Introduction
Cardiovascular diseases are the leading cause of
mortality worldwide and their incidence is increasing concurrently
with the development of society and changes in lifestyle (1). Ischemic heart disease, which can
cause arrhythmia and myocardial infarction, is a current major
focus of the cardiovascular pathologies (2) since it is projected that ischemic
heart disease is likely to be the primary cause of mortality in the
population by 2030 (3,4). Despite advances in understanding the
pathogenesis of this disease, current treatments for ischemic heart
disease are not sufficient (5–7) and,
therefore, the effective relief of the injury caused by myocardial
ischemia is a priority in medical research. Coronary artery
ligation is the most common method of establishing an acute
myocardial ischemia model and is widely used in studies
investigating the mechanisms of myocardial ischemia (8). The method of Langendorff isolated
retrograde heart perfusion is a prominent experimental technique
used in the field of cardiovascular research and provides
stability, reliability and convenience (9,10).
Hydrogen sulfide (H2S) was the third
endogenous signaling gasotransmitter to be identified, following
nitric oxide (NO) and carbon monoxide (11–15).
Endogenous H2S is widely present in mammalian tissues
and has numerous signaling functions, thus contributing to various
physiological and pathological processes (16–21).
It has been shown that H2S can antagonize ischemic
reperfusion injury and therefore exhibit cardioprotective effects
(22–26). However, associations between
H2S and acute myocardial ischemia injury are less well
established, and the mechanisms underlying the effects of
H2S remain unclear.
In this study, an acute myocardial ischemia injury
rat model was established using the method of coronary artery
ligation. The aim of this study was to observe whether
H2S could protect the hearts against ischemic injury and
whether antioxidation was involved in the cardioprotection induced
by H2S.
Materials and methods
Animals
Male Sprague Dawley rats, weighing 250–290 g, were
provided by the Center of Experimental Animals of Hebei Province
(Shijiazhuang, China). Rats were acclimated for one week, with
standard rat chow and water provided ad libitum. All
experimental protocols were approved by the Animal Care and Use
Committee of Hebei Medical University (Shijiazhuang, China) and
were performed in accordance with the Guidelines of Animal
Experiments from the Committee of Medical Ethics, National Health
Department of China (Beijing, China).
Myocardial ischemia injury
Coronary occlusion was performed as previously
described (27). The rats were
anesthetized with 10% chloral hydrate at 350 mg/kg via
intraperitoneal injection. The heart was rapidly excised and washed
with Krebs-Henseleit buffer (K-H buffer), then retrogradely
perfused through the aorta at a constant pressure of 90 cm
H2O with K-H buffer containing 6.91 g/l NaC, 0.35 g/l
KCl, 0.265 g/l CaCl2·2H2O, 2.1 g/l
NaHCO3, 2.2 g/l glucose, 0.296 g/l
MgSO4·7H2O and 0.1632 g/l
KH2PO4 (pH 7.35–7.45 when aerated with 95%
O2/5% CO2 at 37°C). The left anterior
descending (LAD) coronary artery was occluded with a 6.0-silk
suture 2–3 mm from the tip of the left atrium, at the end of the
stabilization period. Successful coronary occlusion was verified by
the development of a pale color in the distal myocardium. Coronary
flow rate (CF) was measured by timed collection of the coronary
effluent for 1 min.
Drug preparation and experimental
protocol
NaHS, 2,3,5-triphenyl tetrazolium and Evans-Blue
were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lactate
dehydrogenase (LDH), mitochondrial malondialdehyde (MDA),
superoxide dismutase (SOD) and glutathione peroxidase
(GSH-Px) detection kits were purchased from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China). NaHS was
dissolved in K-H buffer.
The animals were randomly assigned to five groups:
i) Sham; ii) model; iii) infarct plus NaHS at a concentration of 5
μmol/l; iv) infarct plus NaHS at a concentration of 10 μmol/l; and
v) infarct plus NaHS at a concentration of 20 μmol/l. The three
treatment groups were treated with the H2S-donor (NaHS)
2 h after the induction of ischemia. The LAD coronary artery was
ligated for 4 h in the rats of the model and NaHS-treated groups,
but the rats in the sham group were threaded without ligation. The
sham and model groups were subjected to perfusion with normal
perfusate, and the experimental groups were perfused with NaHS
perfusate 2 h after ischemia (5, 10 or 20 μmol/l, accordingly).
Hemodynamics
A water-filled latex balloon was inserted via the
left atrium into the left ventricle and was inflated to set a left
ventricular end-diastolic pressure of between 6 and 8 mmHg during
the initial equilibration. This allowed monitoring of the left
ventricular systolic pressure, the maximum velocity of left
ventricular systolic pressure (+dP/dtmax) and the
maximum velocity of left ventricular diastolic pressure
(−dP/dtmax). The coronary effluent was collected to
measure coronary flow rate (ml/min).
Tissue H2S concentration
The tissue H2S content was measured as
described previously with modifications (24). Briefly, cardiac tissue was
homogenized in a 10-fold volume (w/v) of 50 mM ice-cold potassium
phosphate buffer (pH 6.8). The cardiac tissue homogenate was then
mixed with 0.5 ml 1% zinc acetate. A total of 0.5 ml 20 mM
N,N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl was
subsequently added, immediately followed by the addition of 0.4 ml
30 mM FeCl3 in 1.2 M HCl. Following 20 min incubation,
0.5 ml 10% trichloroacetic acid was added to the reaction mixture
prior to the addition of 2.5 ml distilled water. The absorbance of
the resulting solution at 670 nm was measured using a BioTek
microplate reader (BioTek Instruments Inc., Winooski, VT, USA). The
H2S content was calculated against a calibration curve
of NaHS, and the H2S concentration was expressed as
micromoles/gram protein.
Tissue cystathionine-γ-lyase (CSE)
activity
The H2S production rate was measured as a
reflection of CSE activity (28).
Myocardial tissue was homogenized in 1:10 (wt/vol) 50 mM ice-cold
potassium phosphate buffer (pH 6.8). The subsequent reactions were
performed in a 25-ml Erlenmeyer flask. The reaction mixture (1 ml)
consisted of 100 mM potassium phosphate buffer (pH 7.4), 10 mM
L-cysteine, 2 mM pyridoxal 5′-phosphate and 10% (w/v) tissue
homogenate. Trapping solution of 0.5 ml 1% zinc acetate was added
to a Cryovial® test tube in the flask, which was used as
the center well, and a small piece of filter paper (2.0×2.5
cm2) was used to increase the air/liquid contact
surface. The flask was flushed with N2 prior to being
sealed, and then was transferred from an ice bath to an agitated
water bath at 37°C to initiate the reaction. After 90 min, the
reaction was terminated with the addition of 0.5 ml 50%
trichloroacetic acid. The flask was incubated further for an hour
at 37°C in order to completely trap the H2S released
from the mixture. The content of the center well was transferred to
a test tube containing 3.6 ml distilled water and 0.5 ml 20 mM
N,N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl was added
immediately, followed by the addition of 0.4 ml 30 mM
FeCl3 in 1.2 M HCl. After 20 min, the absorbance of the
resulting solution at 670 nm was measured with a BioTek microplate
reader. The H2S content was calculated against the
calibration curve of NaHS. The H2S production rates were
expressed as nanomoles per milligram protein per minute.
Infarct size
Myocardial infarction was determined according to a
previously described method (29).
The heart was perfused with 1 ml 1% Evans Blue to stain the
non-ischemic tissue 4 h after ischemia and frozen for between 3 and
24 h. Transverse sections (2-mm) were incubated in 1%
triphenyltetrazolium chloride in phosphate buffer (pH 7.4) for 15
min at 37°C. Non-infarcted tissue was stained red, whereas necrotic
tissue remained unstained. The transverse sections were fixed with
10% formaldehyde and imaged. Images were captured using a digital
camera and were transferred to a computer for subsequent management
with Photoshop CS2 software (Adobe Systems, San Jose, CA, USA).
LDH
Myocardial tissue damage was assessed by determining
the LDH activity in the coronary effluent collected at the end of
stabilization, 3 and 4 h after the induction of ischemia. The
activity of LDH was measured following the manufacturer’s
instructions (Nanjing Jiancheng Bioengineering Institute).
Ultrastructural changes to mitochondria
in myocardial cells
Following 4 h of ischemia, the hearts were rapidly
excised. Transmural tissue samples (1 mm3) were obtained
from the left anterior myocardium and immediately immersed in
ice-cold 4%, 0.1 mol/l phosphate-buffered glutaraldehyde (pH 7.2).
The tissues were washed twice in dimethyl arsenate buffer and the
tissue blocks were then postfixed for 2 h in 1%, 0.1 mol/l
phosphate-buffered OsO4 (pH 7.2). This was followed by a
further two washes in dimethyl arsenate buffer and dehydration. The
tissues were subsequently embedded in araldite. Ultra-thin sections
were cut and double stained in uranyl acetate and lead citrate. The
sections were observed under a transmission electron microscope to
assess the ultrastructural features of the cardiomyocytes.
Isolated mitochondria
Mitochondria were isolated from the adult rat hearts
by homogenization and differential centrifugation, as described
previously (30). Briefly, the
heart was rapidly excised and washed in buffer containing 70 mM
sucrose, 210 mM mannitol, 1 mM EDTA and 50 mM Tris (pH 7.4) at 4°C.
Following changes of buffer, the cardiac samples were cut into
small pieces and homogenized. The homogenate was centrifuged at
1,300 × g for 3 min at 2°C. The supernatant was then collected and
re-centrifuged at 10,000 × g for 8 min at 2°C. The pellet was
resuspended in EDTA-free homogenization buffer [70 mM sucrose, 210
mM mannitol (pH 7.4) with 50 mM Tris] and centrifuged at 10,000 × g
for 10 min at 2°C. The prepared mitochondria were diluted in
isolation medium prior to use.
Mitochondrial MDA, SOD and
GSH-Px
The level of MDA and the activities of SOD and
GSH-Px were determined using reagent kits, in accordance
with the manufacturer’s instructions (Nanjing Jiancheng
Bioengineering Institute).
Statistical analysis
Results are presented as the mean ± standard error
of the mean. All tests were performed using SPSS 13.0 (SPSS, Inc.,
Armonk, NY, USA). Data were analyzed using one-way analysis of
variance followed by Tukey’s post hoc multiple comparison test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of H2S on cardiac
function during ischemia
The cardiodynamic variables are shown in Table I. Subsequent to myocardial ischemia
injury, the values for left ventricular developed pressure (LVDP),
±dP/dtmax and CF were significantly decreased in the
model group relative to those in the sham group (P<0.01), whilst
these parameters showed significant increases in the NaHS low-,
middle- and high-dose groups as compared with those in the model
group (P<0.05 or P<0.01).
 | Table IEffect of hydrogen sulfide on the
cardiac function in rats following ischemia. |
Table I
Effect of hydrogen sulfide on the
cardiac function in rats following ischemia.
| Group | LVDP (mmHg) |
+dP/dtmax (mmHg/sec) |
−dP/dtmax (mmHg/sec) | CF (ml/min) |
|---|
| Sham | 63.12±1.46 | 3189±189 | 1762±122 | 3.65±0.12 |
| Model | 43.27±3.01a | 1174±79a | 861±54a | 2.15±0.21a |
| I+L NaHS | 49.77±2.07b | 1724±126b | 1066±38b | 2.29±0.08c |
| I+M NaHS | 55.50±1.60b | 2298±167b | 1278±45b | 2.95±0.12b |
| I+H NaHS | 61.75±1.67b | 2844±139b | 1587±43b | 3.36±0.11b |
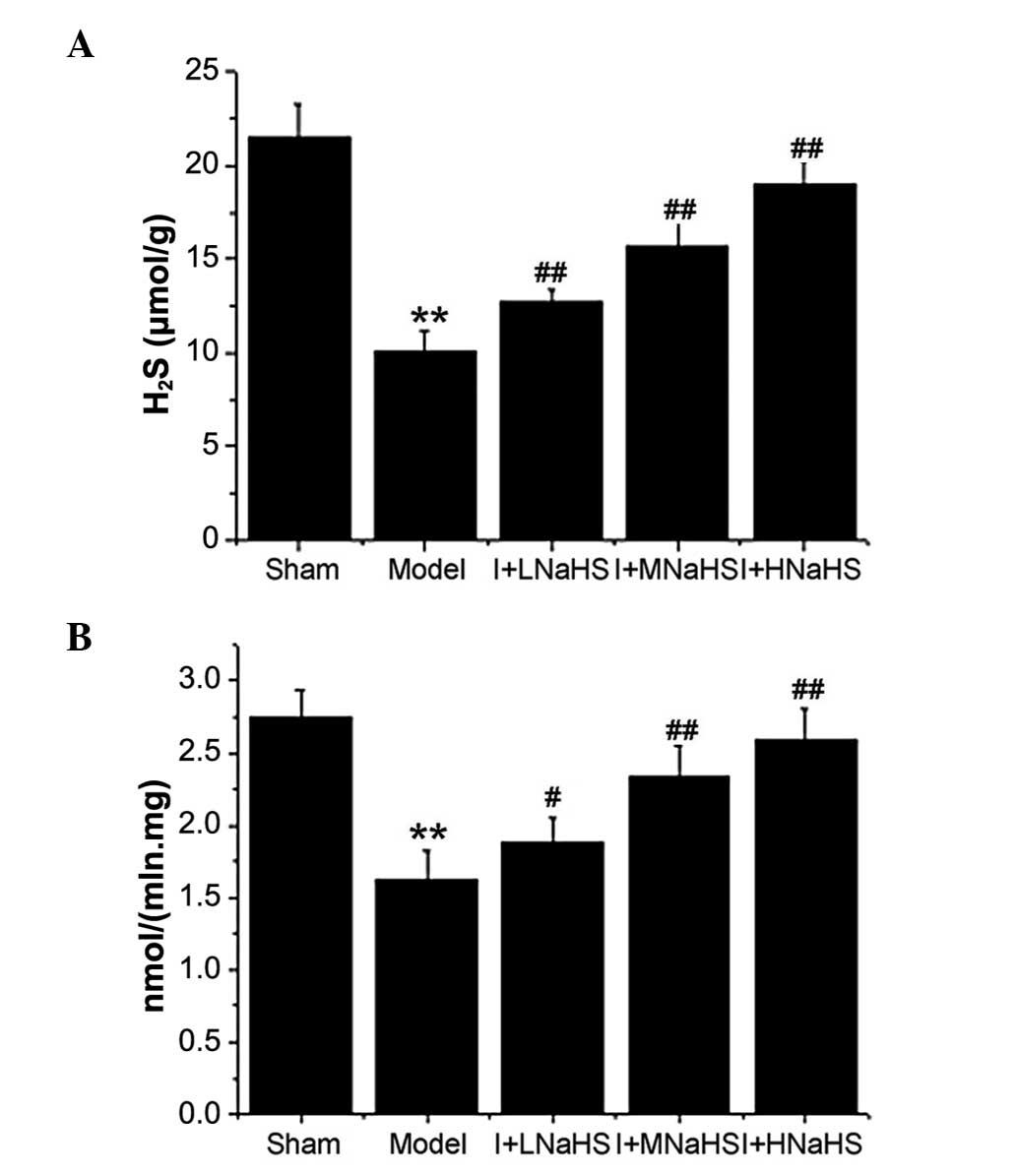
Changes in H2S content and CSE
activity in myocardial tissue
The content of H2S and the activity of
CSE in cardiac tissue were significantly decreased in the model
group as compared with those in the sham group (P<0.01, Fig. 1). However, these parameters showed
significant increases in the NaHS low-, middle- and high-dose
groups compared with those in the model group (P<0.05 or
P<0.01, Fig. 1).
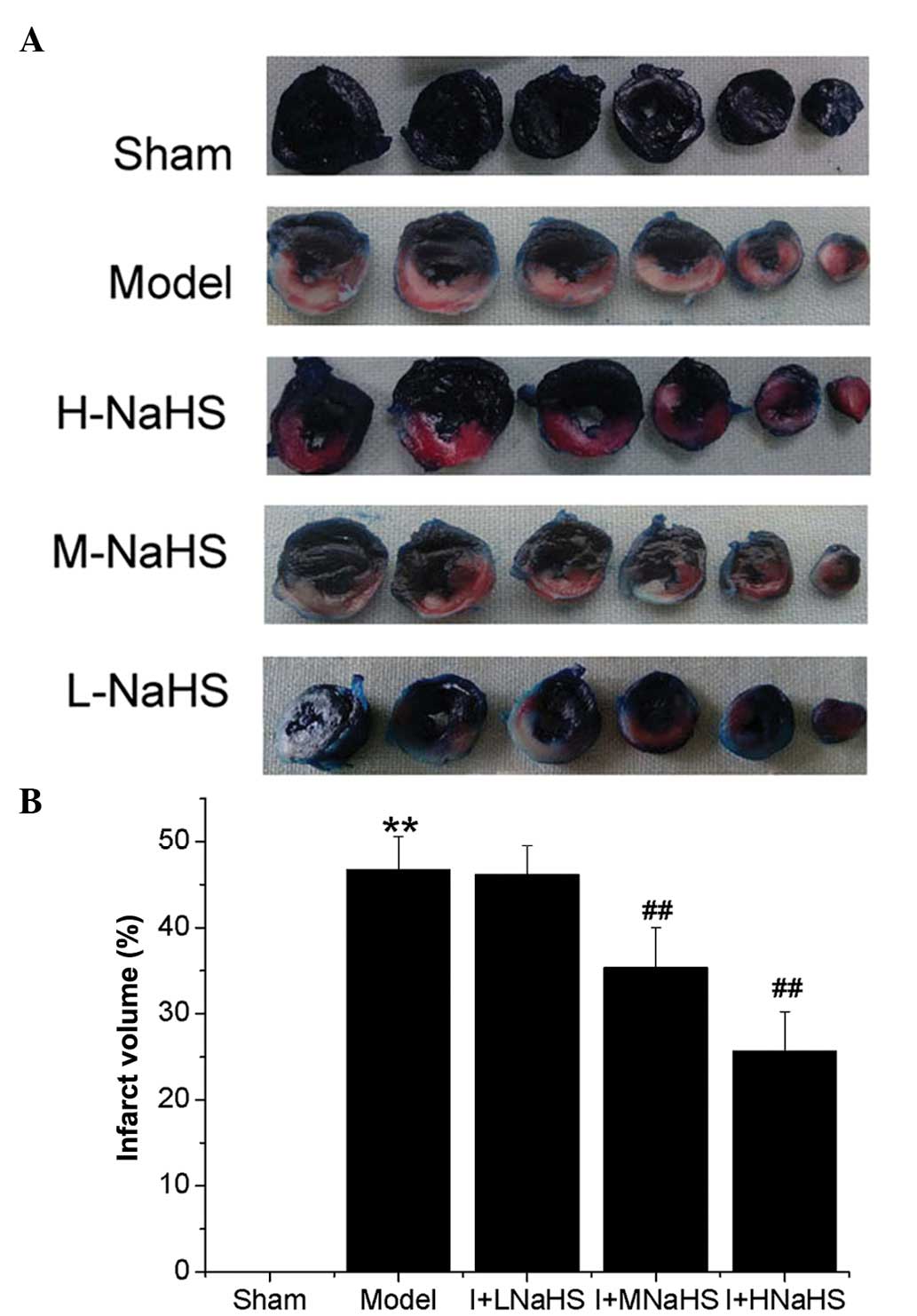
Effect of H2S on infarct
size
The myocardial infarct volume was significantly
increased in the model group compared with that of the sham group
(P<0.01, Fig. 2). No
significant differences were observed in myocardial infarct volume
between the NaHS low-dose group and the model group, whereas the
infarct volumes were significantly decreased in the NaHS middle-
and high-dose groups as compared with those in the model group
(P<0.01, Fig. 2).
Effect of H2S on LDH
activity
No statistically significant differences were
observed in the activity of LDH in the perfusate among the
experimental groups prior to ischemia (Table II). The activity of LDH in the
perfusate was significantly increased in the model group compared
with that of the sham group (P<0.01, Table II). However, the activity of LDH
in the perfusate was significantly decreased in NaHS low-, middle-
and high-dose groups compared with that in the model group
(P<0.01, Table II).
| Table IIEffect of hydrogen sulfide on the
activity of LDH in the coronary effluent of each group. |
Table II
Effect of hydrogen sulfide on the
activity of LDH in the coronary effluent of each group.
| LDH activity
(U/l) |
|---|
|
|
|---|
| Group | BS 20 min | Ischemia 3 h | Ischemia 4 h |
|---|
| Sham | 11.18±0.42 | 31.46±3.97 | 49.68±3.06 |
| Model | 11.24±0.38 | 82.35±4.43a | 103.94±6.22a |
| I+L NaHS | 11.20±0.38 | 66.02±3.69b | 83.49±3.36b |
| I+M NaHS | 11.22±0.35 | 51.32±4.16b | 70.39±4.07b |
| I + H NaHS | 11.17±0.42 | 44.28±2.52b | 60.45±3.89b |
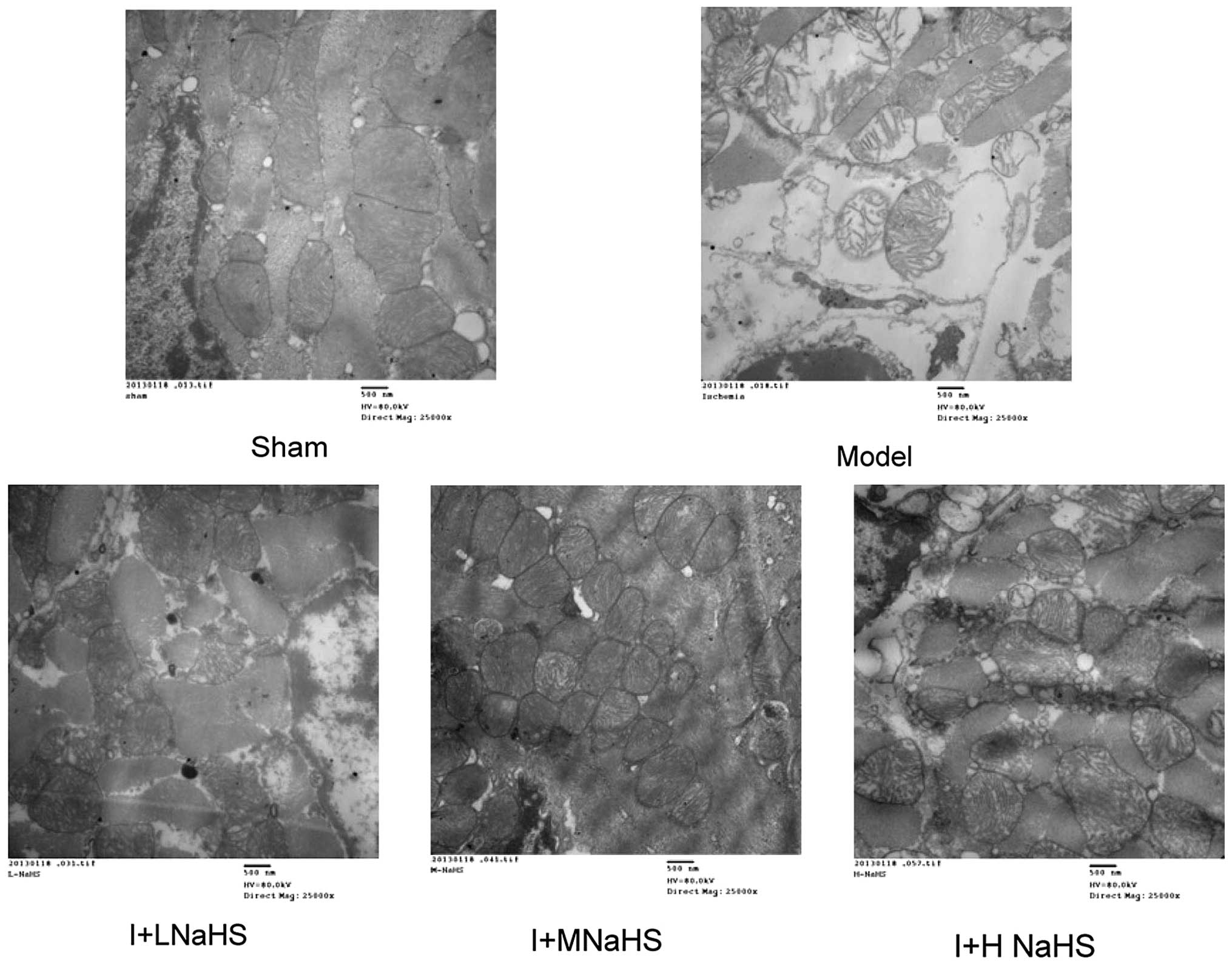
Effect of H2S on the
ultrastructure of mitochondria in myocardial cells
In the sham group, the ultrastructure of the
myocardial cells exhibited regular mitochondria with uniform size,
complete mitochondrial cristae and an intact nuclear membrane. In
the model group, the myocardial cells were characterized by
mitochondrial swelling, disappearance or deformation of
mitochondrial cristae, disruption of the nuclear membrane and
nuclear condensation. The NaHS low-, middle- and high-dose groups
showed less significant pathological changes in the myofilaments,
mitochondria and nuclei as compared with the model group (Fig. 3).
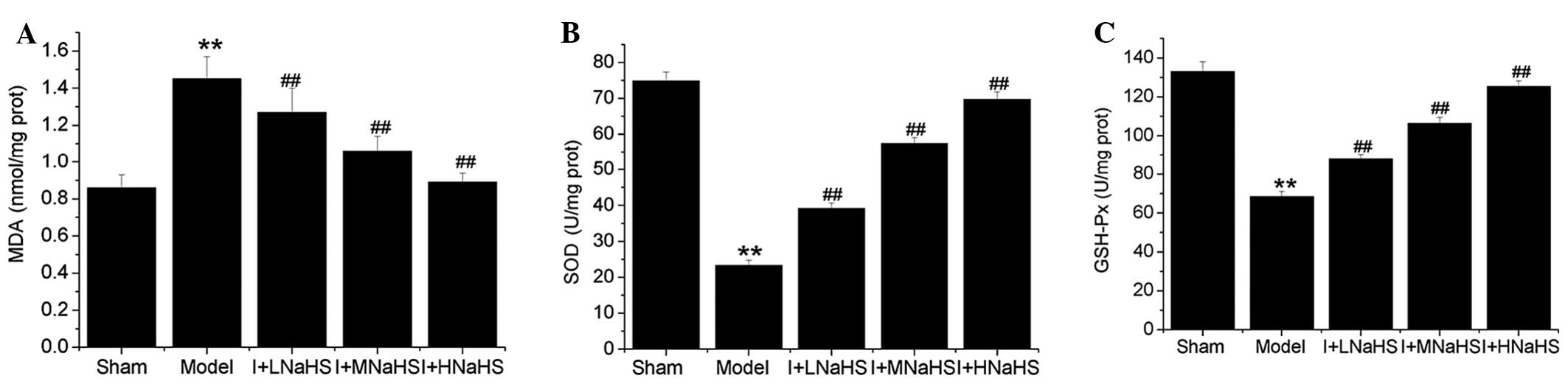
Effects of H2S on
mitochondrial MDA content and SOD and glutathione GSH-Px
activity
The content of MDA in the cardiac mitochondria was
significantly increased and the activities of SOD and
GSH-Px were significantly decreased in the model group
compared with those in the sham group (P<0.01, Fig. 4). By contrast, the content of MDA
was significantly decreased and the activities of SOD and
GSH-Px were significantly increased in the cardiac
mitochondria in the NaHS low-, middle- and high-dose groups
compared with those in the model group (P<0.01, Fig. 4).
Discussion
It has been shown H2S is a novel
gasotransmitter produced in numerous mammalian cells and tissues
(31–33). H2S is generated
primarily from L-cysteine by CSE in the cardiovascular system
(11,34,35);
CSE activity has been detected in the heart (32) and vascular smooth muscle (36). There is a growing evidence to
suggest that there are correlations among CSE activity,
H2S concentration and cardiovascular disease (37–40).
It has been suggested that H2S exerts
antioxidant, anti-inflammatory, and anti-apoptotic effects
(41,42). H2S has been shown to
inhibit lipid peroxidation during heart ischemia-reperfusion and to
decrease the ischemia-induced death of myocardial cells via an
oxygen free radical-reducing mechanism (42). This suggests that H2S
may act as an important modulator in cardiovascular physiology and
pathophysiology. Since the role of H2S in the
pathogenesis of ligature-induced regional myocardial ischemia has
not been investigated in vitro, to the best of our
knowledge, the present study demonstrated for the first time the
role of H2S in a ligature-induced myocardial ischemia
injury model in rats in vitro. An isolated rat heart model
was used such that extrinsic humoral and autonomic nervous system
influences, in addition to the effect of H2S on
peripheral vascular tone, could be excluded (43).
In our previous study, comparisons with the sham
group revealed that the LVDP, ±dP/dtmax and CF were
significantly decreased in the ischemia group at 30 min and 1, 2, 3
and 4 h after ischemia and that the infarct volumes in the ischemia
group were markedly increased between 1 and 4 h after ischemia.
Associated with these injuries, the content of H2S and
the activity of CSE in the cardiac tissue were significantly
decreased compared with those of the sham control group during the
1–4 h after ischemia. These data revealed that the
CSE/H2S pathway may participate in the
pathophysiological regulation of myocardial ischemia injury in
isolated rat hearts. The CSE activity is dependent on pyridoxal
5′-phosphate (PLP), which is reduced in ischemic disease, such as
stroke (44). Low PLP levels may
inhibit the CSE activity and lead to decreased myocardial and
plasma H2S generation. Furthermore, CSE inhibition may
decrease glutathione levels (45),
and this reduction may explain the increase in infarct size
(46).
The cardiodynamic parameters and myocardial
infarction volume were selected as the parameters to assess the
functional performance of the isolated rat heart following
ischemia. To explore the effects of H2S on regional
myocardial ischemia injury, the isolated rat heart was treated with
NaHS following the induction of ischemia. NaHS has a fast releasing
rate in aqueous solution, producing one-third H2S as
compared with the concentration of the salt (47,48),
without changing the pH of the medium (49). In the present study, the content of
H2S and the activity of CSE in the cardiac tissue were
significantly decreased in the model group as compared with those
in the sham group. By contrast, the content of H2S and
the activity of CSE in the cardiac tissue were significantly
increased in the NaHS low-, middle- and high-dose groups as
compared with those in the model group. The LVDP,
±dP/dtmax and CF were significantly increased in the
NaHS low-, middle- and high-dose groups as compared with those in
the model group. The infarct volume is strongly associated with the
prognosis of acute myocardial infarction and negatively correlated
with improvements in the cardiodynamic parameters. In particular,
impairments in the cardiac contractility become increasingly severe
with increasing infarct volume; therefore, a decrease in infarct
volume may be a useful parameter for the evaluation of the
effectiveness of anti-myocardial ischemia drugs (50). In the present study, the infarct
volumes were significantly decreased in NaHS middle- and high-dose
groups compared with those in the model group. Taken together,
these data demonstrate that H2S exerts protective
effects against myocardial ischemia injury in isolated rat hearts.
Similar to NO, H2S has been found to be a vasodilatory
agent that acts through alterations in K+ channel
activity and elevated cyclic guanosine monophosphate levels in
vascular smooth muscle cells (51,52).
Consistent with these observations, our findings revealed that
reduced levels of H2S are associated with the
constriction of blood vessels. Furthermore, the vasodilatory
effects of NaHS can dilate coronary arteries and increase CF in
ischemic diseases, thereby reducing ischemia-induced cellular
damage.
Mitochondrial ultrastructural (53) and functional (54) injury occurs early and progresses
through the course of ischemia (54,55).
Mitochondrial damage results in a loss of mitochondrial function,
impairing energy production and cell physiology, and an enhancement
of pathological function, producing oxidative-, calcium- and
apoptosis-mediated myocyte injury (56). Mitochondrial oxidative damage
participates in a variety of pathologies, including cardiovascular
disorders and neurodegenerative diseases. As such, the protection
of mitochondria from oxidative damage may be an effective
therapeutic strategy. The effect of H2S on myocardial
oxidative stress following ischemia was therefore assessed in the
present study. The production of reactive oxygen species by
mitochondria leads to the formation of lipid peroxidation products
and, in turn, can induce oxidative stress, thereby causing cellular
and mitochondrial damage (57,58).
The release of free radicals, coupled with the ischemia-induced
decrease in antioxidant activity, leaves the myocardium susceptible
to injury. It has been observed that phospholipid peroxidation and
subsequent damage to complex I jointly increase membrane leakage,
mitochondrial swelling, cytochrome c release and caspase
activation, resulting in cell death (59). The leakage of the cytosolic enzyme
LDH is correlated with a loss of cell membrane integrity, and
measuring the change in MDA content indicates the degree of damage
caused by membrane lipid peroxidation. In this study, lipid
peroxidation was observed to increase in the heart upon ligation of
the LAD coronary artery. This was apparent through increases in the
MDA content and LDH level relative to the sham group. However, the
addition of NaHS significantly decreased the MDA content and LDH
level. This result is consistent with the findings on the effects
of H2S on a rat model of myocardial infarction in
vivo (60). In the majority of
mammalian species, SOD and GSH-Px appear to be the most
active antioxidant enzymes in the myocardium to provide defense for
cellular organelles against oxidative damage caused by reactive
oxygen species (61). In the
present study, NaHS administration significantly increased the
activities of SOD and GSH-Px in the cardiac mitochondria
following ischemia. These data demonstrate that treatment with NaHS
enhances the capacity of antioxidant enzymes, and the underlying
mechanism of action is attributed to the protective effects exerted
against the oxidative stress-mediated injury in the mitochondria.
Transmission electron microscopy revealed a marked relief in
mitochondrial swelling and increased matrix density in isolated
ischemic rat hearts receiving NaHS, further suggesting a role for
the preservation of mitochondrial function in the observed
cytoprotection.
In conclusion, the pathophysiological process of
ligature-induced regional myocardial ischemia injury is associated
with the impaired endogenous CSE/H2S pathway.
Exogenously administered H2S via NaHS can effectively
protect myocytes and contractile activity and limit the extent of
myocardial infarction. These protective effects of H2S
against myocardial ischemia injury appear to be mediated by its
antioxidant activities and preservation of mitochondrial function.
Taken together, these data strengthen evidence that H2S
may be a cardiovascular protective regulator for preventing or
treating cardiovascular diseases. However, the exact mechanisms
underlying the action of H2S require further
investigation.
Acknowledgements
This study was supported by the Natural Science
Foundation of Hebei Province (C2009001458) and the Key Basic
Research Program of Hebei Provincial Science and Technology
Department (13967602D).
References
|
1
|
Thom T, Haase N, Rosamond W, et al;
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee. Heart disease and stroke statistics - 2006
update: a report from the American Heart Association Statistics
Committee and Stroke Statistics Subcommittee. Circulation.
113:e85–e151. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nabel EG and Braunwald E: A tale of
coronary artery disease and myocardial infarction. N Engl J Med.
366:54–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4222006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lloyd-Jones D, Adams RJ, Brown TM, et al;
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee. Heart disease and stroke statistics - 2010
update: a report from the American Heart Association. Circulation.
121:e46–e215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Renda G and de Caterina R: Impact of
antiplatelet therapy in heart disease. Adv Cardiol. 47:5–19. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gnecchi M, Danieli P and Cervio E:
Mesenchymal stem cell therapy for heart disease. Vascul Pharmacol.
57:48–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Songco AV and Brener SJ: Initial strategy
of revascularization versus optimal medical therapy for improving
outcomes in ischemic heart disease: a review of the literature.
Curr Cardiol Rep. 14:397–407. 2012. View Article : Google Scholar
|
|
8
|
Bhind R, Witting PK, McMahon AC,
Khachigian LM and Lowe HC: Rat models of myocardial infarction.
Pathogenetic insights and clinical relevance. Thromb Haemost.
96:602–610. 2006.PubMed/NCBI
|
|
9
|
Langendorff O: Untersuchungen am
überlebenden Säugethierherzen. Arch Ges Physiol. 61:291–332.
1895.
|
|
10
|
Skrzypiec-Spring M, Grotthus B, Szelag A
and Schulz R: Isolated heart perfusion according to Langendorff -
still viable in the new millennium. J Pharmacol Toxicol Methods.
55:113–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang R: Two’s company, three’s a crowd:
can H2S be the third endogenous gaseous transmitter?
FASEB J. 16:1792–1798. 2002.
|
|
12
|
Ignarro LJ, Buga GM, Wood KS, Byrns RE and
Chaudhuri G: Endothelium-derived relaxing factor produced and
released from artery and vein is nitric oxide. Proc Natl Acad Sci
USA. 84:9265–9269. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palmer RM, Ferrige AG and Moncada S:
Nitric oxide release accounts for the biological activity of
endothelium derived relaxing factor. Nature. 327:524–526. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marks GS, Brien JF, Nakatsu K, et al: Does
carbon monoxide have a physiological function? Trends Pharmacol
Sci. 12:185–188. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Verma A, Hirsch DJ, Glatt CE, et al:
Carbon monoxide: a putative neural messenger. Science. 259:381–384.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abe K and Kimura H: The possible role of
hydrogen sulfide as an endogenous neuromodulator. J Neurosci.
16:1066–1071. 1996.PubMed/NCBI
|
|
17
|
Pearson RJ, Wilson T and Wang R:
Endogenous hydrogen sulfide and the cardiovascular system-what’s
the smell all about? Clin Invest Med. 29:146–150. 2006.
|
|
18
|
Szabó C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
|
|
19
|
Jha S, Calvert JW, Duranski MR,
Ramachandran A and Lefer DJ: Hydrogen sulfide attenuates hepatic
ischemia-reperfusion injury: role of antioxidant and antiapoptotic
signaling. Am J Physiol Heart Circ Physiol. 295:H801–H806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bos EM, Leuvenink HG, Snijder PM, et al:
Hydrogen sulfide-induced hypometabolism prevents renal
ischemia/reperfusion injury. J Am Soc Nephrol. 20:1901–1905. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y and
Zhu YC: The hydrogen sulfide donor NaHS promotes angiogenesis in a
rat model of hind limb ischemia. Antioxid Redox Signal.
12:1065–1077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Z, Huang H, Liu P, Tang C and Wang
J: Hydrogen sulfide contributes to cardioprotection during
ischemia-reperfusion injury by opening K ATP channels. Can J
Physiol Pharmacol. 85:1248–1253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji Y, Pang QF, Xu G, Wang L, Wang JK and
Zeng YM: Exogenous hydrogen sulfide postconditioning protects
isolated rat hearts against ischemia-reperfusion injury. Eur J
Pharmacol. 587:1–7. 2008. View Article : Google Scholar
|
|
24
|
Sodha NR, Clements RT, Feng J, et al: The
effects of therapeutic sulfide on myocardial apoptosis in response
to ischemia-reperfusion injury. Eur J Cardiothorac Surg.
33:906–913. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sivarajah A, Collino M, Yasin M, et al:
Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in
a rat model of regional myocardial I/R. Shock. 31:267–274. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lavu M, Bhushan S and Lefer DJ: Hydrogen
sulfide-mediated cardioprotection: mechanisms and therapeutic
potential. Clin Sci (Lond). 120:219–229. 2011.PubMed/NCBI
|
|
27
|
Liu H, Yan J, Xu YW, Bai XJ and Wu BW:
Modification of ex-vivo ischemia/reperfusion modeling in isolated
rat heart. Chinese Remedies & Clinics. 7:586–587. 2007.(In
Chinese).
|
|
28
|
Stipanuk MH and Beck PW: Characterization
of the enzymic capacity for cysteine desulphhydration in liver and
kidney of the rat. Biochem J. 206:267–277. 1982.PubMed/NCBI
|
|
29
|
Hwa JS, Jin YC, Lee YS, et al:
2-methoxycinnamaldehyde from Cinnamomum cassia reduces rat
myocardial ischemia and reperfusion injury in vivo due to HO-1
induction. J Ethnopharmacol. 139:605–615. 2012.
|
|
30
|
Shiva S, Brookes PS and Darley-Usmar VM:
Methods for measuring the regulation of respiration by nitric
oxide. Methods Cell Biol. 80:395–416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Abe K and Kimura H: The possible role of
hydrogen sulfide as an endogenous neuromodulator. J Neurosci.
16:1066–1071. 1996.PubMed/NCBI
|
|
32
|
Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J
and Tang C: H2S generated by heart in rat and its
effects on cardiac function. Biochem Biophys Res Commun.
313:362–368. 2004.
|
|
33
|
Li L, Rose P and Moore PK: Hydrogen
sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 51:169–187.
2011. View Article : Google Scholar
|
|
34
|
Chen P, Poddar R, Tipa EV, et al:
Homocysteine metabolism in cardiovascular cells and tissues:
implications for hyperhomocysteinemia and cardiovascular disease.
Adv Enzyme Regul. 39:93–109. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Szabó C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
|
|
36
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP)
channel opener. EMBO J. 20:6008–6016. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Geng B, Chang L, Pan C, et al: Endogenous
hydrogen sulfide regulation of myocardial injury induced by
isoproterenol. Biochem Biophys Res Commun. 318:756–763. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang HL, Wu HC, Li ZL, Geng B and Tang
CS: Changes of the new gaseous transmitter H2S in
patients with coronary heart disease. Di Yi Jun Yi Da Xue Xue Bao.
25:951–954. 2005.(In Chinese).
|
|
39
|
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY,
Zhou S and Moore PK: Role of hydrogen sulfide in the
cardioprotection caused by ischemic preconditioning in the rat
heart and cardiac myocytes. J Pharmacol Exp Ther. 316:670–678.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yong QC, Pan TT, Hu LF and Bian JS:
Negative regulation of beta-adrenergic function by hydrogen
sulphide in the rat hearts. J Mol Cell Cardiol. 44:701–710. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tokuda K, Kida K, Marutani E, et al:
Inhaled hydrogen sulfide prevents endotoxin-induced systemic
inflammation and improves survival by altering sulfide metabolism
in mice. Antioxid Redox Signal. 17:11–21. 2012. View Article : Google Scholar
|
|
42
|
Jiang LH, Luo X, He WA, Huang XX and Cheng
TT: Effects of exogenous hydrogen sulfide on apoptosis proteins and
oxidative stress in the hippocampus of rats undergoing heroin
withdrawal. Arch Pharm Res. 34:2155–2162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ytrehus K: The ischemic heart -
experimental models. Pharmacol Res. 42:193–203. 2000. View Article : Google Scholar
|
|
44
|
Kelly PJ, Kistler JP, Shih VE, et al:
Inflammation, homocysteine, and vitamin B6 status after ischemic
stroke. Stroke. 35:12–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Diwakar L and Ravindranath V: Inhibition
of cystathionine-gamma-lyase leads to loss of glutathione and
aggravation of mitochondrial dysfunction mediated by excitatory
amino acid in the CNS. Neurochem Int. 50:418–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blaustein A, Deneke SM, Stolz RI, Baxter
D, Healey N and Fanburg BL: Myocardial glutathione depletion
impairs recovery after short periods of ischemia. Circulation.
80:1449–1457. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dombkowski RA, Russell MJ and Olson KR:
Hydrogen sulfide as an endogenous regulator of vascular smooth
muscle tone in trout. Am J Physiol Regul Integr Comp Physiol.
286:R678–R685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY,
Zhou S and Moore PK: Role of hydrogen sulfide in the
cardioprotection caused by ischemic preconditioning in the rat
heart and cardiac myocytes. J Pharmacol Exp Ther. 316:670–678.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hosoki R, Matsuki N and Kimura H: The
possible role of hydrogen sulfide as an endogenous smooth muscle
relaxant in synergy with nitric oxide. Biochem Biophys Res Commun.
237:527–531. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nie LH, Zhou YZ, Ding J, et al: Effects of
matrine on infarct size and ultramicrostructure in rats with acute
myocardial ischemic injury. Ningxia Med J. 33:292–294. 2011.(In
Chinese).
|
|
51
|
Yang GD, Wu LY, Jiang B, et al:
H2S as a physiologic vasorelaxant: hypertension in mice
with deletion of cystathionine gamma-lyase. Science. 322:587–590.
2008.
|
|
52
|
Wang R: Hydrogen sulfide: a new EDRF.
Kidney Int. 76:700–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Murry CE, Richard VJ, Reimer KA and
Jennings RB: Ischemic preconditioning slows energy metabolism and
delays ultrastructural damage during a sustained ischemic episode.
Circ Res. 66:913–931. 1990. View Article : Google Scholar
|
|
54
|
Flameng W, Andres J, Ferdinande P,
Mattheussen M and Van Belle H: Mitochondrial function in myocardial
stunning. J Mol Cell Cardiol. 23:1–11. 1991. View Article : Google Scholar
|
|
55
|
Lesnefsky EJ, Tandler B, Ye J, Slabe TJ,
Turkaly J and Hoppel CL: Myocardial ischemia decreases oxidative
phosphorylation through cytochrome oxidase in subsarcolemmal
mitochondria. Am J Physiol. 273:H1544–H1554. 1997.
|
|
56
|
Lesnefsky EJ, Moghaddas S, Tandler B,
Kerner J and Hoppel CL: Mitochondrial dysfunction in cardiac
disease: ischemia - reperfusion, aging, and heart failure. J Mol
Cell Cardiol. 33:1065–1089. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lowell BB and Shulman GI: Mitochondrial
dysfunction and type 2 diabetes. Science. 307:384–387. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Roden M: Muscle triglycerides and
mitochondrial function: possible mechanisms for the development of
type 2 diabetes. Int J Obes (Lond). 29(Suppl 2): S111–S115. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hardy L, Clark JB, Darley-Usmar VM, Smith
DR and Stone D: Reoxygenation-dependent decrease in mitochondrial
NADH:CoQ reductase (Complex I) activity in the hypoxic/reoxygenated
rat heart. Biochem J. 274:133–137. 1991.PubMed/NCBI
|
|
60
|
Liu F, Zhang JX, Li LF, Zhang QZ, Ding YY
and Zhang XY: Effects of hydrogen sulfide on myocardial
mitochondrial injury during acute myocardial ischemia in rats.
Zhongguo Ying Yong Sheng Li Xue Za Zhi. 27:158–162. 2011.(In
Chinese).
|
|
61
|
Crystal GJ, Malik G, Yoon SH and Kim SJ:
Isoflurane late preconditioning against myocardial stunning is
associated with enhanced antioxidant defenses. Acta Anaesthesiol
Scand. 56:39–47. 2012. View Article : Google Scholar
|