Introduction
Myocardial reperfusion injury following transient
focal or systemic ischemia is a complex pathophysiological process
that induces extensive cell death, and a marked portion of which
initiate mitochondrial-mediated apoptosis (1,2). A
previous study has implicated the mitochondrial ultrastructure and
respiratory function involved in myocardial contractility failure
in stimulated ischemia-reperfusion (I/R) (3). However, the mechanisms of molecular
signaling pathways and cytoprotective strategies targeting
mitochondria during I/R-induced apoptosis require further
investigation.
To ameliorate the severity of cardiac I/R, a number
of anti-apoptotic therapies targeting mitochondria are currently
being investigated, including pharmacological and genetic
inhibition (4). Cardiac myocytes
express various members of the BCL-2 family, including
anti-apoptotic and pro-apoptotic BCL-2 proteins, which control a
critical checkpoint in the mitochondria by preventing or promoting
apoptosis (5,6). The anti-apoptotic factors include
BCL-2, BCL-XL and MCL-1. The pro-apoptotic proteins are
further divided into the multi-domain members BAK and BAX, in
addition to a diverse group of ‘BH3-only’ molecules. BAK and BAX
are required for I/R-induced cell death by the mitochondrial or
intrinsic pathway (7). In viable
cells, monomeric BAK is constitutively localized at the
mitochondrial outer membrane (MOM), whereas inactive BAX are
located in the cytosol. Upon stress, active homo-oligomerized BAK
or BAX mediated membrane permeability and the efflux of cytochrome
c from mitochondria, which led to the activation of caspases
(8). Mouse embryonic fibroblasts
(MEFs) with genetic depletion of BAK or BAX were less sensitive to
truncated BID (tBID)-induced MOM permeabilization and apoptosis,
which is due to the reduced recruitment of pro-apoptotic BCL-2
family proteins to MOM (9).
In the view of the mounting evidence supporting a
homeostatic and cytoprotective role of carbon monoxide (CO) in
biological systems, much interest has focused on harnessing the
actions of this molecule for therapeutic purpose (10). Stimulation of endogenously
generated CO or low dose of exogenous CO targeting mitochondria has
shown activity, including oxidative phosphorylation and cell death
inhibition (11). CO treatment
enhanced cytochrome c oxidase (COX) activity and BCL-2
expression as well as their interaction, which protects astrocytes
against oxidative stress-induced apoptosis by improving
mitochondrial oxidative phosphorylation (12). In an oxidative stress model by
Conde de la Rosa et al (13), CO protected primary hepatocytes
against superoxide-anion-induced apoptosis. To the best of our
knowledge, there is little information regarding the effect of CO
on pro-apoptotic BCL-2 protein expression during myocardial I/R
injury. Furthermore, it has been demonstrated that CO-releasing
molecules (CORMs) also deliver CO and these compounds have since
emerged as potential therapeutic agents for the treatment of
various cardiovascular disorders (10,14).
Notably, CORMs treatment is associated with low or minimal
formation of carboxyhemoglobin and is therefore considered a safer
alternative to CO gas inhalation (15). Recent studies reported that the
tricarbonyldichlororuthenium (II) dimer (CORM-2) may attenuate
small bowel and kidney damage caused by I/R (16,17).
Based on the above information, CORM-2 was
hypothesized to ameliorate myocardial apoptosis triggered by I/R
through the mitochondrial pathway. In the current study, myocardial
I/R injury was induced in neonatal rat cardiomyocytes and the
effects of CORM-2 on mitochondrial respiratory function were
investigated, as well as the expression of MOM
permeabilization-related pro-apoptotic protein, BAK/BAX in cellular
extracts.
Materials and methods
Ethics statement
The study was approved by the Animals Ethics
Committee of Sun Yat-sen University (Guangzhou, China) and all
animals received humane care in compliance with the Guide for the
Care and Use of Laboratory Animals published by the National
Institute of Health (NIH publication 86-23, revised 1986).
Reagents
Tricarbonyldichlororuthenium (II) dimer (CORM-2) was
purchased from Sigma-Aldrich (St. Louis, MO, USA); cellular
mitochondria isolation kit from Beyotime Inc. (Jiangsu, China),
respiratory control ratio (RCR) quantitative testing kit from
Genmed Scientifics Inc. (Wilmington, DE, USA), anti-BAK and
anti-BAX antibodies from Abcam (Cambridge, UK) and anti-caspase-3
and anti-cytochrome c antibodies from Santa Cruz
Biotechnology, Inc. (Heidelberg, Germany).
Cell cultures and treatment
In brief, single cells were dissociated from minced
hearts (1 mm3) of Sprague-Dawley (SD) rats (age, 1–3
days) with 0.125% trypsin (fresh preparation; Gibco-BRL, Carlsbad,
CA, USA) and 0.05% type I collagenase (fresh preparation; MP
Biomedicals, Santa Ana, CA, USA). Cell solution was filtered with
100 μm nylon mesh and suspended in low-sugar Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum (both
Gibco-BRL) and 1% (v/v) penicillin-streptomycin solution
(Mediatech, Inc., Manassas, VA, USA) and pre-plated for 60 min to
reduced contamination of non-myocytes. The non-adherent
cardiomyocytes were separated and cultured at a density of
2×105 cells/cm2 with the aforementioned
medium supplemented with 0.1 mM 5′-bromo-2′-deoxyuridine (BrdU;
Sigma-Aldrich, St. Louis, MO, USA) in a 37°C/5% CO2
humidified incubator (Mini Galaxy A, RS Biotech Inc., Irvine, UK).
After a three-day cultivation, cardiac myocytes were randomly
divided into four groups: i) Control: Cells were continuously
cultured for 6 h in the cellular-cultured medium; ii) I/R: Cells
were subjected to 2 h simulated ischemia followed by 4 h of
reperfusion; iii) CORM-2: 20 μM CORM-2 in 0.2% dimethylsulfoxide
(DMSO) was administered to the culture medium at the beginning of
reperfusion and iv) inactive CORM-2 (iCORM-2): 20 μM iCORM-2 (CO
depleted) was added as described previously. iCORM-2 was produced
by preparing CORM-2 in DMSO and leaving it in a sealed, sterile
container exposed to light for 48 h and bubbling N2 gas
to remove the residual solubilized CO. For controls, 0.2% (v/v)
DMEM was added to the medium of the first two groups at the
beginning of reperfusion and equivalent volumes of medium in each
group was added. Only cultures consisting of >95% actin-positive
cells, determined by counting 300 cells in three fields, were
subjected to analysis.
Simulated I/R
In this study, hypoxia in ischemic buffer solution
(serum- and glucose-free DMEM pre-gassed with 95% N2 and
5% CO2 for ≥5 min) was utilized to mimic the ischemic
process in vivo, while oxygen, as well as serum and glucose
restoration stood for reperfusion. The medium was replaced with the
ischemic buffer solution, the cells were placed in a hypoxia
chamber filled with 5% CO2 and 0.3% O2 at
37°C for 2 h. Following hypoxia exposure, the cells were
reoxygenated with 5% CO2 and 19% O2 at 37°C
for a further 4 h in normal cellular-cultured medium.
Cell viability assay
Cell viability was determined based on methyl
thiazolyl tetrazolium (MTT) metabolism (18). The myocytes were seeded in a
96-well plates at a density of 1×105 cells/well and
incubated with 5 g/l MTT for 4 h at 37°C. The reaction was stopped
by the addition of 150 μl diphenylamine solution and the absorbance
of the blue formazan derivative was read at 570 nm using a
spectrophotometer. The percentage of cell viability was calculated
by the following formula: % cell viability = (mean absorbance in
test well)/(mean absorbance in control well) × 100.
Lactate dehydrogenase (LDH) activity
assay
The extent of cellular injury was monitored by
spectrophotometrically measuring LDH activity in the culture medium
with an LDH activity assay kit (Sigma-Aldrich).
Preparation of mitochondria
The mitochondrial isolation procedures were
completed within 1 h of cell collection. Myocardial mitochondria
were extracted by deferential centrifugation in mitochondria
extraction buffer at 4°C, as described previously (3). With a 0.25% solution of crude
trypsin, adherent cells were digested and centrifuged (500 × g).
The cell pellets were mixed with mitochondria extraction buffer and
placed in a glass homogenizer for homogenizing. Following
differential centrifugation (600 × g for 10 min followed by 11,000
× g for 10 min), final deposits were stored within mitochondrial
stocks and the supernatants were cytosol exclusive of
mitochondria.
Mitochondrial respiration
Mitochondrial respiratory function was measured at
25°C using an Oxygen Electrode (Oxygrph™, Hansatech Instruments,
King’s Lynn, UK). Real-time changes of oxygen concentration were
recorded via Oxygraph™ software (Hansatech Instruments). According
to the manufacturer’s instructions, calibration, followed by
zero-oxygen line establishment with sodium hydrosulfite
(NaO2SSO2Na) and sensibility test was
completed in the chamber prior to measurement. While testing, 2.5
ml respiration medium: 225 mM mannitol, 70 mM sucrose, 1 mM EDTA,
10 mM potassium phosphate, 0.1% bovine serum albumin (pH 7.4) was
initially agitated with a magnetic stirrer (Hansatech Instruments)
in the sealed chamber. The medium was saturated with oxygen to
reach an oxygenic concentration of ~280 nmol/ml and mitochondrial
suspension (20 μl) was added to the medium. The rate of state IV
respiration is presented as a gently declined curve and was
initiated by adding 20 μl disodium succinate. The rate of state III
respiration was further initiated by the addition of 20 μl
adenosine 5′-diphosphate (ADP). Real-time oxygen concentration was
recorded for the average oxygen consumption rate. An abrupt drop in
oxygen concentration indicated fast phosphorylation of ADP added to
adenosine 5′-triphosphate (ATP). The RCR, an index of integrality
of membranes and the coupling of oxidative phosphorylation in
mitochondria, was calculated as the ratio of the respiratory rate
in state III to that in state IV.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) double marking flow cytometry
According to the manufacturer’s instructions, cells
were rinsed three times with ice-cold PBS and stained with Annexin
V-FITC (Calbiochem-Merck Co., Darmstadt, Germany) for 15 min at
room temperature in 200 μl binding buffer. Subsequent to this, 300
μl binding buffer was added, the cells were stained with propidium
iodide (PI) for 30 min at 4°C and their fluorescence was analyzed
by flow cytometery (Beckman Coulter, Brea, CA, USA). The percentage
of apoptotic cells was determined using WinMDI version 2.9 (Scripps
Research Institute, San Diego, CA, USA).
Transmission electron microscopy
Ventricular cells were fixed by paraformaldehyde and
glutaraldehyde (2:1 in volume), dehydrated in acetone series and
embedded in epon 812. Ultrathin sections (50–70 nm) were sliced and
stained with uranyl acetate and lead acetate and examined under a
transmission electron microscope (Tecnai G2 Spirit Twin; FEI,
Hillsboro, OR, USA).
Detection of caspase-3 activation
Total proteins (20 μg per lane) were separated by
10% SDS-PAGE and electrophoretically transferred onto
polyvinylidene fluoride membranes. The membranes were blocked with
Tris-buffered saline containing 5% non-fat milk for 2 h at room
temperature and incubated with rabbit anti-caspase-3 monoclonal
antibody (1:500) overnight at 4°C. The membranes were washed three
times in 1X Tris-buffer saline with Tween-20 (TBST) and incubated
with anti-IgG antibody conjugated with alkaline phosphatase,
diluted 1:1,000 in TBST for 1 h at room temperature. β-actin
expression was used as the control. The intensities of the protein
bands were quantified by using the image analysis software BandScan
5.0 (Glyko, Hayward, CA, USA).
Detection of cytochrome c release and
BAK/BAX expression
Cytosol and mitochondria fractions were analyzed by
western blot analysis to quantify cytochrome c and BAK/BAX
release. Mitochondria were isolated as described above and lysed
with lysis solution, and supernatants containing cytosol without
mitochondria were also collected. Blots were probed with primary
mouse anti-cytochrome c, BAK and BAX monoclonal antibody,
respectively and then with the secondary anti-IgG antibody. β-actin
expression was used as a loading control for mitochondria, as well
as cytosol fractions.
Statistical analysis
SPSS 13.0 software (SPSS Inc., Chicago, IL, USA) was
used for statistical analysis. Values are expressed as the mean ±
standard error of the mean for normally distributed data and median
(25th, 75th percentile) for non-normally distributed data. All
groups were performed in triplicate culture wells at least three
times. Comparisons among the groups were performed by
Kruskal-Wallis one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
CORM-2 protects rat neonatal cardiac
myocytes against I/R injury
Cells in either the control or CORM-2 groups showed
an increased survival rate as compared with those subjected to I/R
(P<0.01). I/R induced a decrease in cell viability to ~60% of
control as opposed to the cells treated with CORM-2 where the cell
survival was ~76% of the control. There was a significant increase
of LDH activity in the I/R group compared with the control group
(P<0.01). By contrast, CORM-2 reduced the level of LDH by ~45%
(P<0.01, vs. I/R). However, the iCORM-2 group showed no changes
in MTT assay and LDH level (P>0.05, vs. I/R) (Table I).
 | Table IEffects of CORM-2 on cell viability
and LDH activity in cardiomyocytes subjected to I/R injury. |
Table I
Effects of CORM-2 on cell viability
and LDH activity in cardiomyocytes subjected to I/R injury.
| Groups | MTT (optical
density, %) | LDH
(U/l−1) |
|---|
| Control | 92.6±7.3a | 18.6±1.3. |
| I/R | 56.3±8.8b | 55.4±1.8b |
| CORM-2 | 70.3±9.5a | 30.3±2.5a |
| iCORM-2 | 55.8±9.1c | 56.2±2.1c |
CORM-2 inhibits cardiomyocyte apoptosis
induced by I/R
To evaluate the role of CORM-2 in apoptosis induced
by I/R, apoptosis was assayed with a flow cytometer. Annexin
V-FITC/PI double staining showed that the cardiomyocytes underwent
significant apoptosis when exposed to I/R (P<0.01, vs. control)
and CORM-2 decreased the apoptosis rate markedly (P<0.05, vs.
I/R). However, treatment with iCORM-2 did not alter the apoptotic
ratio (P>0.05, vs. I/R) (Fig.
1).
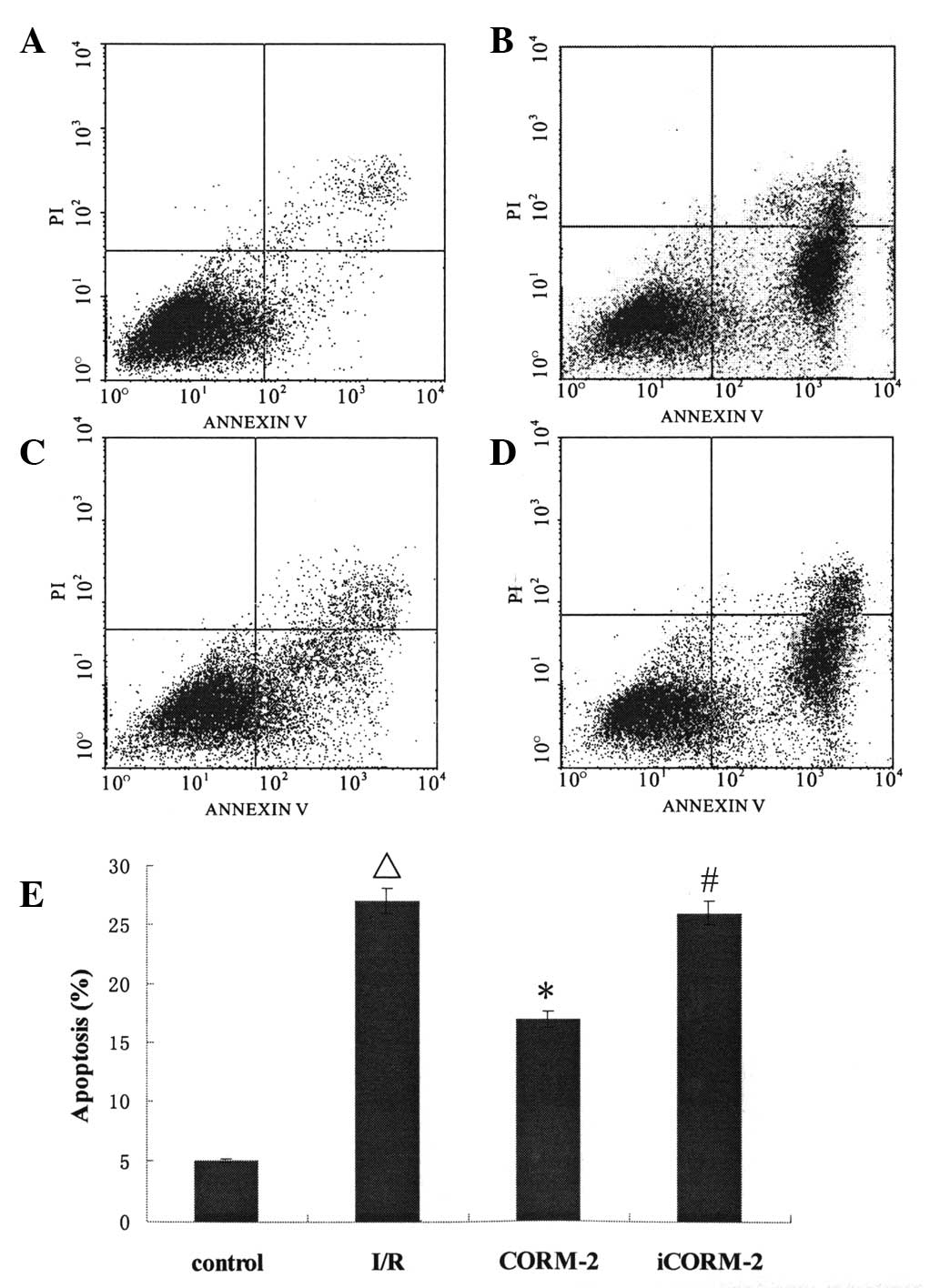 | Figure 1FITC/PI double staining and the
apoptotic rate of cardiomyocytes. (A) Control group: The majority
of the cells emerged at the left lower quadrant, which showed the
cells were viable. (B) I/R group: The quantities of cells in the
right lower quadrant increased compared with that in (A)
(ΔP<0.01), which demonstrated that simulated I/R
triggered apoptosis. (C) CORM-2 group: The quantities of cells in
the right lower quadrant reduced significantly compared with that
in (B) (*P<0.01), suggesting that CORM-2 attenuated
I/R-induced apoptosis. (D) iCORM-2 group: Cell distribution was
similar to that of (B) (#P>0.05). (E) The statistical
data of apoptotic rate. ΔP<0.01, vs. the control
group; *P<0.01, vs. the I/R group;
#P>0.05, vs. the I/R group. FITC, Annexin
V-fluorescein isothiocyanate; PI, propidium iodide; I/R,
ischemia/reperfusion; CORM-2, carbon monoxide releasing molecule-2;
iCORM-2, inactive CORM-2. |
CORM-2 attenuates ultrastructural
deterioration during I/R
Cell apoptosis was measured by transmission electron
microscopy. In the control group, there was no evident
histopathological change. Margination and aggregation of nuclear
chromatin, the swelling of mitochondria, and dissolution and
disappearance of the mitochondrial cristae were observed in the I/R
group. Similar changes were observed when the cells were combined
with iCORM-2 during reperfusion. Notably, the above alterations
were ameliorated in the CORM-2 group (Fig. 2).
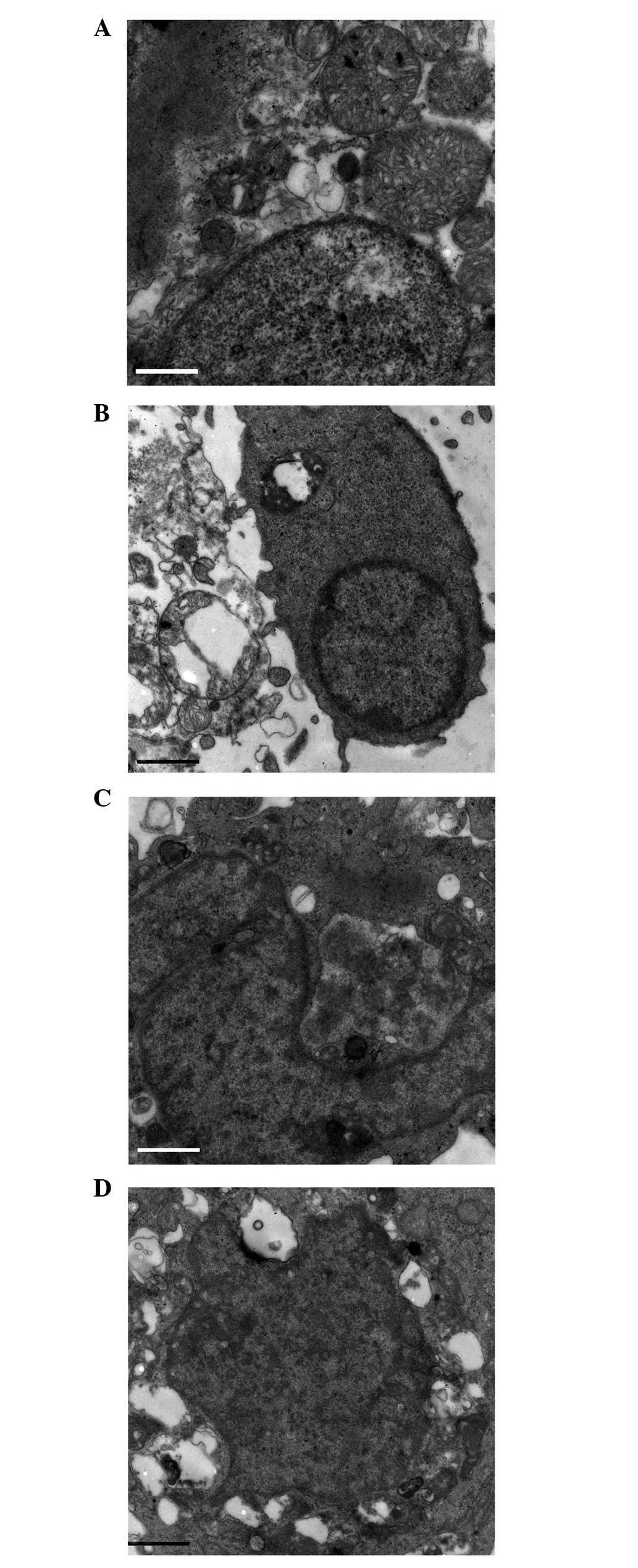 | Figure 2Ultrastructure alteration in
cardiomyocytes. (A) Control, (B) I/R, (C) CORM-2, and (D) iCORM-2
groups (magnification, ×9,700). There was no significant change in
nuclear chromatin, mitochondria and myofilaments in the control
group. In the I/R group, cardiomyocytes were observed in different
degrees of cell shrinkage, nuclear chromatin margination and
karyopyknosis. Mitochondrial turgidity and vacuolization were
observed simultaneously. Similar changes were found in the iCORM-2
group. In the CORM-2 group, local myofilament dissolving,
vacuolization of the mitochondria and disintegrated mitochondrial
cristae were ameliorated to a degree, and the nuclei were almost
normal. I/R, ischemia/reperfusion; CORM-2, carbon monoxide
releasing molecule-2; iCORM-2, inactive CORM-2. |
CORM-2 improves mitochondria respiratory
function
State IV and III respiration in the I/R group
decreased more significantly compared with the control group
(P<0.05), while mitochondrial respiration was partially
normalized by CORM-2 treatment (P<0.05, vs. I/R). In comparison
with the control, the I/R group showed a decreased RCR trend
(P<0.05). CORM-2 treatment further showed a notable recovery of
RCR despite a marked rise in state IV and III respiration
(P<0.05, vs. I/R), indicating that the latter evidently
increased compared with the former. Neither the respiratory rate or
RCR in the iCORM-2 group showed a significant difference when
compared with I/R (P>0.05) (Table
II and Fig. 3).
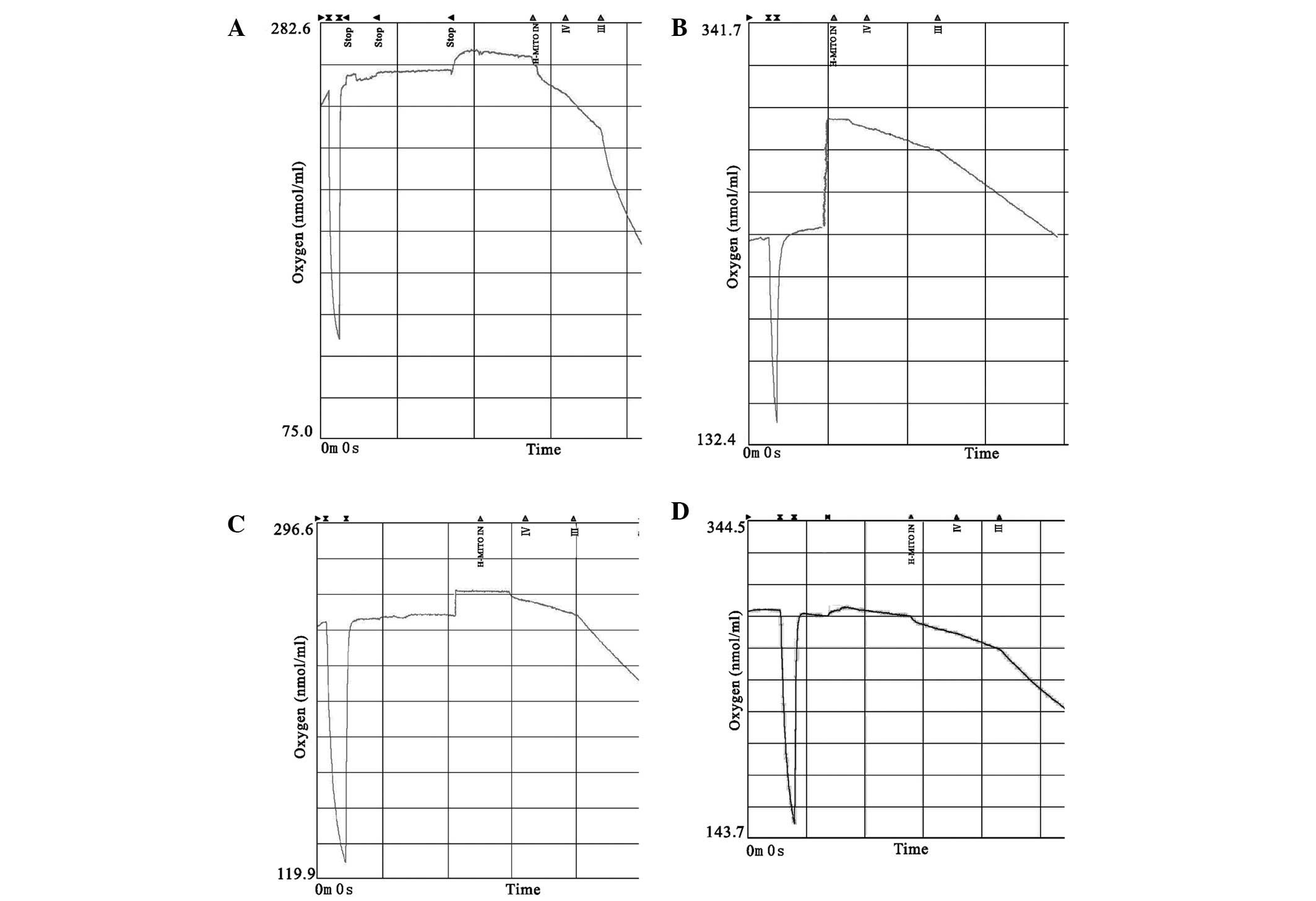 | Figure 3Representative traces of oxygen
consumption of isolated mitochondria by a Clark oxygen electrode.
(A) Control, (B) I/R, (C) CORM-2, and (D) iCORM-2 groups. H-MITO
IN, timestamp of adding mitochondria. III, the rate of state III
respiration (ADP-stimulated) in real time. IV, the rate of state IV
respiration (ADP-limited) in real time. I/R, ischemia/reperfusion;
CORM-2, carbon monoxide releasing molecule-2; iCORM-2, inactive
CORM-2; APD, adenosine 5′-diphosphate. |
| Table IIEffects of CORM-2 on mitochondrial
respiration in cardiomyocytes subjected to I/R injury. |
Table II
Effects of CORM-2 on mitochondrial
respiration in cardiomyocytes subjected to I/R injury.
| Parameter | Control | I/R | CORM-2 | iCORM-2 | P-value |
|---|
| State IV
respiration (nmol of O/min/mg mitochondrial protein) | 35±2.3a | 24±3.1 | 29±3.1a,b | 23±2.9c | <0.05 |
| State III
respiration (nmol of O/min/mg mitochondrial protein) | 221.2±11.2a | 66.7±7.7 | 122±9.8a,b. | 64.4±8.3c | <0.01 |
| RCR | 6.32±0.7a | 2.78±1.1 | 4.2±1.3a,b | 2.8±0.9c | <0.01 |
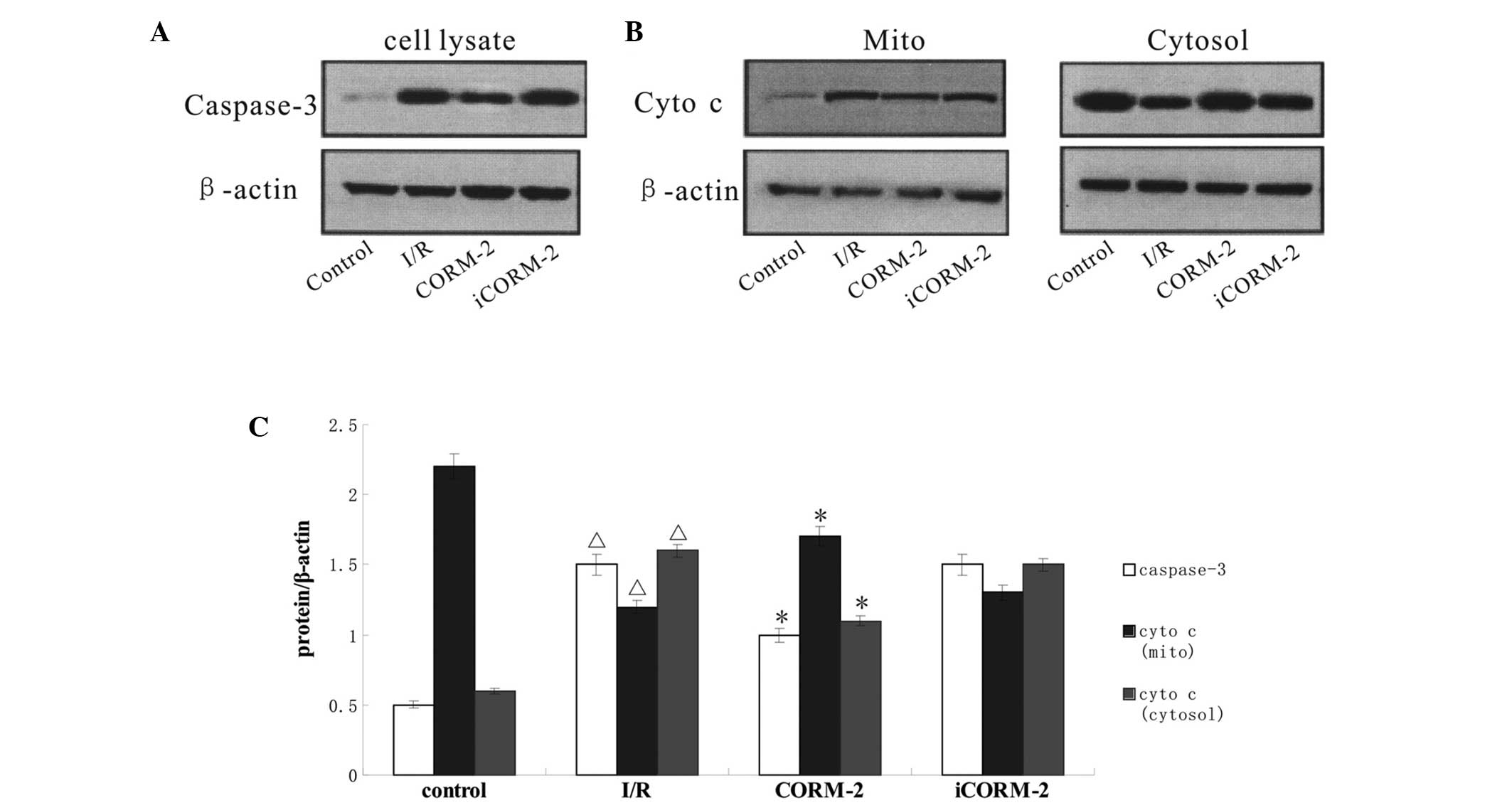
Effect of CORM-2 on the expression of
apoptosis-associated proteins
To further identify the pathway through which CORM-2
suppresses apoptosis, the expression of caspase-3 and the release
of cytochrome c, as well as BAK/BAX translocation were
analyzed by western blot analysis. Compared with the control group,
the expression of caspase-3 and the mitochondrial release of
cytochrome c were markedly increased in the I/R group
(P<0.05) and CORM-2 treatment reversed these increases
(P<0.05, vs. I/R). The cytosol maintained some low-level BAK
content in all groups (P>0.05), while mitochondria contained
medium-level BAK in the control group. The western blot assay
further showed a sharp increase of BAK content evoked by I/R in the
mitochondria (P<0.05, vs. control), where CORM-2 treatment
partially restored the level of BAK (P<0.05, vs. I/R).
Immunoblots in the control group showed the majority of the BAX in
the cytosol, whereas isolated mitochondria showed weakly positive
expression. By contrast, I/R increased the BAX expression level in
the mitochondria and induced a continuous increase in the cytosol
(P<0.05, vs. control). Notably, BAX expression evoked by I/R was
partially inhibited by CORM-2 treatment (P<0.05). iCORM-2 did
not have any effect (P>0.05, vs. I/R) (Figs. 4 and 5).
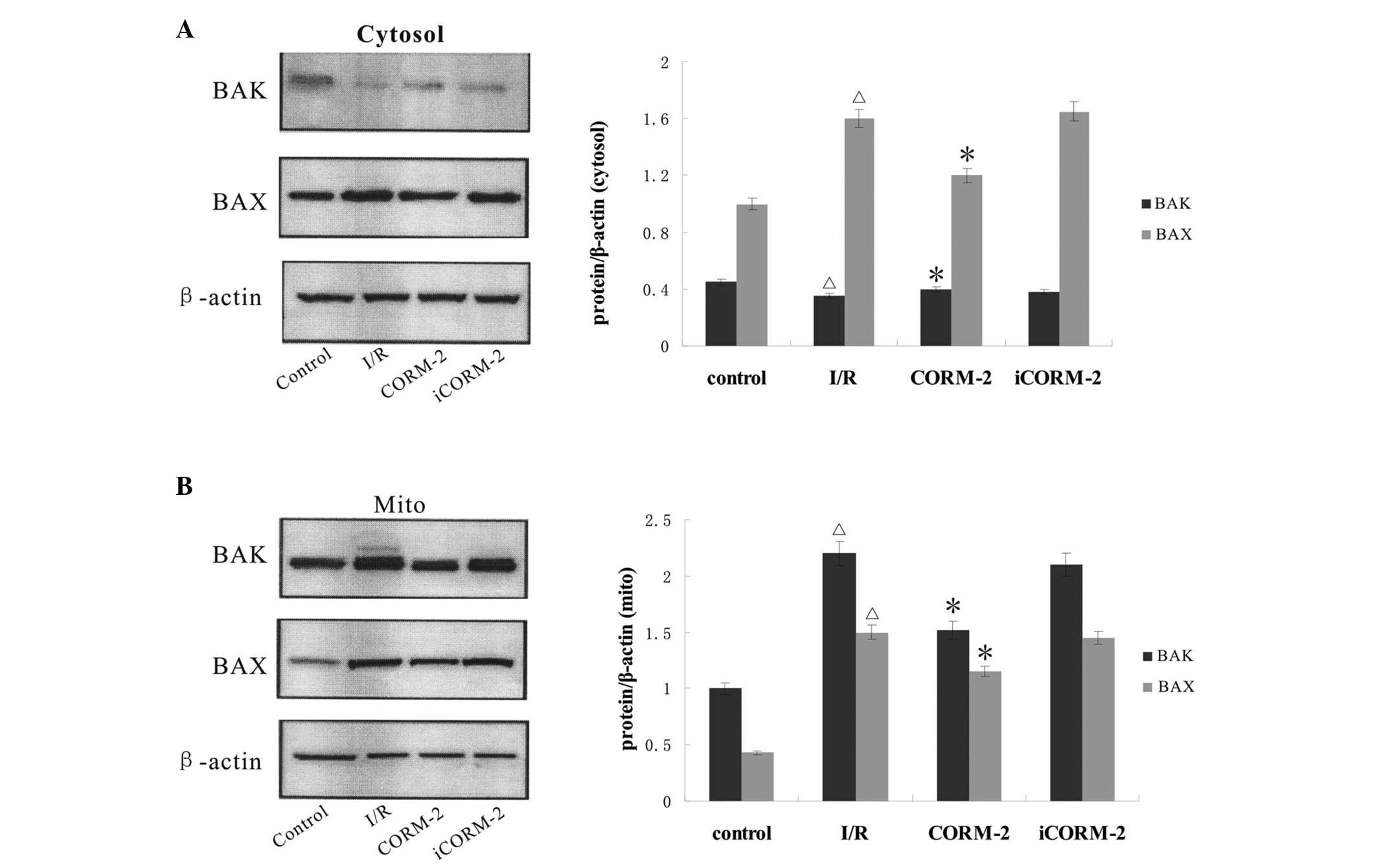 | Figure 5Detection of BAK/BAX expression in
cardiomyocytes by western blot analysis. (A) Immunoblots showed the
presence of BAX and very small amounts of BAK in the cytosol of
control, I/R, CORM-2 and iCORM-2 groups, respectively. (B)
Immunoblots of rapidly separated mitochondria showed that a boost
in BAK/BAX expression suffered from I/R could be reversed by
CORM-2, but not by iCORM-2. Cumulative data of BAK/BAX relative
expression of the cytosol and mitochondria fractions is shown in
the lower panel, respectively. *P<0.05, vs. I/R
group; ΔP<0.05, vs. control. I/R,
ischemia/reperfusion; CORM-2, carbon monoxide releasing molecule-2;
iCORM-2, inactive CORM-2. |
Discussion
The results of five independent assays, biochemical
indexes, Annexin V-FITC/PI double staining using flow-cytometery,
measurement of mitochondrial respiratory rates, the ultrastructure
changes observed by transmission electron microscopy, protein
detection, including BAK/BAX, as well as cytochrome c and
the late apoptotic protease, caspase-3, showed that exogenous CO
released from CORM-2 alleviated I/R-induced apoptosis of primary
cultured myocardial cells from newborn SD rats. The study further
supported the hypothesis that CORM-2 may attenuate myocardial
apoptosis by inhibiting BAK/BAX-mediated intrinsic pathway. Since
iCORM-2, which is chemically identical to CORM-2 but does not
liberate CO, had no effect, the attenuated injury observed in the
CORM-2-treated group must be ascribed to the effect of CO. In
addition, CORM-2 treatment began at the time of reperfusion,
therefore the experimental instructions employed in the present
study have clinical relevance for the treatment of patients with
acute myocardial infarction and cardiac arrest.
Cardiomyocyte apoptosis is a major pathogenesis
during myocardial I/R injury. In the present study, the I/R-induced
cell viability and LDH activity were evaluated, which are
indicators of myocardial membrane injury. Following 2 h ischemia,
followed by 4 h reperfusion, the cell viability was markedly
reduced with LDH levels in the culture medium, 2-fold higher
compared with the control group. These changes were comparable with
flow cytometry data, which indicated myocardial apoptosis.
Furthermore, the marked ultrastructural alterations, as well as
dysfunction of mitochondrial respiration triggered by I/R,
indicated that disrupted energy metabolism and dysregulation of
cell death may target mitochondria. However, CORM-2 treatment
significantly prevented I/R-induced myocardial injury confirmed by
increased viability, reduced apoptosis rate, as well as an
improvement of morphology and respiratory function, suggesting that
CORM-2 may play a positive role in cardioprotection through the
promotion of metabolism and inhibition of mitochondrion-mediated
apoptosis.
To investigate this hypothesis, the expression of
pro-apoptotic molecules BAK and BAX in the mitochondria and
cytoplasm without the mitochondrial part, respectively, was
examined. By contrast with BAX, which is concentrated in the
cytosol in an inactive state and targets to the MOM upon apoptotic
stimuli, monomeric inactive conformer of BAK is detected to
interact specifically with a voltage-dependent anion channel (VDAC)
isoform, VDAC2, within MOM by protein cross-linking
(19). Conditional deletion of BAK
abrogated the increased ionomycin-induced apoptosis of
VDAC2-deficint thymocytes (20). A number of apoptotic agents
activate several BAK/BAX-dependent mechanisms that execute the
permeabilization of mitochondrial membranes (21), which are required for cell death by
mitochondrial or intrinsic pathways. Upon apoptosis, the
‘activator’ BH3-only members, including tBID, BAD, BIM and PUMA,
trigger BAK and BAX to convert from the monomeric form to
homo-oligomers, which mediate the efflux of cytochrome c
from mitochondria and leads to the activation of caspases (22–24).
In the present study, western blot analysis of the cytosolic and
mitochondrial fraction separated at the end of RCR measurement
indicated an increase in BAX expression in the cytosol and
translocation of BAX from the cytosol to the mitochondria following
reperfusion. By contrast, BAK did not alter in the cytosol, but
increased in the mitochondria. The enhanced expression of BAK/BAX
in mitochondria as well increased cytochrome c in the
cytosol suggested that BAK/BAX may be involved in the rupture of
the MOM and may mediate intrinsic apoptosis in triggering I/R. Wei
et al (21) observed
activation and oligomerization of BAK/BAX following tBID treatment,
whereas BAK−/− and BAX−/− MEFs were resistant
to apoptotic death by mitochondria-dependent intrinsic signals. At
present the mechanisms by which BAK and BAX cause the MOM to become
permeable are not well understood. A number of studies suggested
that BAK and BAX regulate mitochondrial morphology machinery to
activate MOM permeabilization (25), form large pores themselves
(26), or change the conformation
of existing pores (27) and may
attribute in part to cytochrome c release and caspase
activation.
Another important finding is that the mitochondrial
respiratory response to simulated ischemia and reperfusion is
involved in decreased respiratory rates of state III
(ADP-stimulated respiration) and IV (non-ADP-stimulated
respiration), as well as depressing RCR, indicating a mitochondrial
proton leak and uncoupling of oxidative phosphorylation. These
findings are consistent with a previous study which demonstrated
that I/R caused a decrease in liver mitochondrial state III
respiration rate, as well as RCR and an increase in state IV
respiration rate in the presence of exogenous cytochrome c,
suggesting the disruption of MOM (28). Previous studies have reported that
a reduction in RCR may be due to no change, or an increase in state
IV respiration, and a decrease in state III respiration (29,30).
The variance of these results may be due to the duration of
ischemia and the degree of reperfusion injury, as well as the
difference between in vitro and in vivo experiments.
It has been also largely accepted that mitochondrial uncoupling
leads to a reduction in mitochondrial ATP synthesis and NADP(H)
content, which were all associated with membrane damage (31). Furthermore, mitochondrial swelling
with membrane permeabilization observed may be partially
responsible for injured mitochondrial respiratory function and
decreased mitochondrial membrane potential.
Increasing evidence has shown the cardioprotective
action of CORMs with transition metal carbonyls as carriers, and
corroborated the hypothesis that CO is a homeostatic and
cytoprotective gaseous mediator required as an adjuvant to
ameliorate apoptotic processes, as well as inflammation and
oxidative stress (32). The
mechanisms underlying CO-induced cardioprotection remain to be
elucidated, despite CO holding beneficial bioactive properties on a
number of tissues. Recent investigations have implicated the
mitochondria as a prime target for cytoprotection and therapy of CO
(11). The present results
indicated that CORM-2 may inhibit simulated I/R-induced apoptosis
via a BAK/BAX-mediated intrinsic pathway. CO has been found to
promote the anti-apoptotic protein BCL-2 expression in lung
ischemia models (33). However, it
was worth noting in the study that the anti-apoptosis effect of CO
may be partly attributed to the inhibition of pro-apoptotic BAK/BAX
protein expression. Another direct method for assessing the effect
of CORM-2 on mitochondria is cellular oxygen consumption. CORM-2,
at a low concentration, was observed to increase respiratory rates
of state III and IV, as well as RCR with an ~20% reduction from
normal level. CO released by CORM-2 is hypothesized to partially
reduce BAK/BAX content in the mitochondria, which contributes to
maintaining MOM integrity and transmembrane potential, as well as
the tightness of coupling between respiration and phosphorylation.
This is in agreement with a previous study where treatment with
CORMs during kidney cold storage increased mitochondrial
respiratory control index at reperfusion (34). However, the results of associated
studies on mitochondrial respiration in response to CO have been
inconsistent. Astrocytes isolated from the cortex presented an
increase of oxygen consumption at 36 h following one brief exposure
to CO (12). Different
experimental conditions, including early or late response and
concentration or period of exposure may alter the mitochondrial
coupling state and oxidative metabolism. Other investigators have
claimed that CO biological function was associated with
mitochondrial mild uncoupling state, which decreased cell
respiration, as well as excessive and toxic mitochondrial reactive
oxygen species production. Inhibition of uncoupling proteins (UCP)
or blockade of adenine nucleotide transporter (ANT) attenuated the
effect, indicating that CO may open UCP and/or ANT at a mild
uncoupling state (35).
It should be noted that the current study may have a
number of limitations. Firstly, CORM-2 did not fully recover the
mitochondrial respiration in the study, thus further experiments
are required to observe whether it maintained the electron
transport chain in a mild uncoupling state or not. Secondly, it may
not directly prove the association between impaired mitochondrial
respiration and BAK/BAX activation, thus, more convincing evidence
is required to support the hypothesis. Finally, further
dose-dependent anti-apoptotic properties of CORM-2, as well as its
side effects should be evaluated in in vitro or in
vivo experiments, although such a dose of CORM-2 appeared to be
effective and safe in this study.
In conclusion, the present study suggested that
exogenous CO released from CORM-2 may protect cardiomyocytes from
I/R-induced apoptosis. It may be associated with the inhibition of
a BAK/BAX-dependent mitochondrial pathway and the improvement of
energy metabolism.
Acknowledgements
The authors would like to thank Jinlang Wu
(Department of Electron Microscopy, Sun Yat-sen University) for his
valuable assistance with the experimental technique and image
analysis.
References
|
1
|
Bialik S, Cryns VL, Drincic A, Miyata S,
Wollowick AL, Srinivasan A and Kitsis RN: The mitochondrial
apoptotic pathway is activated by serum and glucose deprivation in
cardiac myocytes. Circ Res. 85:403–414. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Radhakrishnan J, Wang S, Ayoub IM,
Kolarova JD, Levine RF and Gazmuri RJ: Circulating levels of
cytochrome c after resuscitation from cardiac arrest: a
marker of mitochondrial injury and predictor of survival. Am J
Physiol Heart Circ Physiol. 292:H767–H775. 2007.PubMed/NCBI
|
|
3
|
Li H, Fang X, Yang Z, et al: Ischemia
hypothermia improved contractility under normothermia reperfusion
in the model of cultured cardiomyocyte. In Vitro Cell Dev Biol
Anim. 48:284–292. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Lisa F, Canton M, Menabò R, Kaludercic
N and Bernardi P: Mitochondria and cardioprotection. Heart Fail
Rev. 12:249–260. 2007.
|
|
5
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chipuk JE and Green DR: How do BCL-2
proteins induce mitochondrial outer membrane permeabilization?
Trends Cell Biol. 18:157–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kubli DA, Ycaza JE and Gustafsson AB:
Bnip3 mediates mitochondrial dysfunction and cell death through Bax
and Bak. Biochem J. 405:407–415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ren D, Tu HC, Kim H, et al: BID, BIM, and
PUMA are essential for activation of the BAX- and BAK-dependent
cell death program. Science. 330:1390–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roy SS, Ehrlich AM, Craigen WJ and
Hajnóczky G: VDAC2 is required for truncated BID-induced
mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO
Rep. 10:1341–1347. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Motterlini R and Otterbein LE: The
therapeutic potential of carbon monoxide. Nat Rev Drug Discov.
9:728–743. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Queiroga CS, Almeida AS and Vieira HL:
Carbon monoxide targeting mitochondria. Biochem Res Int.
2012:7498452012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Almeida AS, Queiroga CS, Sousa MF, Alves
PM and Vieira HL: Carbon monoxide modulates apoptosis by
reinforcing oxidative metabolism in astrocytes: role of Bcl-2. J
Biol Chem. 287:10761–10770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Conde de la Rosa L, Vrenken TE, Hannivoort
RA, et al: Carbon monoxide blocks oxidative stress-induced
hepatocyte apoptosis via inhibition of the p54 JNK isoform. Free
Radic Biol Med. 44:1323–1333. 2008.PubMed/NCBI
|
|
14
|
Bannenberg GL and Vieira HL: Therapeutic
applications of the gaseous mediators carbon monoxide and hydrogen
sulfide. Expert Opin Ther Pat. 19:663–682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Motterlini R: Carbon monoxide-releasing
molecules (CO-RMs): vasodilatory, anti-ischaemic and
anti-inflammatory activities. Biochem Soc Trans. 35:1142–1146.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katada K, Bihari A, Mizuguchi S, et al:
Carbon monoxide liberated from CO-releasing molecule (CORM-2)
attenuates ischemia/reperfusion (I/R)-induced inflammation in the
small intestine. Inflammation. 33:92–100. 2010. View Article : Google Scholar
|
|
17
|
Caumartin Y, Stephen J, Deng JP, et al:
Carbon monoxide-releasing molecules protect against
ischemia-reperfusion injury during kidney transplantation. Kidney
Int. 79:1080–1089. 2011. View Article : Google Scholar
|
|
18
|
Gomez LA, Alekseev AE, Aleksandrova LA,
Brady PA and Terzic A: Use of the MTT assay in adult ventricular
cardiomyocytes to assess viability: effects of adenosine and
potassium on cellular survival. J Mol Cell Cardiol. 29:1255–1266.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ
and Korsmeyer SJ: VDAC2 inhibits BAK activation and mitochondrial
apoptosis. Science. 301:513–517. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren D, Kim H, Tu HC, et al: The VDAC2-BAK
rheostat controls thymocyte survival. Sci Signal.
2:ra482009.PubMed/NCBI
|
|
21
|
Wei MC, Zong WX, Cheng EH, et al:
Proapoptotic BAX and BAK: a requisite gateway to mitochondrial
dysfunction and death. Science. 292:727–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei MC, Lindsten T, Mootha VK, et al:
tBID, a membrane-targeted death ligand, oligomerizes BAK to release
cytochrome c. Genes Dev. 14:2060–2071. 2000.PubMed/NCBI
|
|
23
|
Cheng EH, Wei MC, Weiler S, Flavell RA,
Mak TW, Lindsten T and Korsmeyer SJ: BCL-2, BCL-X(L) sequester BH3
domain-only molecules preventing BAX- and BAK-mediated
mitochondrial apoptosis. Mol Cell. 8:705–711. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers
JR, Zambetti GP, Hsieh JJ and Cheng EH: Hierarchical regulation of
mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell
Biol. 8:1348–1358. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karbowski M, Norris KL, Cleland MM, Jeong
SY and Youle RJ: Role of Bax and Bak in mitochondrial
morphogenesis. Nature. 443:658–662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shoshan-Barmatz V, Keinan N and Zaid H:
Uncovering the role of VDAC in the regulation of cell life and
death. J Bioenerg Biomembr. 40:183–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trumbeckaite S, Kincius M, Preidis A,
Preidiene M, Veikutis V, Borutaite V and Gulbinas A: Effects of
ischemia-reperfusion and pretreatment with mildronate on rat liver
mitochondrial function. Pharmacol Rep. 61:859–869. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Venditti P, Masullo P and Di Meo S:
Effects of myocardial ischemia and reperfusion on mitochondrial
function and susceptibility to oxidative stress. Cell Mol Life Sci.
58:1528–1537. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Q, Moghaddas S, Hoppel CL and
Lesnefsky EJ: Reversible blockade of electron transport during
ischemia protects mitochondria and decreases myocardial injury
following reperfusion. J Pharmacol Exp Ther. 319:1405–1412. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elimadi A, Settaf A, Morin D, Sapena R,
Lamchouri F, Cherrah Y and Tillement JP: Trimetazidine counteracts
the hepatic injury associated with ischemia-reperfusion by
preserving mitochondrial function. J Pharmacol Exp Ther. 286:23–28.
1998.PubMed/NCBI
|
|
32
|
Clark JE, Naughton P, Shurey S, et al:
Cardioprotective actions by a water-soluble carbon
monoxide-releasing molecule. Circ Res. 93:e2–e8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Shan P, Alam J, Davis RJ, Flavell
RA and Lee PJ: Carbon monoxide modulates Fas/Fas ligand, caspases,
and Bcl-2 family proteins via the p38alpha mitogen-activated
protein kinase pathway during ischemia-reperfusion lung injury. J
Biol Chem. 278:22061–22070. 2003. View Article : Google Scholar
|
|
34
|
Sandouka A, Fuller BJ, Mann BE, Green CJ,
Foresti R and Motterlini R: Treatment with CO-RMs during cold
storage improves renal function at reperfusion. Kidney Int.
69:239–247. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lo Iacono L, Boczkowski J, Zini R,
Salouage I, Berdeaux A, Motterlini R and Morin D: A carbon
monoxide-releasing molecule (CORM-3) uncouples mitochondrial
respiration and modulates the production of reactive oxygen
species. Free Radic Biol Med. 50:1556–1564. 2011.PubMed/NCBI
|