Introduction
Breast cancer is the most frequently diagnosed type
of cancer and the leading cause of cancer-related mortality in
females. It accounts for ~23% of total cancer cases worldwide
(1). Metastatic breast cancer
often occurs several years after primary breast cancer resection
and it is responsible for the majority of breast cancer-related
fatalities (2). Until recently,
the prognosis of breast cancer and the understanding of the
pathogenesis of this disease have been relatively poor.
Aberrant activation of the WNT signaling pathway is
vital in the development of numerous types of human cancer and the
canonical WNT/β-catenin pathway participates in several biological
processes, such as cell proliferation and differentiation (3). The secreted WNT ligands stimulate
intracellular signaling transduction, which leads to the
stabilization and nuclear translocation of the key effector
β-catenin. β-catenin then binds to lymphoid enhancer-binding
factor-1/T-cell factor (TCF) and activates target gene
transcription (3,4). Deregulation of the WNT pathway
frequently occurs in multiple types of cancer and underlies
hereditary syndromes (5).
Mutations in adenomatous polyposis coli (APC), axin or β-catenin
activate the deregulated WNT signaling pathway and promote the
transcription of target genes encoding colorectal cancer-associated
proteins (6). However, WNT pathway
mutations are rarely detected in breast cancer (7), and deregulation of WNT signaling
occurs by autocrine mechanisms (8,9).
Autocrine WNT signaling has been proposed to promote breast cancer
cell motility and proliferation via the canonical WNT pathway and
epidermal growth factor receptor transactivation (9,10).
β-catenin is a central regulator of the WNT
signaling pathway (11) and it can
also act as a co-activator through interaction with other factors
to regulate downstream gene expression (12). Furthermore, the epigenetic
modification of β-catenin is an important mode of regulation of
β-catenin stability and cellular location, as well as its
transcriptional activity (13,14).
Without WNT stimulation, glycogen synthase kinase 3 phosphorylates
β-catenin and triggers its ubiquitination-depedent degradation
(15–17). β-catenin has also been found to be
acetylated by acetyltransferase CREB-binding protein/p300 at
different residues (K49 and K345) (18,19),
and the deacetylation of β-catenin by HDAC6 is involved in the
interferon regulatory factor 3 (IRF3) signaling pathway (20). However, the association between
epigenetic modification of β-catenin and autocrine WNT signaling is
unclear in breast cancer.
In the present study, the protein level of
acetylated β-catenin was found to be lower in breast cancer tissues
compared with the adjacent normal tissues. In the MCF7 breast
cancer cell line, WNT downregulated the acetylation level of
β-catenin and enhanced the proliferation of breast cancer cells,
which was inhibited by the K345Q mutation in β-catenin. Conversely,
the K345R deacetylation mimic mutant exhibited an increased
proliferation rate of breast cancer cells. WNT-induced axin2 and
TCF7 upregulation was also influenced by the deacetylation of
β-catenin. Further investigation revealed that HDAC6 may be
responsible for this process. These results provide novel insights
for breast cancer therapy through targeting β-catenin deacetylation
to regulate WNT pathway activity.
Materials and methods
Samples and cell culture
Five samples of breast cancer tissue and the
adjacent normal tissue were obtained at the time of surgery from
five patients with breast cancer who were treated at the Department
of Surgery, Nanjing Jinling Hospital (Nanjing, China). MCF7 cells
were maintained in RPMI-1640 medium (Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal calf serum.
Cell fractionation
WNT3A-stimulated MCF7 cells were washed and
collected with cold phosphate-buffered saline (PBS). Cell pellets
were then resuspended in hypotonic lysis buffer [10 mM KCl, 10 mM
Tris (pH 7.5) and 2 mM EDTA] containing protease inhibitor and
phosphatase inhibitor cocktail tablets (Roche Applied Science,
Indianapolis, IN, USA) and histone deacetylase inhibitor cocktail
(including trichostatin A, negative allosteric modulator,
suberoylanilide hydroxamic acid and MS275). Cell suspensions were
incubated on ice for 30 min. Nuclear proteins, including the
unlysed cells, were pelleted by centrifugation at 400 × g for 2 min
at 4ºC. The supernatant that contained cytoplasm and membrane
proteins was then centrifuged at 17,500 × g for 30 min at 4ºC. The
supernatant was collected and analyzed by western blotting.
Anti-Phospho-β-catenin S33/S37/T41 (1:1,000; Cell Signaling
Technology, Inc., Beverly, MA, USA), anti-Ace-β-catenin K345
antibody (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), anti-Ace-β-catenin K49 antibody (1:400; Cell Signaling
Technology, Inc.), anti-β-catenin (1:1,000; BD Transduction
Laboratories, San Jose, CA, USA) and anti-GAPDH (1:10,000; Abcam,
Cambridge, MA, USA) were used for immunoprecipitation or western
blot analysis.
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA was isolated from MCF7 cells with TRIzol
(Invitrogen Life Technologies). Total RNA (2.5 μg) was used for
cDNA synthesis using Superscript II (Invitrogen Life Technologies),
according to the manufacturer’s instructions. Each PCR cycle was
conducted in a volume of 25 μl reaction mixture (HotStarTaq Master
mix; Qiagen, Hilden, Germany). The reverse-transcribed cDNA
products were directly analyzed by qPCR using SYBR-Green (Sigma,
Oakville, ON, Canada) as a label. The primers were used as follows:
Sense: 5′-GAAGGTGAAGGTCGGAGT-3′ and antisense:
5′-GAAGATGGTGATGGGATTTC-3′ for GAPDH; sense:
5′-TACACTCCTTATTGGGCGATCA-3′ and antisense:
5′-TTGGCTACTCGTAAAGTTTTGGT-3′ for axin2; and sense:
5′-TGGAGGGCTCTTTAAGGGG-3′ and antisense: 5′-GATCCGTTGGGGAGGTAGG-3′
for TCF7.
In vitro tumor cell growth assay
The in vitro growth of MCF7 cells was
determined by measuring increases in cell number using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
(Promega Corporation, Madison, WI, USA) according to the
manufacturer’s instructions. Three independent experiments were
performed. Cell-mediated reaction products were then recorded by a
enzyme-linked immunosorbent assay reader (SpectraMax 250; Molecular
Devices, Sunnyvale, CA, USA) at a wavelength of 450 nm.
Immunoprecipitation
At 80% confluence, MCF7 cells were treated with
either WNT3A or control-conditioned medium as indicated. Cells were
washed and collected with cold PBS, lysed in cold lysis buffer
containing 150 mM NaCl, 30 mM Tris (pH 7.5), 1 mM EDTA, 1% Triton
X-100, 10% glycerol, 0.1 mM phenylmethylsulfonyl fluoride, 0.5 mM
dithiothreitol, protease inhibitor cocktail tablets (EDTA-free) and
phosphatase inhibitor cocktail tablets (both from Roche Applied
Science, Indianapolis, IN, USA) and histone deacetylase inhibitor
cocktail (including TSA, NAM, SAHA and MS275). Following separation
by centrifugation (14,000 rpm for 30 min at 4ºC), the cellular
lysates were precleared with IgG-agarose beads (Sigma, St. Louis,
MO, USA) for at least 6 h at 4ºC. Immunoprecipitation of endogenous
β-catenin was conducted by incubating the cellular lysates with
anti-β-catenin antibody, using mouse IgG (BD Transductions
Laboratories, Lexington, KY, USA) as a blank control.
Immunoproteins were washed with cold lysis buffer six times,
resuspended in 2X SDS sample buffer, and subjected to SDS-PAGE and
western blot analysis.
HDAC6 knockdown and transfection
The synthesized HDAC6 and control (scramble) small
hairpin RNAs (shRNAs) were cloned into pSuper vectors (Oligoengine,
Seattle, WA, USA). The HDAC6 knockdown efficiency was determined by
western blot analysis. HDAC6 shRNAs sequences were shown as
follows: 5′-TACAACAGCC ACAACGTCTAT-3′ for control shRNA; and
5′-CATCCCATC CTGAATATCCTT-3′ for HDAC6 shRNA. Plasmids were
transfected with Lipofectamine 2000 (Invitrogen Life Technologies)
into MCF7 cells at 50% confluency.
Statistical analysis
Each experiment was performed at least three times.
Data are presented as the mean ± standard deviation. Statistical
significance was determined by Student’s t-test and P<0.05 was
considered to indicate a statistically significant difference.
Results
β-catenin acetylation is downregulated in
breast cancer tissues
Aberrant WNT signaling is activated in breast cancer
in an autocrine manner to regulate breast cancer cell proliferation
(9). To investigate the
association between the epigenetic modification of β-catenin and
breast cancer, the protein level of acetylated β-catenin was
analyzed by western blotting in five samples of breast cancer
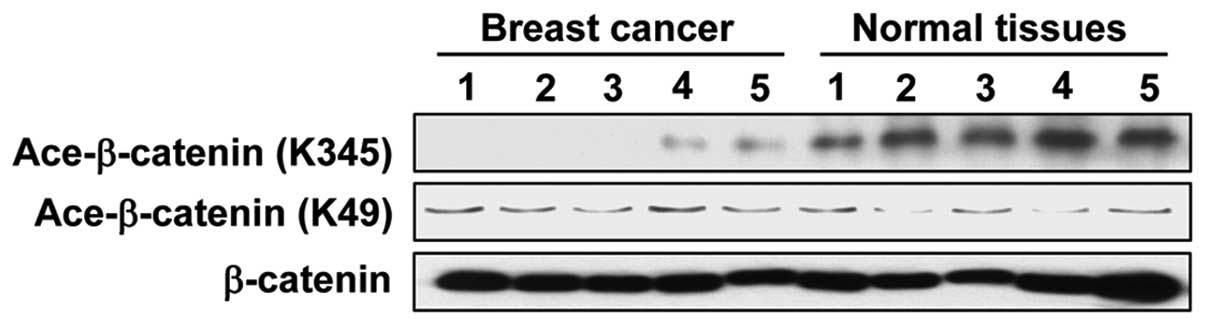
tissue and the corresponding adjacent normal tissue (Fig. 1). The results showed that the level
of acetylated β-catenin at the K345 site in the breast cancer
tissues was markedly lower than in normal tissues. Notably, K49
acetylated β-catenin was maintained at a relatively stable level.
This suggests that the modification of β-catenin at the K345 site
may be associated with the development of breast cancer.
WNT signaling decreases the β-catenin
acetylation level and promotes breast cancer cell
proliferation
The acetylation level of β-catenin is downregulated
in breast cancer, while the association between β-catenin
deacetylation and autocrine WNT signaling remains unknown. To
explore this question, the MCF7 breast cancer cell line was used as
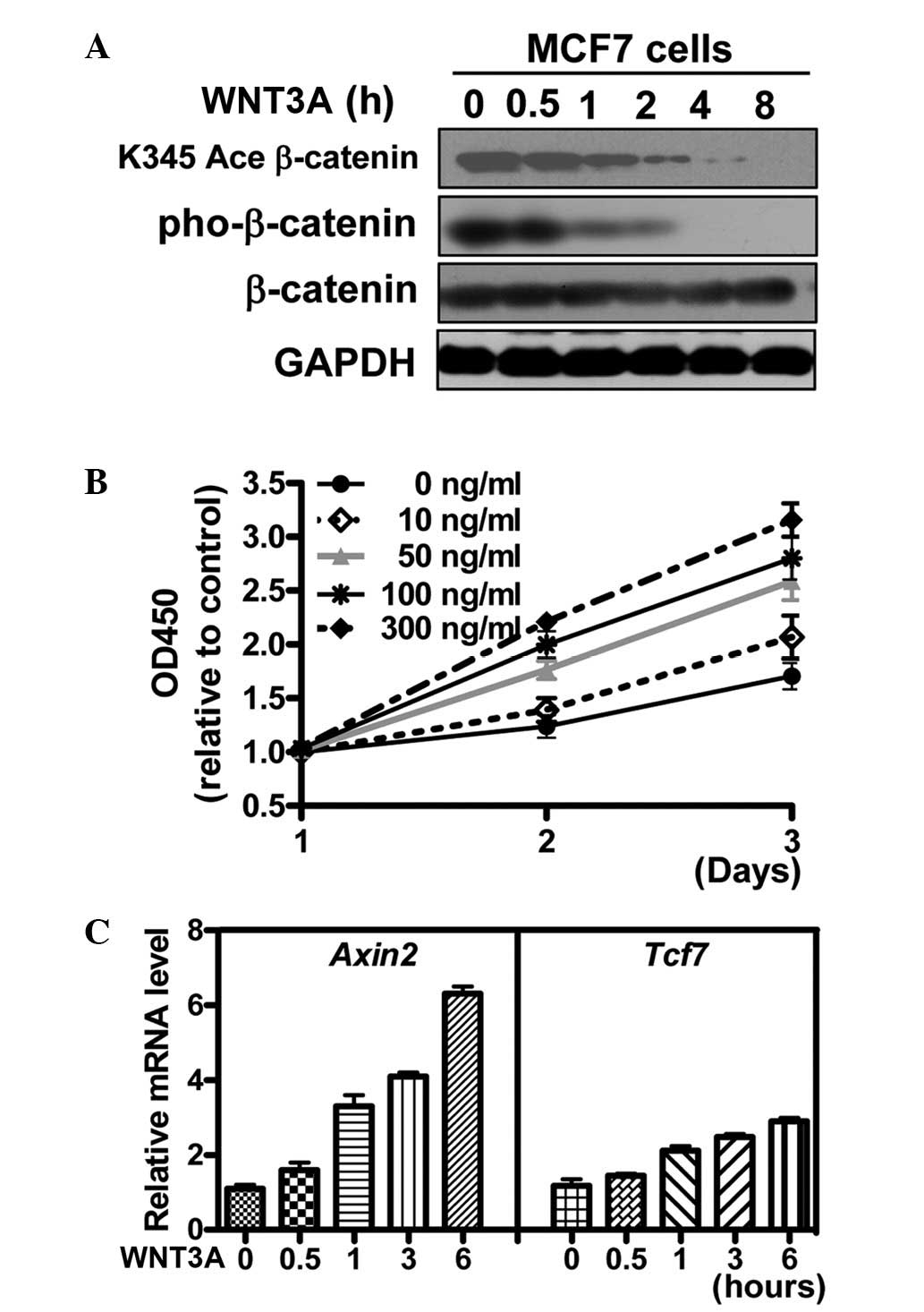
an in vitro model. In MCF7 cells, WNT3A treatment
downregulated the acetylation level of β-catenin (K345) in a
time-dependent manner (Fig. 2A).
The proliferation rate of MCF7 cells was futher determined under
the stimulation of WNT3A and it was found that WNT promoted the
proliferation of the breast cancer cells in a dose-dependent manner
(Fig. 2B), which is consistent
with a previous study (9). The
expression level of WNT target genes axin2 and TCF7 (21,22)
was examined by qPCR and the results showed that the mRNA level was
markedly elevated by WNT treatment (Fig. 2C). These observations suggest that
WNT-induced β-catenin deacetylation and cell proliferation may be
associated.
K345 mutants affect the growth of breast
cancer cells by influencing β-catenin phosphorylation
β-catenin acetylation has been proposed as a novel
mechanism for the regulation of WNT/β-catenin transcriptional
activity (19); therefore the
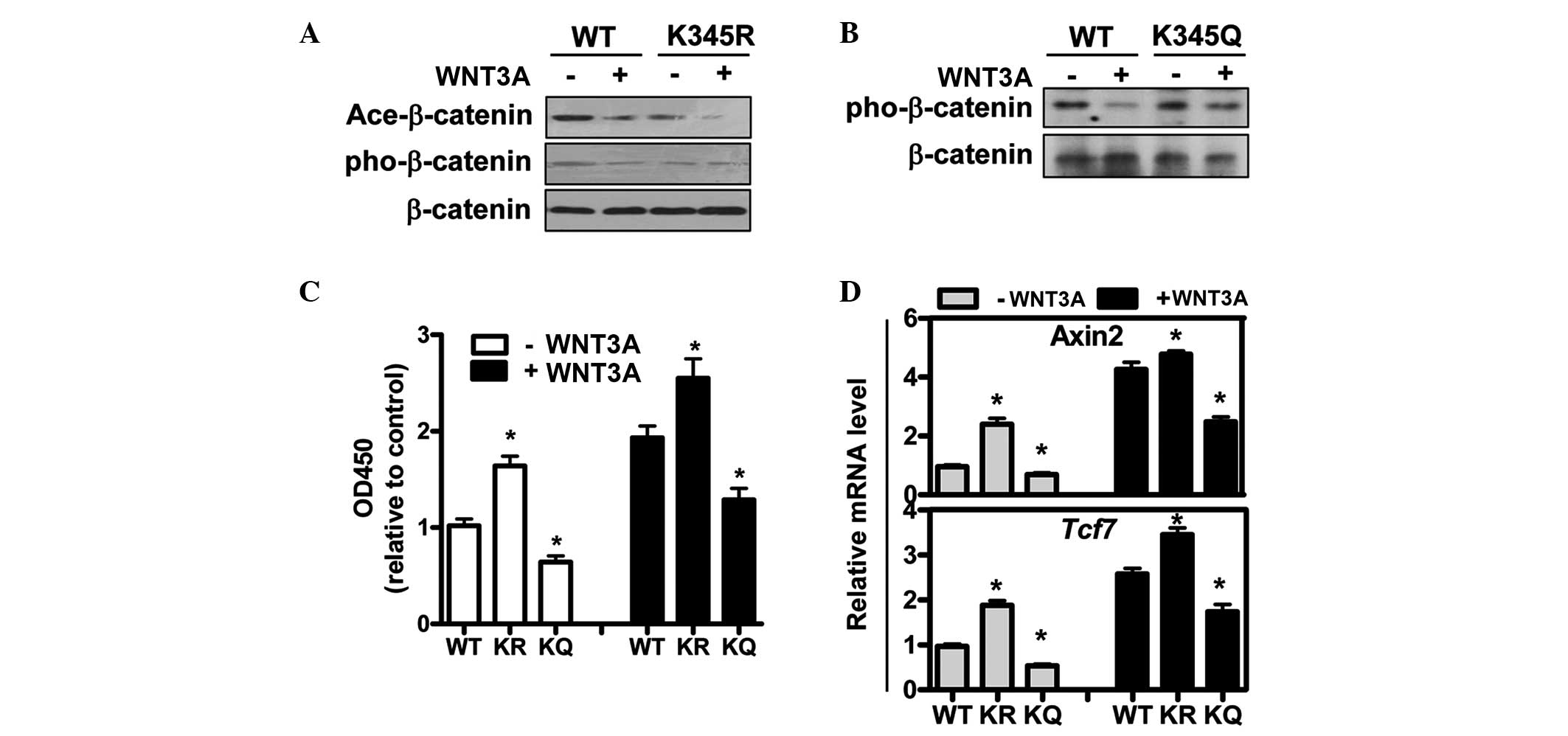
effects of K345 mutation on the epigenetic modification of
β-catenin in 293T cells, in which the transfected β-catenin can be
normally acetylated, were examined (Fig. 3A). Two β-catenin mutants (K345R and
K345Q), utilized to mimic acetylation modification, were
transfected into the MCF7 cells. K345R mutants inhibited the
acetylation of β-catenin and impaired WNT-induced downregulation of
β-catenin phosphorylation in 293T cells (Fig. 3A). Conversely, the K345Q mutation,
which could mimic the acetylation modification, promoted the
phosphorylation of β-catenin even in the presence of WNT3A
(Fig. 3B). These results
demonstrated that the acetylation at residue K345 may be the
triggering signal for β-catenin phosphorylation, which can be used
to mark the molecule for subsequent degradation.
It was assessed whether the K345 mutation in
β-catenin affects WNT-induced cell proliferation and WNT responsive
gene expression in MCF7 cells. The deacetylation mutation K345R
promoted the proliferation of the breast cancer cells (Fig. 3C), similar to the effects of WNT3A
(Fig. 2B). The acetylation mimic
mutant K345Q suppressed the growth of MCF7 cells and also impaired
the promotional effects of WNT3A on the proliferation rate of
breast cancer cells (Fig. 3C). In
addition, the expression of WNT-responsive genes axin2 and MCF7 was
upregulated in K345R mutants and decreased in K345Q mutants
(Fig. 3D). It was hypothesized
that β-catenin undergoes acetylation and phosphorylation as well as
further degradation without WNT stimulation, and that WNT pathway
activation could promote the deacetylation of β-catenin and the
downregulation of phosphorylation, and subsequently activate the
canonical WNT/β-catenin pathway.
HDAC6 is responsible for the WNT-induced
decrease in β-catenin acetylation in breast cancer cells
Previously, HDAC6 has been proposed as a requirement
for β-catenin deacetylation on Lys-49 and its nuclear translocation
in the epidermal growth factor receptor or the interferon
regulatory transcription factor 3 signaling pathway (20,23).
However, it is unknown whether the deacetylation of β-catenin on
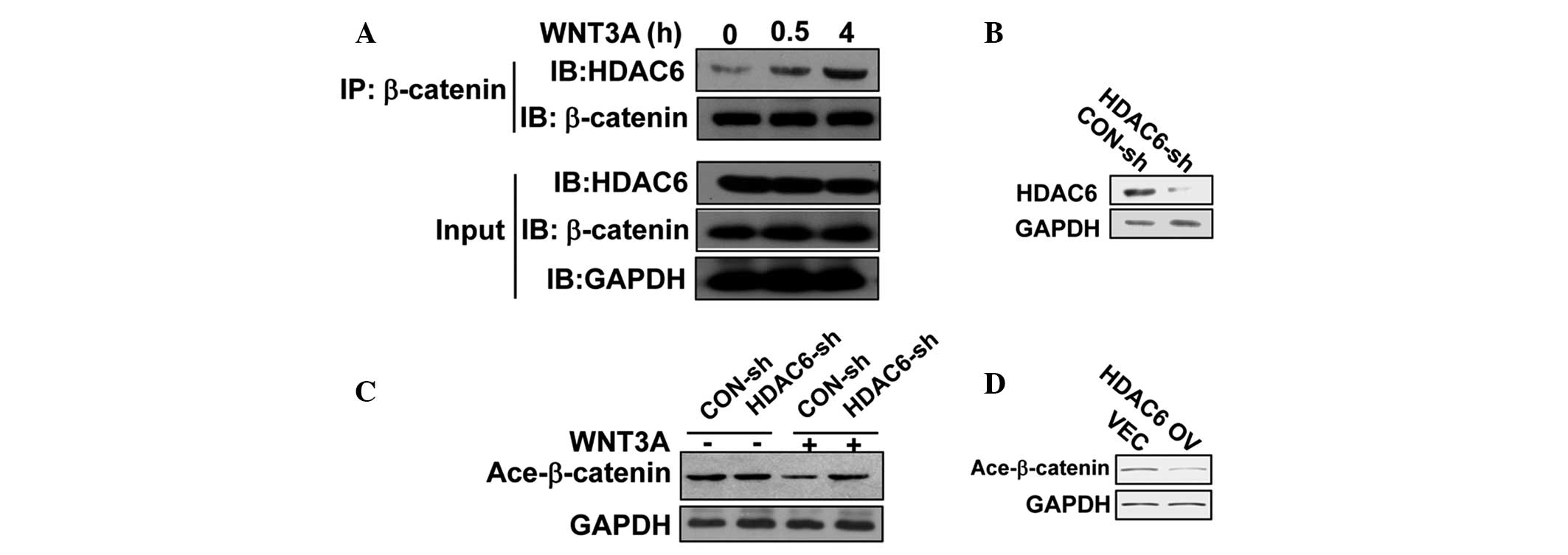
Lys-345 is also exerted by HDAC6. Thus an immunoprecipitation assay
was performed using an anti-β-catenin antibody and a strong
interaction between endogeous HDAC6 and β-catenin under the
stimulation of WNT3A was observed, indicating that the two proteins
were present in a complex (Fig.
4A). In order to establish the link between HDAC6 and
β-catenin, HDAC6 shRNA was delivered into MCF7 cells, which
efficiently knocked down the expression of HDAC6 at the protein
level (Fig. 4B). Without WNT
treatment, HDAC6 knockdown marginally enhanced the acetylation
level of β-catenin (K345). However, WNT-induced downregulation of
the acetylated β-catenin level was significantly rescued by HDAC6
shRNA (Fig. 4C), suggesting that
HDAC6 affects the acetylation level of β-catenin. To further
confirm this, HDAC6 was overexpressed in MCF7 cells, which resulted
in the reduction of the acetylated β-catenin level (Fig. 4D). In conclusion, HDAC6 is
sufficient and required for the deacetylation of β-catenin in
breast cancer cells.
Discussion
Aberrant activation of WNT signaling frequently
occurs in numerous types of human cancer and targeting this pathway
may be a promising therapeutic approach (24). The deregulation of the WNT
signaling pathway can occur by different mechanisms, including
APC/β-catenin mutations in colorectal cancer and autocrine WNT
signaling in breast cancer (8,9,25).
Protein post-translational modifications have also been reported in
human cancers. Previously, the global loss of monoacetylation and
trimethylation of histone H4 has been established as a common
hallmark of human cancer cells (26), demonstrating that cancer
progression is associated with deregulation of biological events at
the transcriptional level as well as the post-translational level.
In the present study, it was demonstrated that the protein level of
acetylated β-catenin (K345) is markedly lower in breast cancer
tissues than in the adjacent normal tissues (Fig. 1). It is possible that β-catenin is
deacetylated during the progression of breast cancer. Although the
quantity of specimens used in the present study was limited, the
data indicate that the epigenetic modification of β-catenin may be
correlated with breast cancer.
Autocrine WNT activity in human breast cancer cells
with diverse genetic alterations has been identified in a previous
study (9). In the present study,
the possibility that the downregulation of acetylated β-catenin
level is caused by WNT ligands was analyzed in MCF7 cells. As
expected, WNT3A stimulation leads to a reduction in the acetylated
and phosphorylated β-catenin levels. Consistent with previous
findings (9), WNT signaling
promoted the proliferation capacity of breast cancer cells in a
dose-dependent manner. In addition, the expression of two important
WNT downstream targets, axin2 and TCF7, was markedly elevated under
the WNT3A treatment (Fig. 2),
which may result in autocrine-activated WNT signaling in the
nucleus to promote cell proliferation in breast cancer.
In the present study, the alterations of
residue-specific acetylation of β-catenin in breast cancer cells
was notable. The level of acetylated β-catenin at K345 was
decreased in cancer tissues whereas K49-acetylated β-catenin was
expressed at a relatively stable level. Previous studies show that
the mutation of β-catenin at K49 neither impaired the
WNT-responsive reporter TOPFLASH transactivation nor modulated the
interaction of β-catenin with TCF (18,19).
Conversely, the K345A mutation abolished the interaction between
β-catenin and TCF4, and K345 mutations affected their ability to
transactivate the TOPFLASH reporter (19).
To explore the functional association between
β-catenin acetylation and WNT signaling in breast cancer, the
effects of K345 mutated β-catenin were examined with and without
WNT treatment. The K345R mutation repressed the acetylation and
phosphorylation of β-catenin, and also promoted the proliferation
of MCF7 cells. Furthermore, K345Q β-catenin mutants antagonized
WNT-induced downregulation of phosphorylated β-catenin and
increased the cell proliferation rate (Fig. 3). The change in the phosphorylation
level in K345-mutated β-catenin suggests that the phosphorylation
of β-catenin may be dependent on its acetylation step. This view is
supported by previous data establishing the link between
phosphorylation and acetylation in PRKAA1 (27), and explains how K345A (similar to
K345Q) β-catenin was shown to be efficiently degraded by APC
(28). K345Q β-catenin mutants may
constitutively phosphorylate β-catenin, lead to its degradation and
further inhibit WNT signaling activity. The marginal downregulation
of total β-catenin is observed in Fig.
3B. In previous studies (20,23),
HDAC6 has been identified as the deacetylase that regulates the
aectylation level of β-catenin. HDAC6 is sufficient and required
for WNT-stimulated deacetylation of β-catenin in breast cancer
cells, but the specific correlation between HDAC6 and WNT signaling
requires further investigation.
In conclusion, the present study demonstrated that
downregulation of the level of acetylated β-catenin is associated
with breast cancer. In vitro assays in MCF7 breast cancer
cells reveal that autocrine WNT signaling may result in the
deacetylation of β-catenin and activate the downstream signal
transduction. In the absence of WNT ligands, acetylated β-catenin
may mediate the phosphorylation and degradation of β-catenin under
normal conditions. The deacetylation of β-catenin by HDAC6
modulates the in vitro proliferation of breast cancer cells
through responding to WNT stimulation. The inhibition of
WNT-elicited deacetylation of β-catenin exhibited a strong effect
on the growth of breast cancer cells and it may provide a valid
therapeutic approach in breast cancer.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Amerongen R, Mikels A and Nusse R:
Alternative wnt signaling is initiated by distinct receptors. Sci
Signal. 1:re92008.PubMed/NCBI
|
|
5
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Howe LR and Brown AM: Wnt signaling and
breast cancer. Cancer Biol Ther. 3:36–41. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van de Wetering M, Barker N, Harkes IC, et
al: Mutant E-cadherin breast cancer cells do not display
constitutive Wnt signaling. Cancer Res. 61:278–284. 2001.PubMed/NCBI
|
|
8
|
Bafico A, Liu G, Goldin L, Harris V and
Aaronson SA: An autocrine mechanism for constitutive Wnt pathway
activation in human cancer cells. Cancer Cell. 6:497–506. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schlange T, Matsuda Y, Lienhard S, Huber A
and Hynes NE: Autocrine WNT signaling contributes to breast cancer
cell proliferation via the canonical WNT pathway and EGFR
transactivation. Breast Cancer Res. 9:R632007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsuda Y, Schlange T, Oakeley EJ, Boulay
A and Hynes NE: WNT signaling enhances breast cancer cell motility
and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231
xenograft growth. Breast Cancer Res. 11:R322009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y
and Cao X: The cytosolic nucleic acid sensor LRRFIP1 mediates the
production of type I interferon via a beta-catenin-dependent
pathway. Nat Immunol. 11:487–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu C, Li Y, Semenov M, et al: Control of
beta-catenin phosphorylation/degradation by a dual-kinase
mechanism. Cell. 108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ikeda S, Kishida S, Yamamoto H, Murai H,
Koyama S and Kikuchi A: Axin, a negative regulator of the Wnt
signaling pathway, forms a complex with GSK-3beta and beta-catenin
and promotes GSK-3beta-dependent phosphorylation of beta-catenin.
EMBO J. 17:1371–1384. 1998. View Article : Google Scholar
|
|
15
|
Kitagawa M, Hatakeyama S, Shirane M, et
al: An F-box protein, FWD1, mediates ubiquitin-dependent
proteolysis of beta-catenin. EMBO J. 18:2401–2410. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yost C, Torres M, Miller JR, Huang E,
Kimelman D and Moon RT: The axis-inducing activity, stability, and
subcellular distribution of beta-catenin is regulated in Xenopus
embryos by glycogen synthase kinase 3. Genes Dev. 10:1443–1454.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu C, Kato Y, Zhang Z, Do VM, Yankner BA
and He X: beta-Trcp couples beta-catenin
phosphorylation-degradation and regulates Xenopus axis formation.
Proc Natl Acad Sci USA. 96:6273–6278. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wolf D, Rodova M, Miska EA, Calvet JP and
Kouzarides T: Acetylation of beta-catenin by gapdh (CBP). J Biol
Chem. 277:25562–25567. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levy L, Wei Y, Labalette C, Wu Y, Renard
CA, Buendia MA and Neuveut C: Acetylation of beta-catenin by p300
regulates beta-catenin-Tcf4 interaction. Mol Cell Biol.
24:3404–3414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu J, Coyne CB and Sarkar SN: PKC alpha
regulates Sendai virus-mediated interferon induction through HDAC6
and β-catenin. EMBO J. 30:4838–4849. 2011.PubMed/NCBI
|
|
21
|
Jho EH, Zhang T, Domon C, Joo CK, Freund
JN and Costantini F: Wnt/beta-catenin/Tcf signaling induces the
transcription of Axin2, a negative regulator of the signaling
pathway. Mol Cell Biol. 22:1172–1183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roose J, Huls G, van Beest M, et al:
Synergy between tumor suppressor APC and the beta-catenin-Tcf4
target Tcf1. Science. 285:1923–1926. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Zhang X, Polakiewicz RD, Yao TP and
Comb MJ: HDAC6 is required for epidermal growth factor-induced
beta-catenin nuclear localization. J Biol Chem. 283:12686–12690.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takahashi-Yanaga F and Sasaguri T: The
Wnt/beta-catenin signaling pathway as a target in drug discovery. J
Pharmacol Sci. 104:293–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fraga MF, Ballestar E, Villar-Garea A, et
al: Loss of acetylation at Lys16 and trimethylation at Lys20 of
histone H4 is a common hallmark of human cancer. Nat Genet.
37:391–400. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin YY, Kiihl S, Suhail Y, et al:
Functional dissection of lysine deacetylases reveals that HDAC1 and
p300 regulate AMPK. Nature. 482:251–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
von Kries JP, Winbeck G, Asbrand C, et al:
Hot spots in beta-catenin for interactions with LEF-1, conductin
and APC. Nat Struct Biol. 7:800–807. 2000.PubMed/NCBI
|