Madison, WI, USA). U0126 (a MEK inhibitor) and PD098059 (an
ERK inhibitor) were purchased from Promega (Madison, WI, USA). The
P38MAPK inhibitor, SB203580, was purchased from Merck Millipore
(Billerica, MA, USA). PhosStop tablets were purchased from Roche
(Basel, Switzerland). Recombinant human OPG was purchased from
R&D Systems (Minneapolis, MN, USA). Toluidine blue was provided
by Sigma-Aldrich (St. Louis, MO, USA), and
5-(and-6)-carboxyfluorescein diacetate succinimidyl ester (CFSE)
was provided by Invitrogen Life Technologies (Carlsbad, CA,
USA).
Animals
Twelve 1-week-old, six 12-week-old, and six
36-week-old specific pathogen-free (SPF) Sprague-Dawley (SD) rats
used in this study were purchased from the Zhejiang Academy of
Medical Sciences (Huangzhou, China). Animals were fed and treated
in accordance with the Guidelines for the Care and Use of
Laboratory Animals 2006, administered by the First Affiliated
Hospital of College of Medicine, Zhejiang University (Hangzhou,
China).
Culture and identification of
chondrocytes
The rats were sacrificed by cervical dislocation,
soaked in 75% alcohol for 10 min and their knee joints were then
separated and the superficial cartilage was removed by a sterile
surgical knife on the clean bench. The cartilage was washed five
times with ice-cold phosphate-buffered saline (PBS) containing 1%
ampicillin and streptomycin. Subsequent to this, the cartilage was
sectioned into several 1-mm3 slices which were placed on
a plate containing 0.2% type II collagenase and transferred to a
37°C incubator. Supernatant was collected every 60 min and
centrifuged at 250 × g for 5 min to collect the cell pellet. These
procedures were repeated four times. The cells were re-suspended in
DMEM complete culture medium (containing 10% FBS, 100 U/ml
ampicillin and 100 U/ml streptomycin). Cells were filtered through
200 mesh stainless steel filters and seeded in flasks at a density
of 1×105/cm2 and cultured at 37°C in a 5%
CO2 incubator. When the cells reached 80% confluence,
the primary cultured cells were passaged. Chondrocytes were
identified with toluidine blue staining, as described previously
(13).
Treatment and inhibition
The cells were seeded in 35 mm2-wells at
a density of 1×105 cells/cm2. Following 24 h
of starvation, the chondrocytes were treated with OPG at
concentrations of 5, 10, 25, 50, 100 and 200 ng/ml. The maximum
effective dose was selected and used for the rest of the
experiments. RNA was extracted for reverse transcription-polymerase
chain reaction (RT-PCR) analysis after 24 h of OPG treatment and
protein for western blot analysis after 48 h of treatment.
Chondrocytes were incubated with various inhibitors for 30 min
prior to the addition of OPG. PD098059 was used at 10 μM, U0126 at
5 μM and SB203580 at 20 μM.
Assessment of cell proliferation
Passage 1 chondrocytes were plated at a density of
5×103 cells/well in a 96-well plate in 0.1 ml DMEM
complete culture medium. The cells were cultured for 24 h and then
starved for 24 h in serum-free DMEM. The chondrocytes were
incubated with or without U0126 (a MEK inhibitor), PD098059 (an ERK
inhibitor) or SB203580 (a P38MAPK inhibitor). After 30 min, OPG was
added (final concentrations of 5, 10, 25, 50, 100, 200 and 500
ng/ml) and the cells were incubated for an additional 48 h at 37°C.
The cells were then counted using CCK-8. Briefly, 10 μl of the kit
reagent was added to each well and the cells were incubated for 2
h. Cell viability was determined by measuring the absorbance at 450
and 655 nm with a microplate reader (Microplate Reader 680; Bio-Rad
Laboratories, Hercules, CA, USA). Each experimental condition was
analyzed in five wells. Chondrocytes used were isolated from 1-,
12- and 36-week-old SD rats.
Western blot analysis detection of MEK,
ERK1/2, P38MAPK, JNK, NF-κBp65, TIMP-4 and ADAMTS-5 in chondrocytes
following OPG treatment
Subsequent to OPG stimulation for 0, 15, 30 and 60
min, the total protein was extracted from each group according to
the manufacturer’s instructions. For the determination of TIMP-4
and ADAMTS-5 the cells were incubated with OPG for 2 days. A
bicinchoninic acid assay was used to determine the protein
concentration. PhosStop was used to inhibit the degradation of
phosphoprotein. Samples from different groups containing 20 or 30
μg proteins were loaded onto a 10% sodium dodecyl
sulfate-polyacrylamide gel, electrophoresed and transferred to a
polyvinylidene fluoride (PVDF) membrane. Following blocking with 5%
non-fat milk, the PVDF membranes were incubated with primary
antibodies in a Tris-buffered saline with Tween 20 (TBST) buffer
overnight at 4°C. The primary antibodies used were against p-MEK,
MEK, p-ERK1/2, ERK1/2, p-P38MAPK, P38MAPK, p-JNK, JNK, p-NF-κBp65,
NF-κBp65, TIMP-4 and ADAMTS-5. GAPDH served as an internal control.
On the following day, subsequent to three washes with TBST, the
membranes were incubated with the appropriate secondary antibodies
for 1 h at room temperature. The membranes were washed another
three times when the membranes had been soaked in an enhanced
chemiluminescence reagent for 5 min, and the bands were visualized
with X-ray film (VersaDoc Imaging system; Bio-Rad, Hercules, CA,
USA). The intensity of the bands was analyzed by Quantity One
software (Bio-Rad). Data were expressed in arbitrary units.
qPCR detection for anabolic and catabolic
factors expressed by chondrocytes
The total RNA in each group was extracted with
TRIzol reagent according to the manufacturer’s instructions. RNA (1
μg) was reverse transcribed into cDNA with the RevertAid First
Strand cDNA Synthesis kit. The obtained cDNA was amplified by qPCR
by an ABI VII Real-Time PCR system (Applied Biosystems, Carlsbad,
CA, USA) and SYBR-Green Real-Time PCR Master mix. The primers used
for qPCR are listed in Table I,
with GAPDH serving as an internal control. The conditions for the
qPCR amplification reaction were 95°C for 10 sec, 95°C for 30 sec,
60°C for 40 sec and 72°C for 45 sec, for a total of 40 cycles. The
dissolution curve was analyzed to determine the specificity of the
qPCR amplification. Quantification of the relative expression
levels of the target genes was achieved using the formula:
2−ΔΔCt, where ΔΔCt = (Ct of the target gene − Ct of
GAPDH) × treatment − (Ct of the target gene − Ct of GAPDH) ×
control. Data were presented in arbitrary units relative to the
control, which was defined as a value of 1.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Sense (5′→3′) | Antisense
(5′→3′) |
|---|
| IGF-I |
cttttacttcaacaagcccaca |
tacatctccagcctcctcaga |
| TGF-β |
tggaagtggatccacgcgcccaagg |
gcaggagcgcacgatcatgttggac |
| bFGF |
tactgcaagaacggcggcttcttc |
tactggccagttcgtttcagtgcc |
| BMP-2 |
ggaaaacttcccgacgcttct |
cctgcatttgttcccgaaaa |
| TIMP-1 |
tggcatcctcttgttgctatc |
cgaatcctttgagcatcttagtc |
| TIMP-2 |
aaccccatcaagaggattcaa |
cagggcacaataaagtcacaga |
| TIMP-3 |
gcgtgtatgaaggcaagatgta |
gcgtagtgtttggactgatagc |
| TIMP-4 |
cgtctgccactctgctttagta |
ctgcttctgactgttggtttct |
| ADAMTS-4 |
cccggaatggtggaaagtatt |
tcttcacggaaggtcaatgct |
| ADAMTS-5 |
ctacagcaactccgtgtgtgtc |
agtctggtctttggctttgaac |
| Collagen II |
ctcaagtcgctgaacaacc |
ctatgtccacaccaaattcc |
| Aggrecan |
aggatggcttccaccagtgc |
tgcgtaaaagacctcaccctcc |
| GADPH |
acagcaacagggtggtggac |
tttgagggtgcagcgaactt |
Detection of cell proliferation by CFSE
using fluorescence-activated cell sorting (FACS)
Passage 1 chondrocytes were collected and incubated
with CFSE for 15 min at 37°C in the dark, and were then placed in
cold DMEM containing 10% ice-cold FBS. The cells were seeded in
6-well plates (density, 1×105 cells/well). When the
cells had been cultured for 24 h, they were treated with or without
OPG for 4 days in an incubator. The cells were then digested with
0.25% trypsin, collected and suspended in FACS buffer. For CFSE
detection, the cells were analyzed directly by FACS. The results
were interpreted by FlowJo (Tree Star, Inc., Ashland, OR, USA).
Statistical analysis
Data were expressed as the mean ± SEM. Statistical
significance was assessed by a two-tailed Student’s t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Growth and identification of
chondrocytes
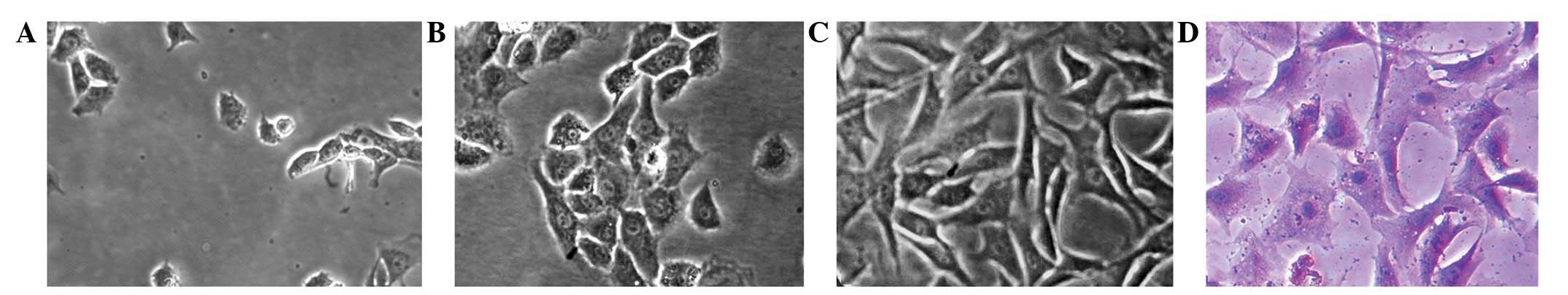
Chondrocytes were small and round when initially
seeded, and began to adhere and gradually spread out to form
pseudopods after ~24 h (Fig. 1A).
With an appropriate seeding density of
1×105/cm2 in the present study, the cells
grew in clusters and completely covered the culture flask within
3–4 days. The primary chondrocytes were oval and formed paving
stone-like arrangement (Fig. 1B)
and the passage 1 chondrocytes were trilateral or polygonal with
more pseudopods (Fig. 1C). The
chondrocytes were identified by toluidine blue staining and purple
metachromatic granules were observed within and around the cells
(Fig. 1D).
Effect of OPG on chondrocyte viability
and proliferation
Cell viability and proliferation were analyzed by
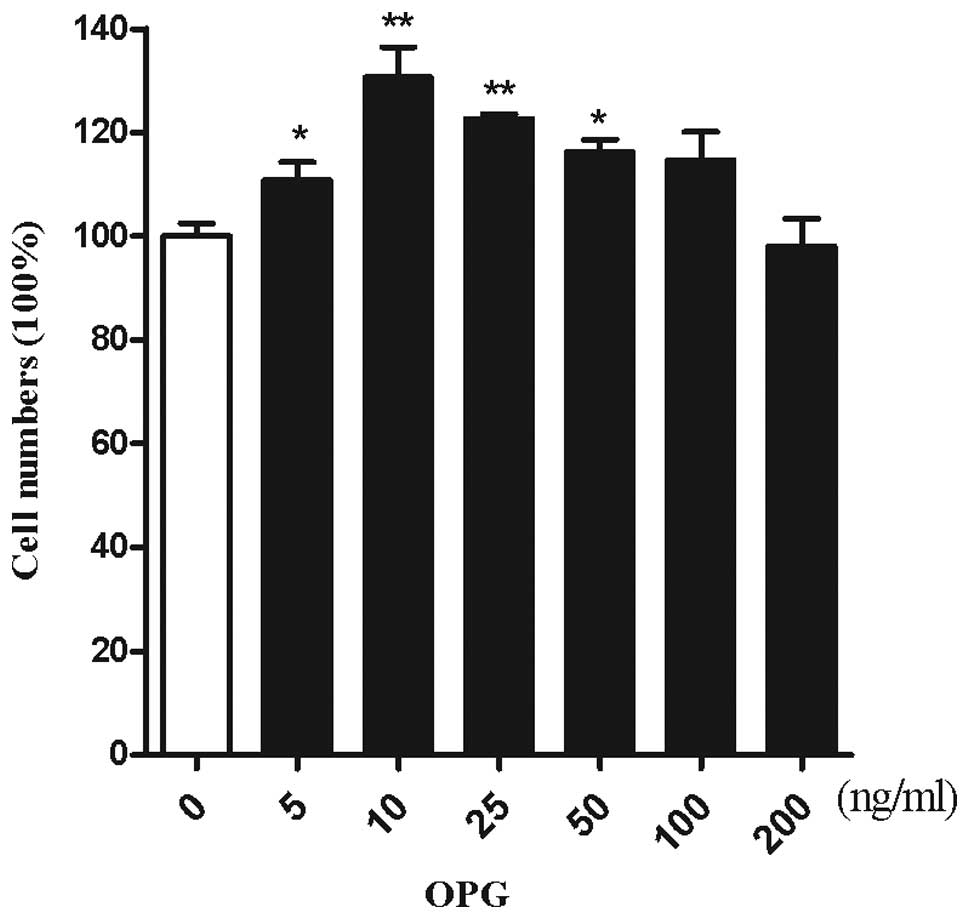
CCK-8. To investigate the effect of OPG on the number of
chondrocytes, chondrocytes were treated with increasing doses of
OPG. As demonstrated by Kotake et al(14), OPG concentrations were 5–35 ng/ml
in the synovial fluid. The cartilage was surrounded by the synovial
fluid, thus chondrocytes are exposed to an in vitro
concentration range of 5–200 ng/ml OPG. OPG at a concentration of
10 ng/ml exerted the maximum effect on the proliferation of
chondrocytes (Fig. 2).
OPG-induced chondrocyte proliferation
through the MEK/ERK signaling pathway
The effect of OPG was analyzed on the
phosphorylation of three MAPKs, ERK, P38MAPK and JNK.
Phosphorylation of ERK1/2 was increased to 4.12-fold that of
untreated samples at 15 min and 2.12-fold at 30 min following
treatment with OPG. However, P38MAPK and JNK signaling were not
influenced by OPG during the treatment period (Fig. 3A–C).
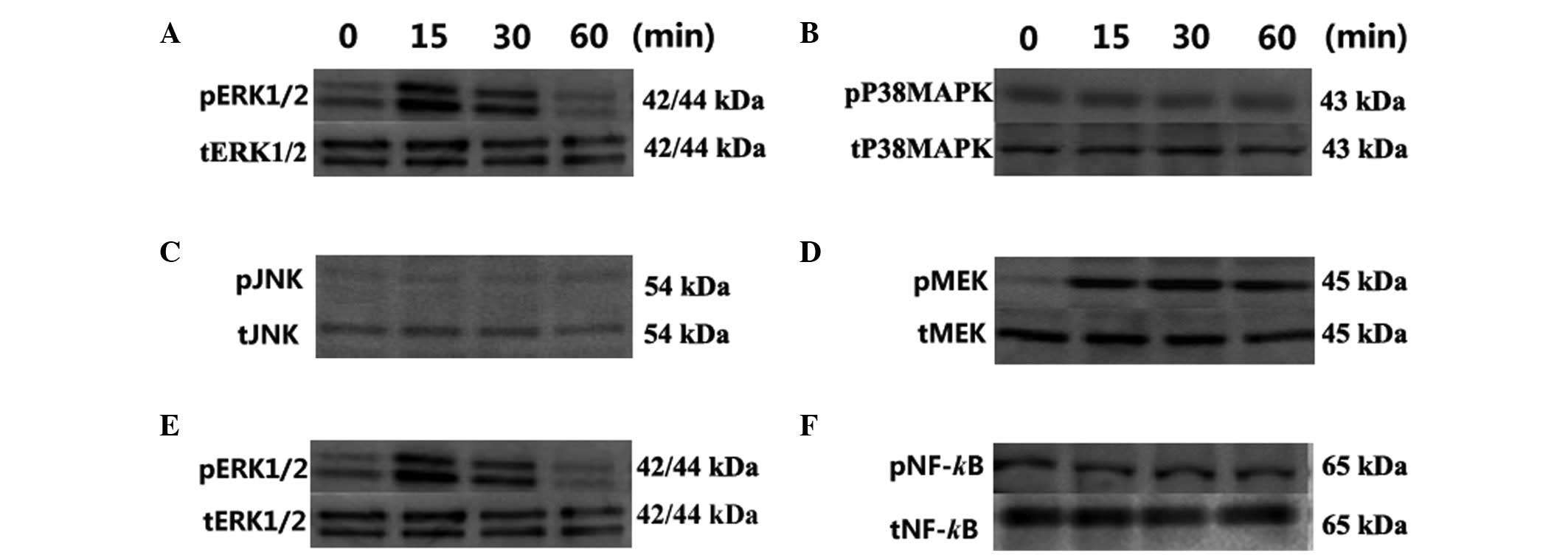 | Figure 3Effect of OPG on MEK, ERK1/2, P38MAPK,
JNK and NF-κBp65 phosphorylation analyzed by western blot analysis.
Phosphorylation of the MAPK family following treatment: (A) ERK1/2;
(B) P38MAPK and (C) JNK. Phosphorylation of the MEK/ERK/NF-κB
cascade: (D) MEK, (E) ERK1/2 and (F) NF-κBp65. Chondrocytes were
treated with 10 ng/ml OPG for 0, 15, 30 and 60 min. The results
shown represent three independent experiments. OPG,
osteoprotegerin; MEK, mitogen-activated/extracellular
signal-regulated kinase kinase; ERK, extracellular signal-regulated
kinase; P38MAPK, P38 mitogen-activated protein kinase; JNK, c-Jun
N-terminal protein kinase; NF-κB, nuclrear factor-κB. |
The upstream and downstream signaling of ERK1/2
signaling pathways was then analyzed in the chondrocytes. As
confirmed by previous studies, extracellular signals are
transmitted by MEK1/2 in ERK1/2 signaling cascades (15–17).
To elucidate the ERK1/2 signaling cascades involved in OPG-induced
proliferation, the phosphorylation of MEK was analyzed
simultaneously with ERK1/2. The 10 ng/ml concentration was selected
to be used in all experiments based on the CCK-8 assay. As shown in
Fig. 3D, the level of p-MEK was
3.5- and 3.0-fold higher in the OPG 15 and 30 min groups,
respectively, than in the control group (Fig. 3D).
Previous studies have demonstrated that NF-κB is
involved in the proliferation of tumor and other cells (18–21),
and acts as a downstream effector of ERK1/2. Thus, ERK1/2 signaling
was analyzed by determining the phosphorylation of ERK1/2 and NF-κB
simultaneously. As shown in Fig.
3F, the NF-κB phosphorylation level was not affected by OPG
stimulation during the exposure times used (Fig. 3F).
Inhibition of OPG-induces chondrocyte
proliferation following ERK activity suppression
To further determine whether the MAPK pathway was
involved in the OPG-induced proliferation of chondrocytes.
Chondrocytes were pretreated with U0126 (a MEK inhibitor), PD098059
(an ERK inhibitor) or SB203580 (a P38MAPK inhibitor) for 30 min
prior to OPG stimulation. As shown in Fig. 4, when incubated with U0126 or
PD98067, cell proliferation induced by OPG was suppressed
significantly. While SB203580 only exhibited marginal influence on
the cell proliferation induced by OPG (Fig. 4).
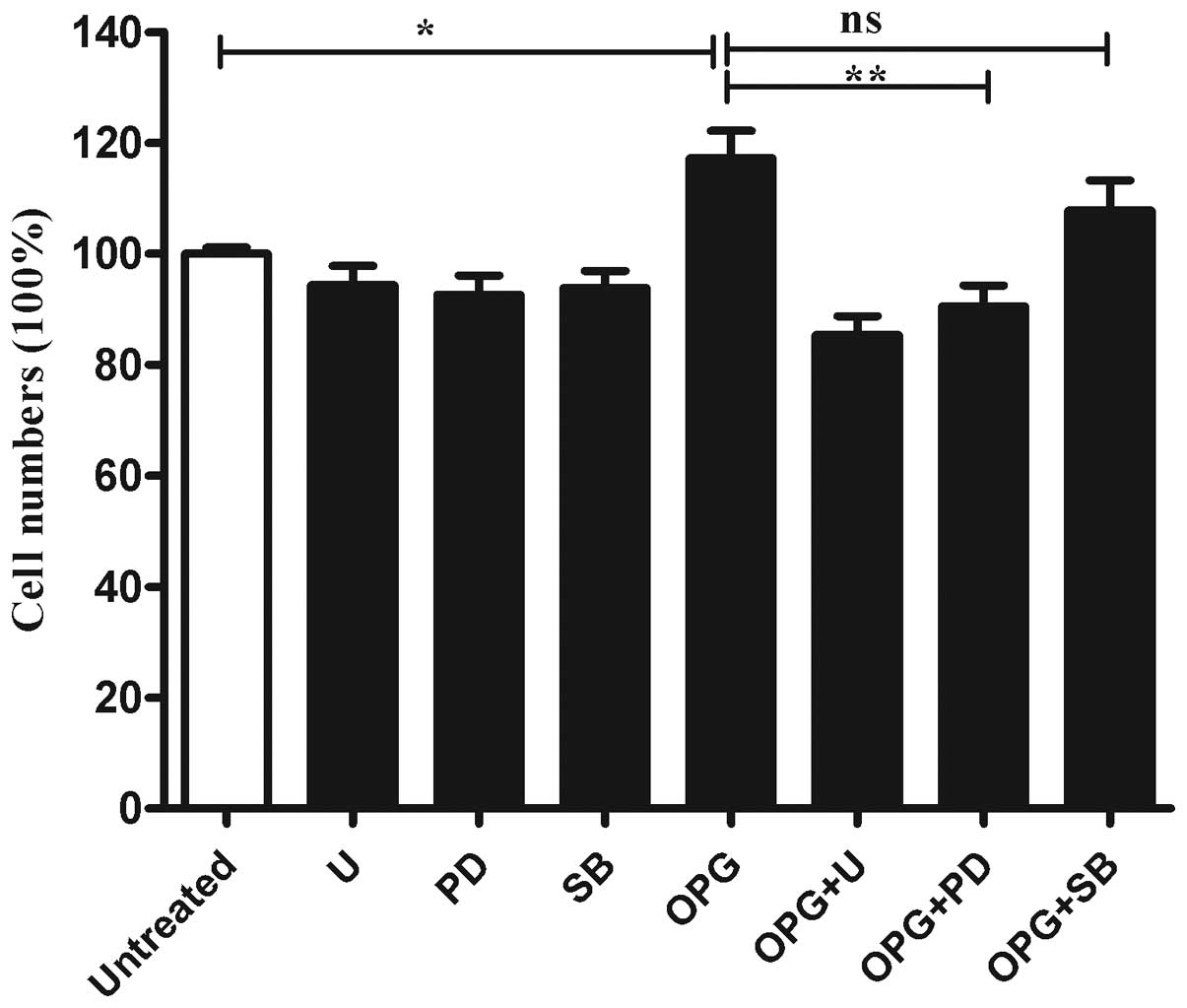 | Figure 4Inhibition of MEK or ERK rather than
P38MAPK activation suppressed OPG-mediated chondrocyte
proliferation. Relative cell proliferation was analyzed by CCK-8.
Columns show the percentage of the values obtained from cells
without treatment. Untreated, treated without OPG; U, cells treated
with 5 μM U0126 (MEK inhibitor); PD, cells treated with 10 μM
PD098095 (ERK inhibitor); OPG, cells treated with 10 ng/ml OPG; OPG
+ U, OPG + PD, OPG + SB, cells pretreated with the indicated
concentration of U0126, PD098095, SB203580, then treated with 10
ng/ml OPG. MEK, mitogen-activated/extracellular signal-regulated
kinase kinase; ERK, extracellular signal-regulated kinase; P38MAPK,
P38 mitogen-activated protein kinase; OPG, osteoprotegerin; CCK-8,
Cell Counting kit-8; ns, not significant. Data are presented as the
mean ± SEM of four independent wells value. *P<0.05
and **P<0.01 vs. the control group. |
Effect of OPG on chondrocyte
proliferation detected by CFSE
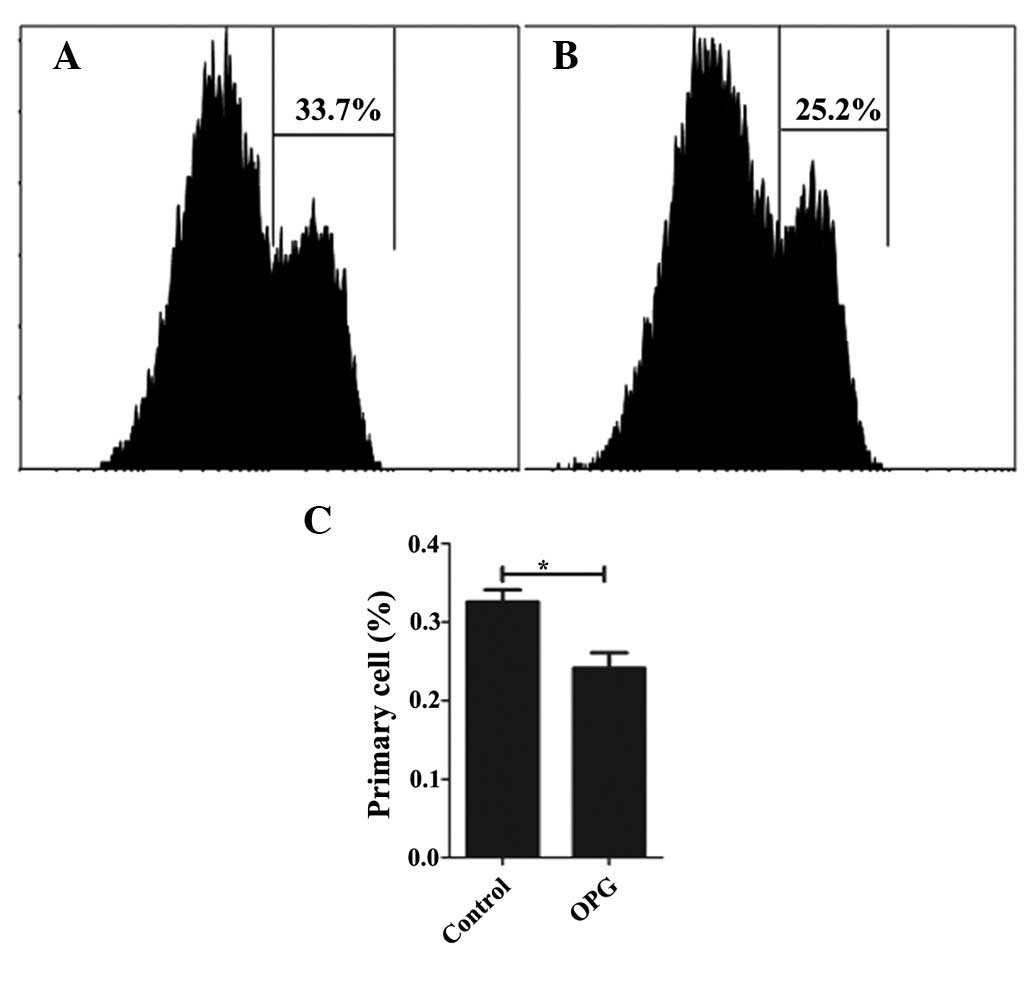
Following confirmation of the effect of OPG on
chondrocyte proliferation by CCK-8, the proliferation effect was
analyzed in greater detail using CFSE. While CCK-8 only reflects
the number of live cells, CFSE remains in the cell even when the
cells have died or undergone apoptosis. Thus, CFSE shows a
comprehensive profile of the cell proliferation. Consistent with
the result of the CCK-8 assay, OPG at the concentration of 10 ng/ml
increases the proliferation rate of chondrocytes, with 75.9 and
67.2% of the cells undergoing proliferation in the OPG and control
groups (Fig. 5), respectively.
Alterations of expression levels of
catabolic and anabolic factors following OPG treatment
Komuro et al(3) demonstrated that RANKL has no effect
on the expression of proinflammatory mediators, including IL-1β,
IL-6, cyclooxygenase-2, TNF-α, RANTES or inducible nitric oxide
synthase, and collagenase activity was not affected by the
treatment of OPG and RANKL. Kwan Tat et al(8) analyzed other factors, including
IL-1β, IL-6, IL-17, TNF-α, MMP-1, -2, -9, and -13, TIMP-1 and
PAR-2, and observed that OPG significantly promoted the expression
of MMP-13 and PAR-2. Thus, in the present study, the gene
expression levels of the anabolic factors containing TIMP-1, -2, -3
and -4, IGF-I, TGF-β, bFGF, and BMP-2, as well as catabolic
factors, including ADAMTS-4, ADAMTS-5 and constitutively expressed
factors collagen II α1 and aggrecan were analyzed. OPG treatment
significantly downregulated the gene expression of ADAMTS-5
(Fig. 6A) and upregulated the gene
expression level of TIMP-4 (Fig.
6B). None of the other factors analyzed were affected by OPG
treatment (data not shown).
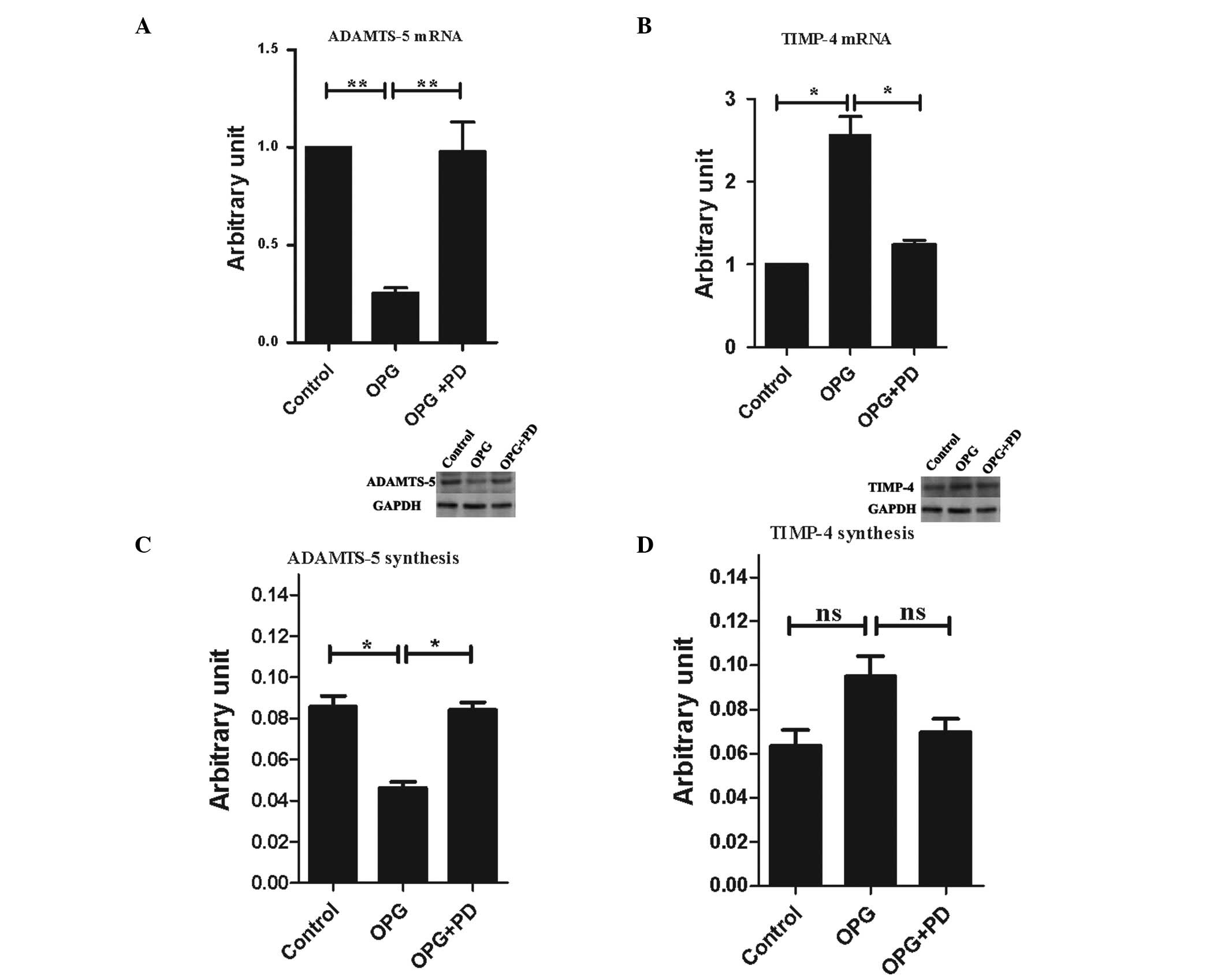 | Figure 6Alterations of expression levels of
the catabolic and anabolic factors following OPG treatment. Gene
expression level of (A) ADAMTS-5 and (B) TIMP-4, and protein
production of (C) ADAMTS-5 and (D) TIMP-4 on chondrocytes. Control,
untreated cells; OPG, cells treated with 10 ng/ml OPG; OPG + PD,
cells pretreated with the indicated concentration of PD098095, then
treated with 10 ng/ml OPG. The gene expression level is presented
as arbitrary units compared with the control, which was attributed
a value of 1. The protein production is presented as arbitrary
units compared to the internal control (GAPDH). The western blot
analysis figures shown represents three independent experiments.
Data are presented as the mean ± SEM of three independent
experiments. *P<0.05 and **P<0.01; vs.
the control group. OPG, osteoprotegerin; TIMP-4, tissue inhibitor
of metalloproteinase-4; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; ns, not significant. |
The protein production of TIMP-4 and ADAMTS-5 were
also analyzed following treatment with OPG. Subsequent to OPG
treatment 48 h, ADAMST-5 protein production exhibited the same
profile of gene expression: 2.2-fold greater in the OPG group than
in the control group, which was statistically significant (Fig. 6C). TIMP-4 production was 1.5-fold
higher in the OPG group than the control group, while it did not
reach statistical significance (P=0.55 between the OPG and control
groups, P=0.08 between the OPG and OPG + PD groups; Fig. 6D).
Inhibition of ERK1/2 activation by PD098095,
suppressed OPG-mediated alteration of ADAMTS-5 and TIMP-4
expression of the gene and protein levels.
Discussion
OPG is predominantly expressed by osteoblasts, and
its involvement in inhibiting osteoclastogenesis has been well
documented; however, there are few studies concerning the effects
of OPG on cartilage. In an OA mouse model, treatment with OPG
exhibited a beneficial effect against the progression of cartilage
destruction (2,5), but the exact mechanism has not been
fully elucidated. Moreover, little is known about the direct effect
of OPG on chondrocytes. The present study suggests that OPG exerted
a direct effect on chondrocytes, and it upregulated the expression
of TIMP-4 and downregulated the expression of ADAMTS-5
(aggrecanase-2). This is partly consistent with a previous study,
in which the number of ADAMTS-4 and -5 expressing cells was
decreased by OPG treatment in an OA mouse model (5). The two aggrecanases have been
identified to be essential factors in aggrecanolysis and cartilage
destruction (12). It was
demonstrated that ADAMTS-5 was predominant, and ADAMTS-5-knockout
mice were protected against cartilage destruction in an unstable
joint model (22). In the present
study, ADAMTS-5 was decreased by ~5-fold in RNA expression and
2.2-fold in protein secretion. The expression profile of ADAMTS-4
was not changed by OPG treatment. Moreover, TIMP-4, an inhibitor of
MMPs, was upregulated by OPG stimulation. However, Kwan Tat et
al(8) demonstrated that OPG
increased the gene expression level of MMP-13 and PAR-2, which were
two catabolic factors in cartilage. This may be attributable to the
different source of chondrocytes used in the studies, as OA
chondrocytes were used in the previous study and normal
chondrocytes were used in the present study. Alternatively, OPG
changes the expression profile of catabolic and anabolic factors,
with complex effects on the chondrocytes. In brief, the results of
the present study aid in the understanding of OPG protection of
cartilage in RA and OA mouse models. Other factors expressed by
chondrocytes were not affected by OPG treatment.
Initially, the number of chondrocytes isolated from
1-week-old rats was increased by OPG treatment, as confirmed by a
CCK-8 assay. It was investigated whether there was any difference
among the effects of OPG on chondrocytes isolated from different
aged rats. Therefore, chondrocytes isolated from 12- and
36-week-old rats were used to perform CCK-8 tests; however, no
difference was observed (data not shown). This finding suggests OPG
has a permanent and stable effect on chondrocytes as long as OPG
within an appropriate concentration range is used.
There are a large number of signaling pathways that
participate in the biological activity and behavior of
chondrocytes. Signaling via JNK (23), phosphoinositol 3-kinase (PI3k)/AKT
(24), Src (25), Wnt/β-catenin (26) is important in maintaining the
self-renewal of cartilage. MAPK signaling was regarded as important
in varied cell responses, such as the proliferation,
differentiation and apoptosis of chondrocytes. ERK1/2 and P38MAPK
pathways have been confirmed to be involved in the cell
proliferation and differentiation of chondrocytes (27–29).
It was demonstrated that soluble OPG activated several signals in
different cell types, inducing cytoskeleton reorganization through
FAK, Src and ERK1/2 signaling in endothelial cells (1), promoting endothelial cell
proliferation and migration partly via ERK1/2 signaling (30), increasing periodontal ligament
cells expressing osteopontin via syndecan-1 and PI3k/AKT (9), altering the morphology and function
of pancreatic islets possibly via the renin-angiotensin system
(31) and inducing proliferation
of rodent vascular smooth muscle cells (32). In the present study, MAPK signaling
molecules were analyzed as candidate downstream effectors of OPG.
Accumulation of phosphorylation ERK1/2 is observed following OPG
treatment. It was assumed that NF-κB was the downstream effector of
ERK1/2, as NF-κB participated in the differentiation of
chondrocytes (33). However, the
NF-κB phosphorylation level was not affected by OPG stimulation.
The ERK1/2 cascade was also investigated. MEK, one of the upstream
factors of ERK1/2, was analyzed simultaneously with ERK1/2 and was
activated by OPG treatment. Data collectively demonstrated that OPG
promoted chondrocyte proliferation through the MEK/ERK pathway but
not P38MAPK and JNK, and it was independent of NF-κB.
In conclusion, OPG exerted a direct effect on
chondrocytes. It increased the expression of TIMP-4 and decreased
the expression of ADAMTS-5. Moreover, it promoted the proliferation
of chondrocytes through the MEK/ERK pathway, independent of the
activation of NF-κB.
Acknowledgements
This study was supported by the Natural Science Fund
of Ningbo Science and Technology Bureau (grant no.
201101A6110108).
References
|
1
|
Kobayashi-Sakamoto M, Isogai E and Holen
I: Osteoprotegerin induces cytoskeletal reorganization and
activates FAK, Src, and ERK signaling in endothelial cells. Eur J
Haematol. 85:26–35. 2010.PubMed/NCBI
|
|
2
|
Kadri A, Ea HK, Bazille C, Hannouche D,
Lioté F and Cohen-Solal ME: Osteoprotegerin inhibits cartilage
degradation through an effect on trabecular bone in murine
experimental osteoarthritis. Arthritis Rheum. 58:2379–2386. 2008.
View Article : Google Scholar
|
|
3
|
Komuro H, Olee T, Kühn K, et al: The
osteoprotegerin/receptor activator of nuclear factor
kappaB/receptor activator of nuclear factor kappaB ligand system in
cartilage. Arthritis Rheum. 44:2768–2776. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kong YY, Yoshida H, Sarosi I, et al: OPGL
is a key regulator of osteoclastogenesis, lymphocyte development
and lymph-node organogenesis. Nature. 397:315–323. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimizu S, Asou Y, Itoh S, et al:
Prevention of cartilage destruction with intraarticular
osteoclastogenesis inhibitory factor/osteoprotegerin in a murine
model of osteoarthritis. Arthritis Rheum. 56:3358–3365. 2007.
View Article : Google Scholar
|
|
6
|
Kong YY, Feige U, Sarosi I, et al:
Activated T cells regulate bone loss and joint destruction in
adjuvant arthritis through osteoprotegerin ligand. Nature.
402:304–309. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwan Tat S, Pelletier JP, Lajeunesse D,
Fahmi H, Lavigne M and Martel-Pelletier J: The differential
expression of osteoprotegerin (OPG) and receptor activator of
nuclear factor kappaB ligand (RANKL) in human osteoarthritic
subchondral bone osteoblasts is an indicator of the metabolic state
of these disease cells. Clin Exp Rheumatol. 26:295–304. 2008.
|
|
8
|
Kwan Tat S, Amiable N, Pelletier JP, et
al: Modulation of OPG, RANK and RANKL by human chondrocytes and
their implication during osteoarthritis. Rheumatology (Oxford).
48:1482–1490. 2009.PubMed/NCBI
|
|
9
|
Yongchaitrakul T, Manokawinchoke J and
Pavasant P: Osteoprotegerin induces osteopontin via syndecan-1 and
phosphoinositol 3-kinase/Akt in human periodontal ligament cells. J
Periodontal Res. 44:776–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goldring MB and Marcu KB: Cartilage
homeostasis in health and rheumatic diseases. Arthritis Res Ther.
11:2242009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lories RJ and Luyten FP: The
bone-cartilage unit in osteoarthritis. Nat Rev Rheumatol. 7:43–49.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Verma P and Dalal K: ADAMTS-4 and
ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem.
112:3507–3514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Peng J, Wu M, et al: BMP2 promotes
chondrocyte proliferation via the Wnt/β-catenin signaling pathway.
Mol Med Rep. 4:621–626. 2011.PubMed/NCBI
|
|
14
|
Kotake S, Udagawa N, Hakoda M, et al:
Activated human T cells directly induce osteoclastogenesis from
human monocytes: possible role of T cells in bone destruction in
rheumatoid arthritis patients. Arthritis Rheum. 44:1003–1012. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rubinfeld H and Seger R: The ERK cascade:
a prototype of MAPK signaling. Mol Biotechnol. 31:151–174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dai Y, Rahmani M, Pei XY, et al:
Farnesyltransferase inhibitors interact synergistically with the
Chk1 inhibitor UCN-01 to induce apoptosis in human leukemia cells
through interruption of both Akt and MEK/ERK pathways and
activation of SEK1/JNK. Blood. 105:1706–1716. 2005. View Article : Google Scholar
|
|
17
|
Kimata M, Michigami T, Tachikawa K, et al:
Signaling of extracellular inorganic phosphate up-regulates cyclin
D1 expression in proliferating chondrocytes via the
Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway.
Bone. 47:938–947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng JQ, Xing L, Zhang JH, et al:
NF-kappaB specifically activates BMP-2 gene expression in growth
plate chondrocytes in vivo and in a chondrocyte cell line in vitro.
J Biol Chem. 278:29130–29135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun HZ, Yang TW, Zang WJ and Wu SF:
Dehydroepiandrosterone-induced proliferation of prostatic
epithelial cell is mediated by NFKB via PI3K/AKT signaling pathway.
J Endocrinol. 204:311–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu S, Tan WY, Chen QR, et al:
Daintain/AIF-1 promotes breast cancer proliferation via activation
of the NF-kappaB/cyclin D1 pathway and facilitates tumor growth.
Cancer Sci. 99:952–957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Massoumi R, Chmielarska K, Hennecke K,
Pfeifer A and Fässler R: Cyld inhibits tumor cell proliferation by
blocking Bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Glasson SS, Askew R, Sheppard B, et al:
Deletion of active ADAMTS5 prevents cartilage degradation in a
murine model of osteoarthritis. Nature. 434:644–648. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou Y, Millward-Sadler SJ, Lin H, et al:
Evidence for JNK-dependent up-regulation of proteoglycan synthesis
and for activation of JNK1 following cyclical mechanical
stimulation in a human chondrocyte culture model. Osteoarthritis
Cartilage. 15:884–893. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kita K, Kimura T, Nakamura N, Yoshikawa H
and Nakano T: PI3K/Akt signaling as a key regulatory pathway for
chondrocyte terminal differentiation. Genes Cells. 13:839–850.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bursell L, Woods A, James CG, Pala D,
Leask A and Beier F: Src kinase inhibition promotes the chondrocyte
phenotype. Arthritis Res Ther. 9:R1052007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong YF, Soung do Y, Chang Y, et al:
Transforming growth factor-beta and Wnt signals regulate
chondrocyte differentiation through Twist1 in a stage-specific
manner. Mol Endocrinol. 21:2805–2820. 2007. View Article : Google Scholar
|
|
27
|
Ryan JA, Eisner EA, DuRaine G, You Z and
Reddi AH: Mechanical compression of articular cartilage induces
chondrocyte proliferation and inhibits proteoglycan synthesis by
activation of the ERK pathway: implications for tissue engineering
and regenerative medicine. J Tissue Eng Regen Med. 3:107–116. 2009.
View Article : Google Scholar
|
|
28
|
Yonekura A, Osaki M, Hirota Y, et al:
Transforming growth factor-beta stimulates articular chondrocyte
cell growth through p44/42 MAP kinase (ERK) activation. Endocr J.
46:545–553. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yosimichi G, Nakanishi T, Nishida T,
Hattori T, Takano-Yamamoto T and Takigawa M: CTGF/Hcs24 induces
chondrocyte differentiation through a p38 mitogen-activated protein
kinase (p38MAPK), and proliferation through a p44/42
MAPK/extracellular-signal regulated kinase (ERK). Eur J Biochem.
268:6058–6065. 2001. View Article : Google Scholar
|
|
30
|
Kobayashi-Sakamoto M, Isogai E, Hirose K
and Chiba I: Role of alphav integrin in osteoprotegerin-induced
endothelial cell migration and proliferation. Microvasc Res.
76:139–144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toffoli B, Bernardi S, Candido R, et al:
Osteoprotegerin induces morphological and functional alterations in
mouse pancreatic islets. Mol Cell Endocrinol. 331:136–142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Candido R, Toffoli B, Corallini F, et al:
Human full-length osteoprotegerin induces the proliferation of
rodent vascular smooth muscle cells both in vitro and in vivo. J
Vasc Res. 47:252–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bradley EW and Drissi MH: WNT5A regulates
chondrocyte differentiation through differential use of the
CaN/NFAT and IKK/NF-kappaB pathways. Mol Endocrinol. 24:1581–1593.
2010. View Article : Google Scholar : PubMed/NCBI
|