Introduction
Bone marrow mesenchymal stem cells (BMSCs) are
self-renewable stem cells that may be differentiated into various
cell types in vitro(1).
BMSCs have also been identified as an essential component in the
hematopoietic stem cell (HSC) niche, as in vivo BMSC
depletion reduces HSC content (2).
The majority of the HSC niche is comprised of BMSCs, osteoblasts,
adipocytes, vascular endothelial cells and the extracellular matrix
(3). The HSC niche is essential
for the maintenance of HSCs (4).
Following niche disruption, HSCs may proliferate and differentiate
abnormally.
Acute myeloid leukemia (AML) is the most common type
of malignant myeloid disorder in adults and there is evidence to
suggest that, apart from gene mutations in HSCs, alterations in the
HSC niche have an important role in the proliferation,
differentiation and migration of malignant stem cells (5). However, little is known with regard
to the alteration of BMSCs in the HSC niche of AML patients, which
limit the understanding of AML pathology.
In this study, BMSCs were isolated from AML patients
and controls and their morphologies, differentiation capabilities
and adhesion molecule expression, which were hypothesized to be
associated with the function of BMSCs in the HSC niche, were
compared. These results may provide evidence for the pathological
study of the HSC niche.
Materials and methods
Clinical characteristics of patients
In total, 10 AML patients (age, 19–60 years; median
age, 44 years; six males and four females) and 13 control patients
(age, 19–77 years; median age, 48 years; 10 males and three
females) were enrolled from the Department of Hematology, Huashan
Hospital (Shanghai, China). AML patients included four cases of M2,
one of M3, two of M4, one of M5, one cases of M6 and one case that
was unclassified. Control patients included eight cases of phase
I/II group A B-cell lymphoma, two of phase I/II group A T-cell
lymphoma, one of vasculitis, one of splenic hyperfunction and one
of bronchitis. All conditions were confirmed by pathological
examination and all patients provided informed consent. The
experiments met the ethical standards of human experimentation and
were approved by the ethics committee of Huashan Hospital.
Isolation and culture of human BMSCs
Under sterile conditions, 2.5 ml bone marrow was
extracted using a 20 ml syringe from the posterior superior iliac
spine of the AML and control patients. Subsequently, the bone
marrow was mixed and diluted with 5 ml phosphate-buffered saline
(PBS). The cell suspension was added to 3.5 ml Ficoll extraction
liquid (Pharmacia, Uppsala, Sweden)and centrifuged at 432 × g for
20 min. The single nucleus layer was transferred to another
centrifuge tube, and washed twice with 10 ml Dulbecco’s modified
Eagle’s medium-low glucose (DMEM-LG; Gibco, Grand Island, NY, USA)
medium (containing 10% fetal bovine serum (Gibco) and 100 U/ml
penicillin-streptomycin; Gibco). The cells were cultured at a
density of 5×105/cm2 in 5% CO2
incubator (Heal Force, Hong Kong, SAR, China) at 37°C. The medium
was changed after 48 h and non-adherent cells were removed gently.
Subsequently, the media were changed every 2–3 days. When cells
covered ~90% of the dish the cells were passaged.
Identification of human BMSCs
The P4 human BMSCs were digested for 2 min with
0.25% Trypsin/EDTA (Gibco), 10-fold volume of DMEM-LG medium was
used to terminate the digestion, the solution was centrifuged at
260 × g for 5 min and the supernatant was removed. The cells were
washed and resuspended with 0.5 ml cold PBS at a density of
106/ml. Human CD73-peridinin chlorophyl (PerCP,
CD105-phycoerythrin (PE), CD90-allophycocyanin (APC),
CD45-fluorescein isothiocyanate (FITC) monoclonal antibodies
(eBioscience, San Diego, CA, USA) and their isotype controls were
added to the cell suspension and incubated for 30 min on ice, then
washed and resuspended with 0.5 ml cold PBS. The samples were
detected by flow cytometry and analyzed by Cell Quest software
(Beckman Coulter, Brea, CA, USA). Each sample included a minimum of
10,000 cells.
Differentiation of human BMSCs
P4 human BMSCs were digested for 2 min with 0.25%
Trypsin/EDTA, then resuspended by adipose induction medium
(containing DMEM-LG medium, 4 mM L-glutamine, 10 μg/ml bovine
insulin, 0.25 μM dexamethasone, 0.5 mM IBXM, 50 μM indomethacin) or
osteoblast induction medium (containing DMEM-LG medium, 0.2 mM
vitamin C, 0.1 uM dexamethasone, 10 mM β-glycerophosphate),
respectively. All buffer components were from Sigma (St. Louis, MO,
USA) The cells were seeded in 6-well plates at a density of
5×104 per well. The induction medium was changed every
2–3 days and photographed.
Oil red O staining
Following 14 days of adipose induction, the medium
was abandoned and washed twice with PBS. Subsequently the cells
were frozen at −20°C for 20 min, the cells were fixed with 4%
paraformaldehyde (PFA) for 20 min and washed twice with PBS. To
this 0.5% Oil Red O solution (Sigma)was added for 20 min and the
cells were subsequently washed twice with PBS. The cells were
stained with hematoxylin for 5 min and washed twice with PBS.
Alizarin red staining
Following 14 days of osteoblast induction, the
medium was abandoned and washed twice with PBS. The cells were
fixed with 95% ethanol for 10 min and washed twice with PBS.
Alizarin red (1%; Sigma) was added to 2% ethanol for 5 min and
washed twice with PBS.
Quantitative polymerase chain reaction
(qPCR)
TRIzol (1 ml; Invitrogen Life Technologies,
Carlsbad, CA, USA) was added to P4 cultured cells at room
temperature for 5 min, subsequently 0.2 ml chloroform was added for
3 min. The solution was centrifuged at 12,000 × g for 15 min and
the supernatant was removed. Isopropanol (0.5 ml) was added at room
temperature for 10 min, centrifuged at 12,000 × g for 10 min and
the supernatant was subsequently removed. In order to wash the
precipitate, 1 ml 75% ethanol was added, centrifuged at 7500 × g
for 5 min and the supernatant was subsequently removed and
dissolved in RNAase free H2O. Reverse transcription was
performed using an RT-PCR kit (Takara, Otsu, Japan) in accordance
with the manufacturers instructions. cDNA (2 μl) was obtained and 1
μM each of the upstream and downstream primers (Invitrogen Life
Technologies), 10 μl master mix and 6 μl DEPC water were added.
Amplification was performed for 40 cycles (GeneAmp PCR system 9700;
Applied Biosystems, Foster City, CA, USA) and the data was analyzed
using the relative quantification method (UV-2000
spectrophotometer; Unico, Franksville, WI, USA). Primers sequences
used are as follows: Sense: 5′-GAGA TTTCTCTGTATGGCACC-3′ and
antisense: 5′-CTGCAAAT GAGACACTTTCTC-3′ for Lipoprotein lipase
(LPL); sense: 5′-TTGCAGCCTTCTCAGCCAA-3′ and antisense: 5′-GGAG
GCAAAAGCAAATCACTG-3′ for osteopontin (OPN); sense:
5′-TTCCCTCGACACCCGATTC-3′ and antisense: 5′-TAGGTGGAGTCCCAGGCGTA-3′
for E-Cadherin; sense: 5′-GGTGGAGGAGAAGAAGAC-3′ and antisense:
5′-GGCATCAGGCTCCACAGT-3′ for N-Cadherin; and sense:
5′-GGATTTGGTCGTATTGGG-3′ and antisense: 5′-GGAAGATGGTGATGGGATT-3′
for GAPDH.
Western blotting
P4 cultured cells were lysed in 0.2 ml lysis buffer
[0.1% SDS, 1% NP-40, 50 mM HEPES, pH 7.4, 2 mM EDTA, 100 mM NaCl, 5
mM sodium orthovanadate (all Sigma), 40 μM PNPP (Santa Cruz
Biotechnology, Inc., Dallas, TX, US) and 1% Protease Inhibitor
Cocktail set I (Calbiochem, La Jolla, CA, USA)]. Lysates were
centrifuged at 4,000 × g for 25 min. The supernatant was obtained
and denatured directly for western blotting. Subsequently, samples
were separated by electrophoresis in 10% SDS-PAGE and transferred
to a polyvinylidene fluoride membrane. The membranes were blocked
for 2 h at room temperature in 5% bovine serum albumin, followed by
overnight incubation at 4°C with the primary antibody, then rinsed
and incubated for 1 h with the peroxidase-conjugated goat
anti-rabbit secondary antibody (Sigma). Membranes were then rinsed
in Tris-buffered saline with Tween-20 three times and visualized
using enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech,
Piscataway, NJ, USA). The primary antibodies used were as follows:
Anti-LPL (polyclonal, Santa Cruz Biotechnology Inc., 1:500),
anti-OPN (polyclonal, Santa Cruz Biotechnology Inc., 1:500),
anti-tubulin (monoclonal, Sigma, 1:5,000).
Statistical analysis
All data were obtained from a minimum of three
independent experiments. A non-parametric test was performed
between the different groups. P<0.05 was considered to indicate
a statistically significant difference. Excel (Microsoft, Redmond,
Washington, USA) and SPSS 5.0 software (SPSS Inc., Chicago, IL,
USA) was used for the statistical tests.
Results
Morphology and phenotype of BMSCs
BMSCs were collected from patients diagnosed with
AML (n=10) and controls (n=13). BMSCs derived from the AML patients
and controls were cultured in the same culture medium and were
capable of being passaged several times.
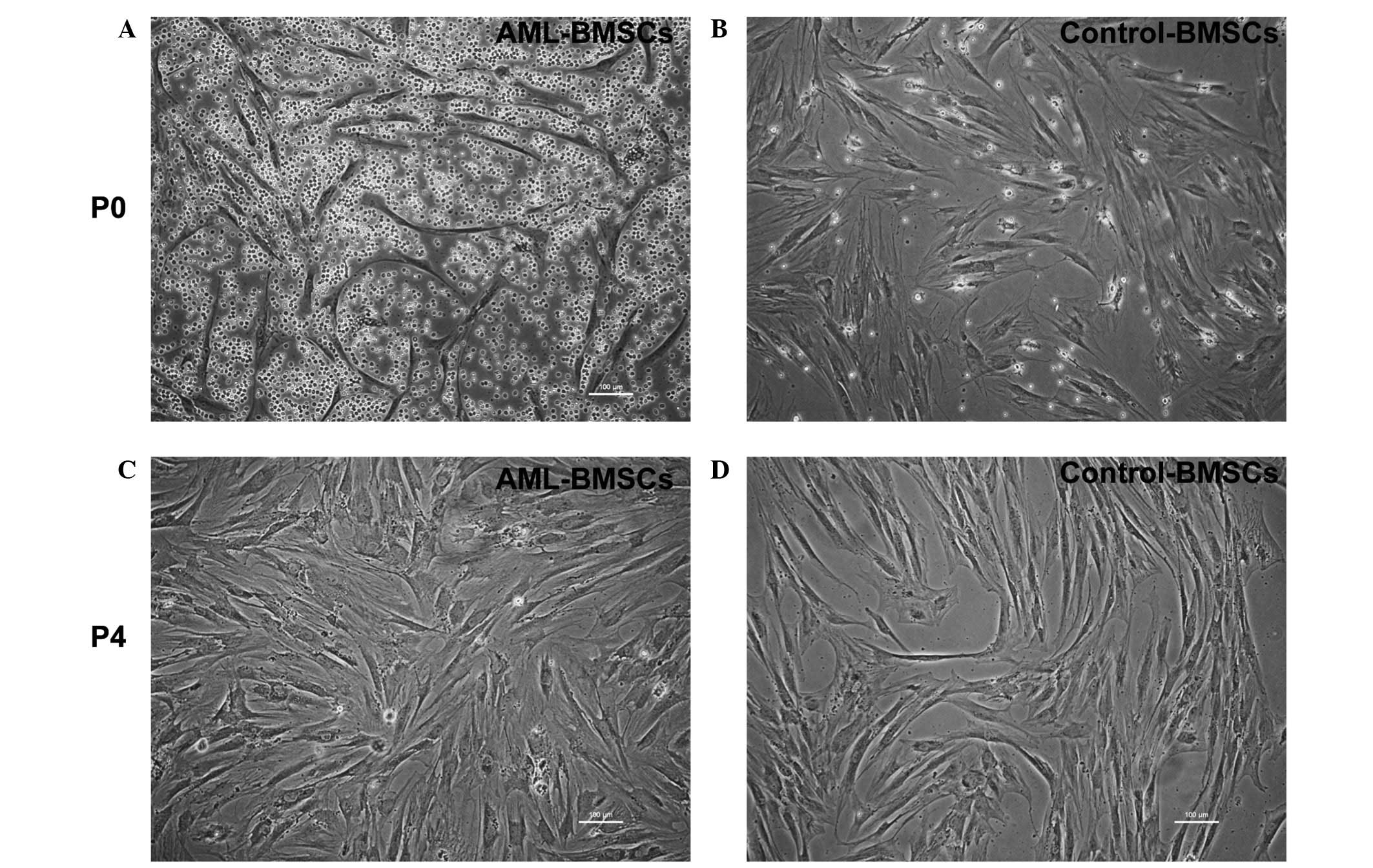
Normally, primary cultured BMSCs exhibit a
spindle-shaped morphology with a few round suspended cells.
However, the BMSCs from the AML patients exhibited a more irregular
morphology, with an increased number of branches and a greater
number of round cells compared with those from the controls
(Fig. 1A and B). Following
passaging four times, the two groups of cells presented with a
similar normal, long spindle shape. (Fig. 1C and D)
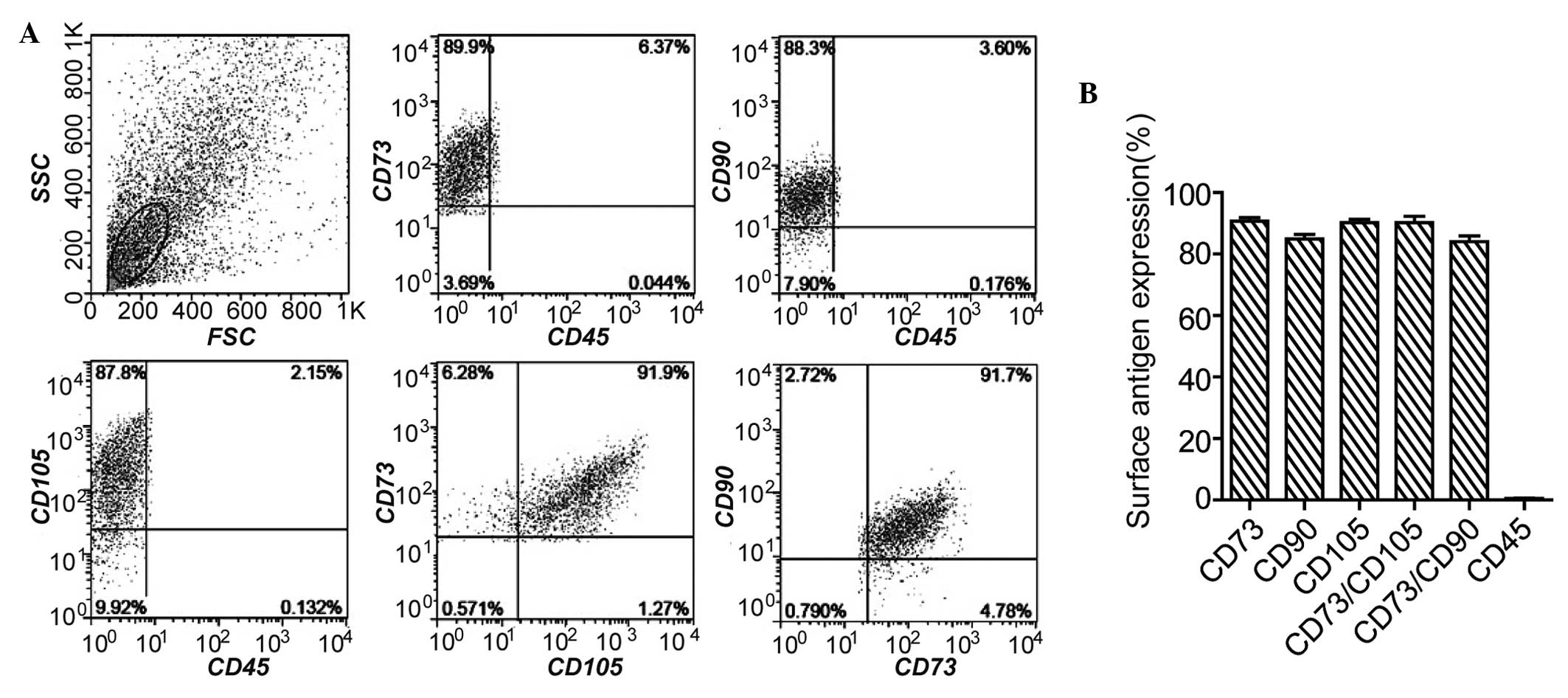
When the results were sorted by flow cytometry, it
revealed that these BMSCs expressed positive surface markers CD105,
CD73 and CD90, and the negative surface marker, CD45 (Fig. 2A and B). Furthermore, following
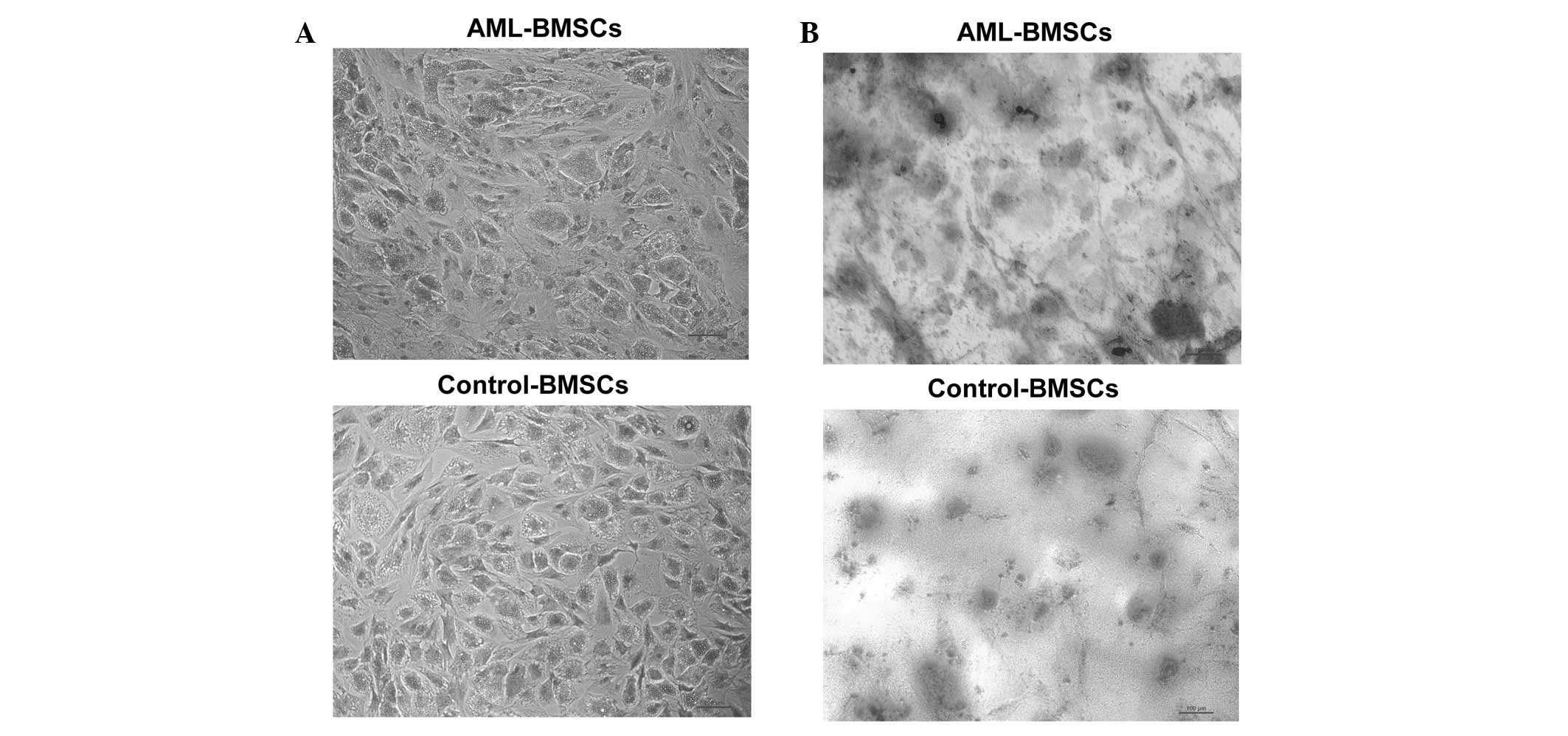
differentiation, the two groups of BMSCs may differentiate into
adipocytes and osteocytes (Fig.
3). Taken together, these results confirmed that these isolated
cells were BMSCs.
Comparison of adipogenic and osteogenic
differentiation ability
Subsequently, BMSCs from patients with AML and the
controls were tested for their differentiation ability. Prior to
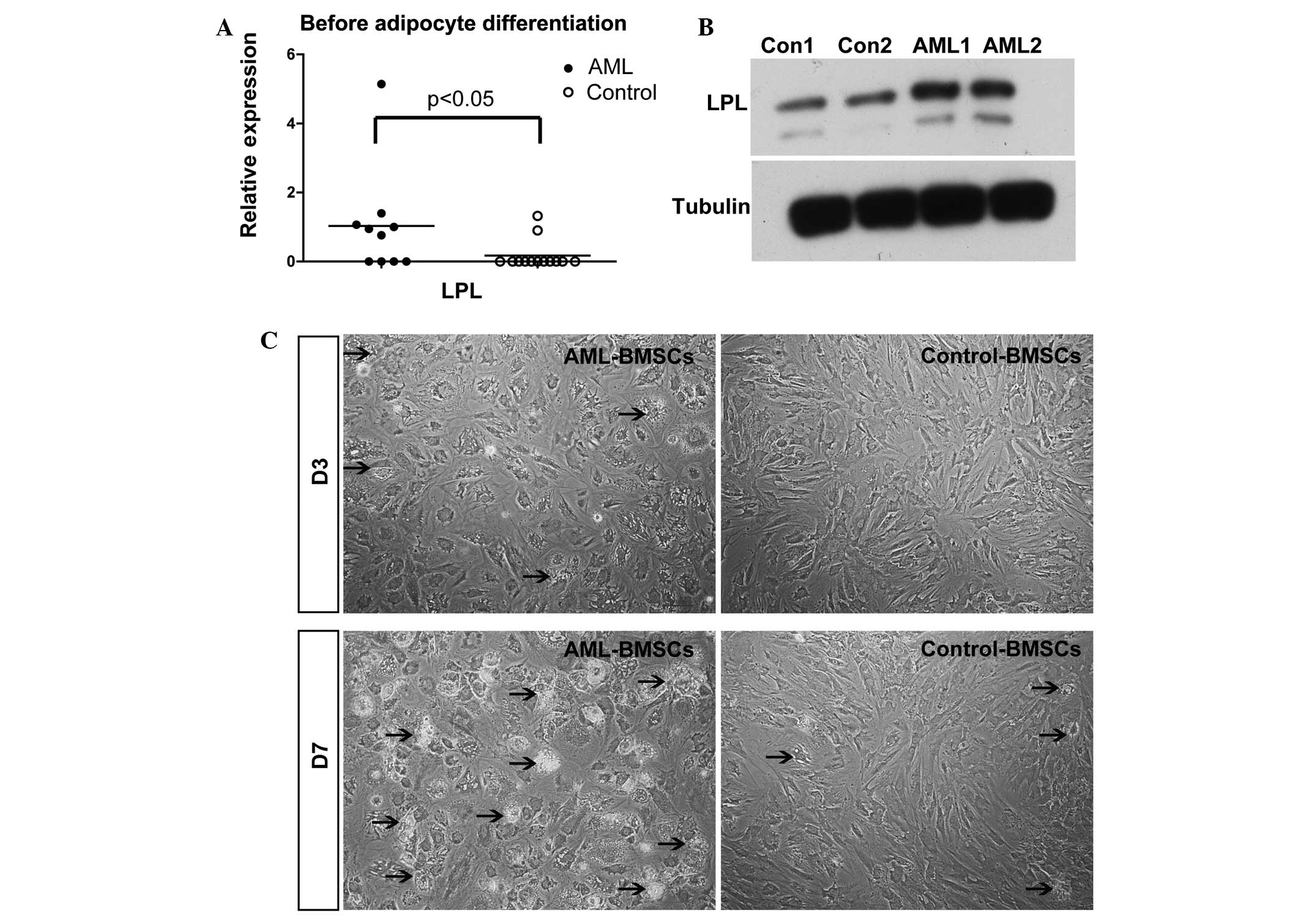
induction, the qPCR results revealed that the mRNA levels of the
lipid cell marker LPL in the AML group were significantly higher
than those identified in the controls (P<0.05, Fig. 4A). The results for the protein
levels revealed similar results (Fig.
4B). Following three days of adipogenic induction, lipid drops
were only detected in the BMSCs of AML patients. Furthermore, there
were more lipid droplets in the BMSCs of AML patients than in those
of the controls following seven days of adipogenic induction
(Fig. 4C). These results
demonstrated that the BMSCs of AML patients tended to
pre-differentiate to adipocytes.
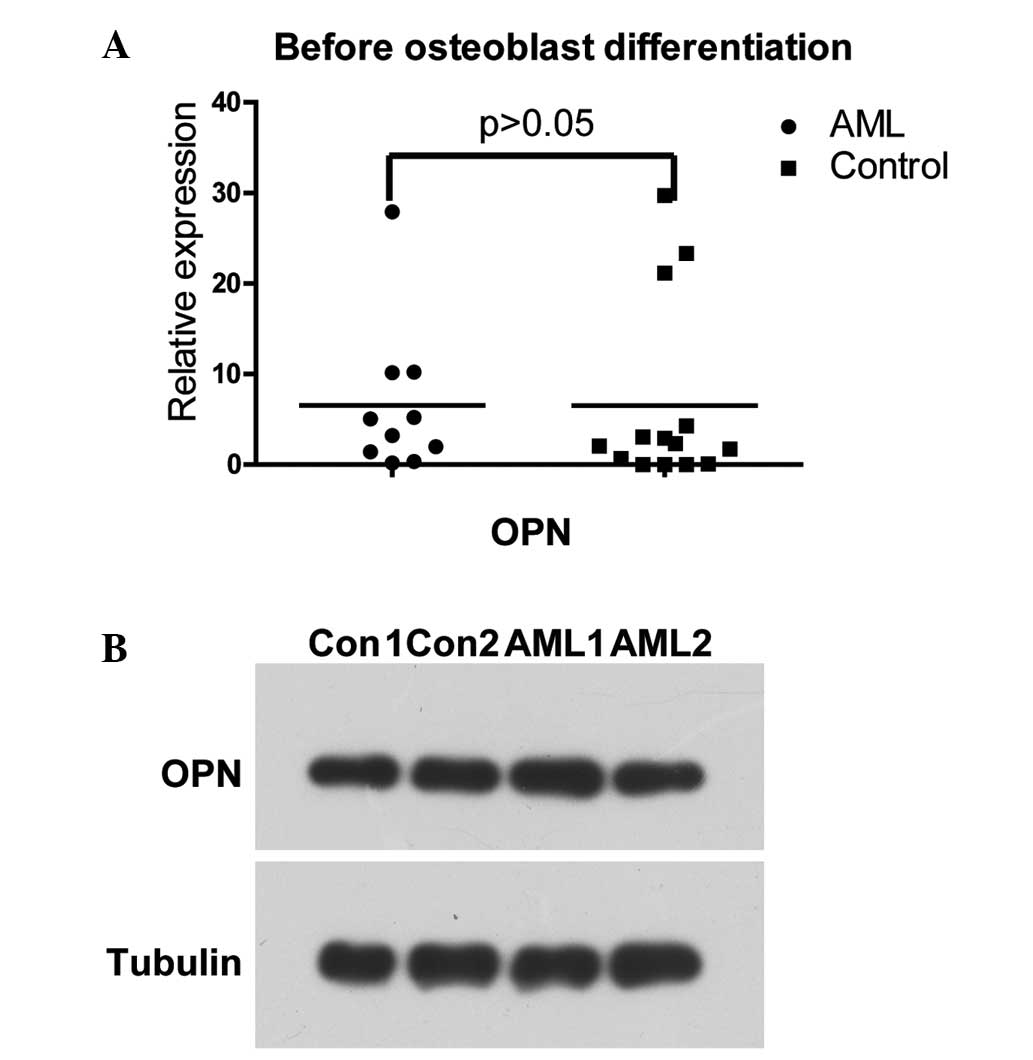
Prior to osteogenic differentiation, the mRNA and
protein levels of OPN exhibited no differences between the two
groups (Fig. 5A and B).
Furthermore, no differences in the expression of OPN were observed
between the two groups following osteogenic differentiation (data
not shown).
Following 14 days of adipogenic and osteogenic
differentiation, oil red O staining and alizarin red staining
revealed that the BMSCs of AML patients were capable of
differentiating into adipocytes and osteocytes. The staining
revealed no differences between the BMSCs of the AML patients and
those of the controls (Fig.
3).
mRNA levels of N- and E-Cadherin in the
BMSCs of AML patients and controls prior to and following
differentiation
The BMSCs from AML patients and the controls
expressed N-Cadherin/E-Cadherin and their mRNA levels revealed no
significant differences prior to differentiation (data not shown).
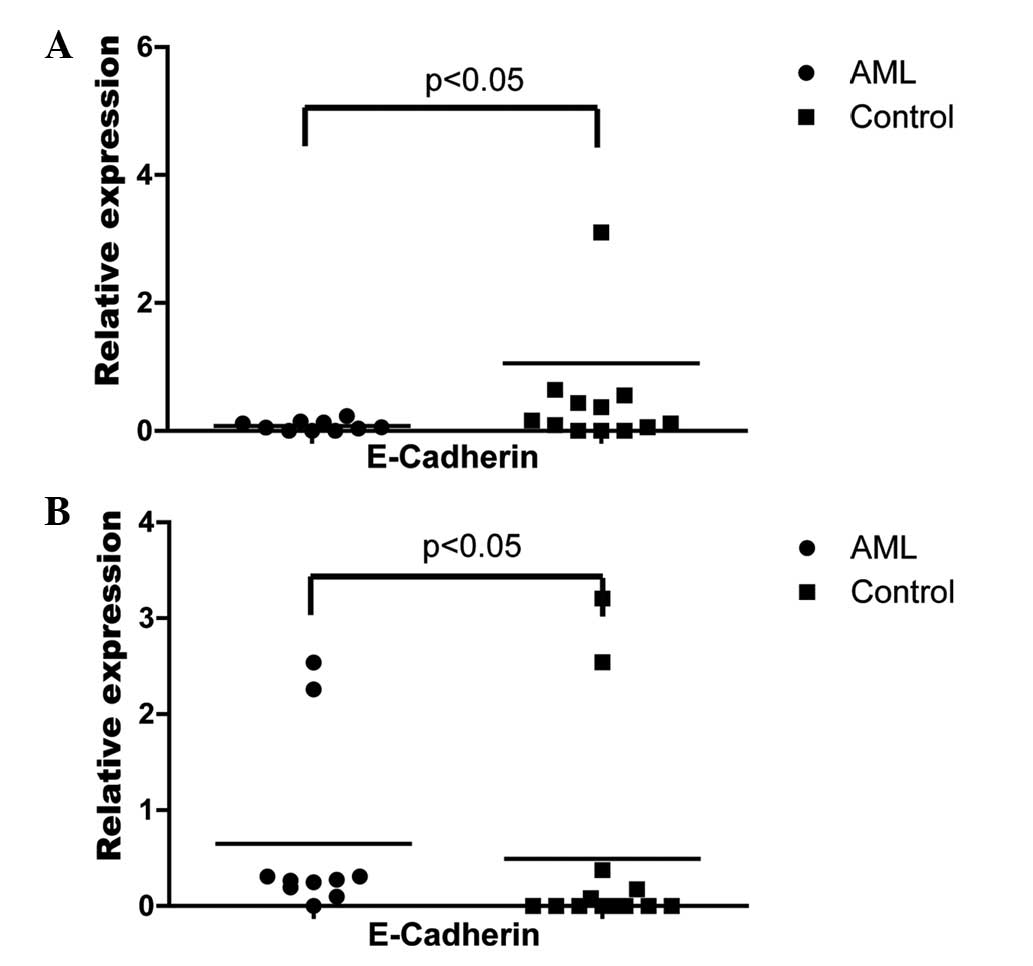
Following adipogenic induction, the mRNA level of E-cadherin in the
BMSCs of AML patients was significantly lower than that of the
controls following adipogenic induction (Fig. 6A). Following osteogenic induction,
the mRNA level of E-cadherin in the BMSCs of the AML patients was
significantly higher compared with the controls following
osteogenic induction (Fig. 6B). By
contrast, N-cadherin was not identified to be differentially
expressed between the two groups following differentiation (data
not shown).
Discussion
The bone marrow HSC niche is essential for the
maintenance of HSCs. The HSC niche alternation was hypothesized to
be the major cause of the malignant myeloid disorder AML. However,
little is known with regard to the alteration of BMSCs in AML
patients. The results of the present study demonstrated that BMSCs
from AML patients exhibited certain differing characteristics
compared with the controls. Primary cultured BMSCs from AML
patients exhibited an irregular morphology compared with those from
the controls. In addition, BMSCs from AML patients exhibited higher
lipid marker LPL expression prior to adipogenic differentiation and
pre-differentiated to adipocytes. Furthermore, following
differentiation, the mRNA levels of E-cadherin but not N-cadherin
in the BMSCs of AML patients were identified to be significantly
different from the controls.
The morphological changes of primary BMSCs may
influence the function of BMSCs in the HSC niche as the support
cells. This difference disappeared following several generations in
an in vitro culture, which suggests that the morphological
changes may arise from in vivo pathological conditions.
The results of the present study demonstrated that
BMSCs from AML patients were capable of differentiation into
osteoblasts and adipocytes following 14 days of in vitro
induction. However, BMSCs of AML patients tended to
pre-differentiate into adipocytes. While osteocytes were
hypothesized to be positive regulators of the HSC niche (6,7),
adipocytes were identified as negative regulators of the HSC niche
(8). The expansion of adipocytes
harmed the maintenance of the HSC niche. Therefore, BMSCs from the
AML patients may pre-differentiate to adipocytes in vivo,
leading to HSC depletion. As the results did not exhibit any
difference in the capacity for osteogenic differentiation, the HSC
niche alternation in AML patients may not be derived from the
increasing or depletion of BMSCs-derived osteocytes.
Cell-cell adhesion mediates cell migration and
signaling transduction, which is vital for HSC niche maintenance
(9). Several reports have
emphasized the function of N-cadherin in the homing and maintenance
of HSCs (10,11). However, knocking out N-cadherin in
mice did not lead to HSC depletion (12,13).
One potential explanation is that another redundant adhesion
molecule compensates for the function of N-cadherin. The current
results also exhibited no differences in the expression of
N-cadherin between BMSCs from AML patients and those from the
controls. However, the mRNA from E-caderin was detected in the
BMSCs from AML patients and controls and the mRNA levels of
E-cadherin in the BMSCs of AML patients were significantly
different from the controls following differentiation. As a result,
adipocytes and osteocytes derived from BMSCs may have different
signaling transduction and motility capabilities.
The mechanism for the alteration of BMSCs may arise
from a factor secreted from malignant cells, such as stem cell
factor (SCF), tumor necrosis factor (TNF) or macrophage colony
stimulating factor (M-CSF) in the bone marrow (14).
In conclusion, it was identified that BMSCs from AML
patients exhibited different morphologies, differentiation
capability and adhesion molecular expression from the controls
in vitro. These results may further the understanding of the
HSC niche in pathological conditions.
Acknowledgements
The authors would like to thank the patients and
staff of the Department of Hematology of Huashan Hospital
(Shanghai, China) for the donation and collection of bone marrow
samples. This study was supported by the National Basic Research
Program of China (grant no. 2011CB910404), grants from National
Natural Science Foundation of China (grant no. 81070399, 31371480),
and Foundation from Science and Technology Commission of Shanghai
Municipality (grant no. 13JC1406404) to T.C.
References
|
1
|
Pittenger MF, Mackay AM, Beck SC, et al:
Multilineage potential of adult human mesenchymal stem cells.
Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Méndez-Ferrer S, Michurina TV, Ferraro F,
Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A,
Enikolopov GN and Frenette PS: Mesenchymal and haematopoietic stem
cells form a unique bone marrow niche. Nature. 466:829–834.
2010.PubMed/NCBI
|
|
3
|
Wang LD and Wagers AJ: Dynamic niches in
the origination and differentiation of haematopoietic stem cells.
Nat Rev Mol Cell Biol. 12:643–655. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kiel MJ and Morrison SJ: Uncertainty in
the niches that maintain haematopoietic stem cells. Nat Rev
Immunol. 8:290–301. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steffen B, Müller-Tidow C, Schwäble J,
Berdel WE and Serve H: The molecular pathogenesis of acute myeloid
leukemia. Crit Rev Oncol Hematol. 56:195–221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calvi LM, Adams GB, Weibrecht KW, et al:
Osteoblastic cells regulate the haematopoietic stem cell niche.
Nature. 425:841–846. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang J, Niu C, Ye L, et al:
Identification of the haematopoietic stem cell niche and control of
the niche size. Nature. 425:836–841. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Naveiras O, Nardi V, Wenzel PL, Hauschka
PV, Fahey F and Daley GQ: Bone-marrow adipocytes as negative
regulators of the haematopoietic microenvironment. Nature.
460:259–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takeichi M: Cadherins: A molecular family
important in selective cell-cell adhesion. Annu Rev Biochem.
59:237–252. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hosokawa K, Arai F, Yoshihara H, et al:
Cadherin based adhesion is a potential target for niche
manipulation to protect hematopoietic stem cells in adult bone
marrow. Cell Stem Cell. 6:194–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hosokawa K, Arai F, Yoshihara H, et al:
Knockdown of N-cadherin suppresses the long-term engraftment of
hematopoietic stem cells. Blood. 116:554–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kiel MJ, Radice GL and Morrison SJ: Lack
of evidence that hematopoietic stem cells depend on
N-cadherin-mediated adhesion to osteoblasts for their maintenance.
Cell Stem Cell. 1:204–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kiel MJ, Acar M, Radice GL and Morrison
SJ: Hematopoietic stem cells do not depend on N-cadherin to
regulate their maintenance. Cell Stem Cell. 4:170–179. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wetzler M, Kurzrock R, Estrov Z, Estey E
and Talpaz M: Cytokine expression in adherent layers from patients
with myelodysplastic syndrome and acute myelogenous leukemia.
Leukemia Res. 19:23–34. 1995. View Article : Google Scholar : PubMed/NCBI
|