Introduction
Tumor necrosis factor-α (TNF-α) is a key cytokine
that initiates and regulates the cytokine cascade during an
inflammatory response in various tissues, including those of the
central nervous system (CNS). In the brain tissue of humans, it is
secreted from microglia, astrocytes and neurons to maintain
homeostatic conditions, but also, to induce pathophysiological
responses (1–3). TNF-α binding to receptors and the
downstream signaling cascade regulate fundamental processes in
brain function, such as the formation and regulation of synapses,
regeneration, neurogenesis and maintenance of CNS functions
(3). However, the expression of
TNF-α in healthy brains is very low, rendering its role under
physiological conditions hard to assess (4). In the brain of patients with
Alzheimer’s disease (AD), TNF-α, inflammatory cytokines and
neurotoxic molecules are predominantly produced from activated
neuronal cells, although most brain-derived TNF-α is synthesized by
glial cells (5). TNF-α can also
induce a variety of physiological processes including fever,
apoptotic cell death, sepsis (through the production of
interleukins 1 and 6), cachexia and inflammation, while it inhibits
tumorigenesis and viral replication. Therefore, dysregulation of
the expression of TNF-α is tightly related to a number of human
diseases such as AD, depression, cancer and inflammatory bowel
disease (6–9).
A number of studies have suggested novel strategies
for the development of medical compounds to cure and/or relieve the
symptoms of AD. Among these compounds, selenium has been identified
as a candidate agent for the prevention and treatment of AD.
Selenite, long been known as an ubiquitous trace compound in
nature, has been shown to be essential to animal and human health
(10). Sodium selenate was
recently found to contribute to the mitigation of pathological
symptoms of AD in animal models. Specifically, sodium selenate was
shown to reduce tau phosphorylation in SH-SY5Y neuroblastoma cells
that stably express human tau carrying the frontotemporal lobar
degeneration pathogenic mutation P301L (11). Furthermore, this compound activated
the serine-threonine protein phosphatase 2A (PP2A), a key
phosphatase implicated in tau protein phosphorylation. Following
sodium selenate treatment, TAU441 transgenic mice exhibited reduced
levels of phosphorylated and total tau in the hippocampus and
amygdala compared to the vehicle-treated group (12). Selenium deficiency promoted the
onset and progression of AD pathological phenotypes in the brain of
Tg2576 mice (13). However, few
studies have investigated whether selenium treatment can affect tau
hyperphosphorylation induced by TNF-α treatment in neuroblastoma
cells.
As demonstrated by our data, the increase in cell
viability, tau phosphorylation and activity of tau kinases induced
by TNF-α is significantly reduced by sodium selenite treatment in
neuroblastoma cells, suggesting that sodium selenite treatment can
contribute to the recovery of TNF-α-induced tau
hyperphosphorylation, and ultimately attenuate AD pathological
phenotypes, including the accumulation of neurofibrillary tangles
(NFT) and memory defects.
Materials and methods
Cell culture and selenium treatment
SH-SY5Y cells that originated from human
neuroblastoma were obtained from the Korean Food and Drug
Administration (Osong, Korea). The cell line was maintained for
24–36 h in HyClone™ RPMI-1640 medium containing 10% HyClone™ fetal
bovine serum (both from Thermo Fisher Scientific Inc., Waltham, MA,
USA), 100 IU/ml of penicillin and 100 μg/ml of streptomycin. The
cells were maintained in a humidified incubator at 37°C and 5%
CO2. Sodium selenite (Na2SeO3),
hereafter termed as selenite, was purchased from Sigma-Aldrich
(cat. no. S5261; St. Louis, MO, USA) and was dissolved in distilled
water to a 5 μM final concentration.
Experimental groups
Wells in a 96-well plate were randomly divided into
two groups. The first group was not treated with Sel or TNF-α and
served as the control (non-treated group). The second group was
exposed to 25 ng/ml of TNF-α for 24 h to induce cell death
(TNF-α-treated). After 24 h, this group was further divided into
four subgroups, a vehicle-treated group and three groups treated
with different concentrations of selenite. The first subgroup of
cells (TNF-α+vehicle-treated group) received a comparable volume of
distilled water, whereas the other three (TNF-α+selenite-treated
groups) received different concentrations of selenite (1.25, 2.50
and 5.00 μM) for 24 h in order to determine its optimal
concentration. From all the above groups, three (non-treated,
TNF-α+vehicle and TNF-α+5.0 μM selenite-treated) were retained for
further analyses.
Cell viability assay
For the cell viability assay, SH-SY5Y cells were
seeded in 96-well plates at a density of 4×104 cells/200
μl and were grown for 24 h in a 37°C incubator. Upon reaching
70–80% confluence, cells were exposed to RPMI-1640 medium
containing distilled water (vehicle) or TNF-α dissolved in
distilled water along with different concentrations of selenite for
another 24 h. Cell proliferation was then determined using the
tetrazolium compound MTT assay (cat. no. M2128; Sigma-Aldrich).
Next, the supernatants from the vehicle or the selenite-treated
cells were discarded, 200 μl of fresh RPMI-1640 medium and 50 μl of
MTT solution (2 mg/ml in phosphate-buffered saline) were added to
each well, and the samples were incubated at 37°C. Reduction of MTT
by viable cells to insoluble purple formazan dye crystals was
evaluated in a 220-μl sample recovered after 4 h. The formazan
precipitate was dissolved in dimethyl sulphoxide, and the
absorbance of the samples was directly read at 570 nm using a
SoftMax Pro 5 spectrophotometer (Molecular Devices, Sunnyvale, CA,
USA). The cell number was indirectly evaluated from the absorbance
values, allowing to quantify changes in cell proliferation.
Immunofluorescence and
4′,6-diamidino-2-phenylindole (DAPI) staining
SH-SY5Y cells were initially seeded in 24-well
plates at a density of 1×105 cells/ml, and then grown
for 24 h in a 37°C incubator. Upon reaching 70–80% confluence,
cells were exposed to RPMI-1640 medium containing distilled water
(vehicle) or selenite dissolved in distilled water for 24 h after
TNF-α treatment. Cells were next fixed with formaldehyde (Junsei
Chemical Co. Ltd., Tokyo, Japan) for 1 h and then permeabilized
with 1% Triton X-100 for 5 min. After blocking with 0.5% bovine
serum albumin for 1 h, cells were incubated overnight at 4°C with
an antibody targeting the p-tau protein at the Ser404 site (cat.
no. sc-12952; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Cells in each well were then washed with washing buffer (137 mmol/l
NaCl, 2.7 mmol/l KCl, 10 mmol/l Na2HPO4, 2
mmol/l KH2PO4 and 0.05% Tween-20) and
incubated at room temperature for 2 h with fluorescein
isothiocyanate-conjugated goat anti-rabbit IgG (Zymed Laboratories
Inc., South San Francisco, CA, USA) or with 350.25 mg of Molecular
Probes® DAPI (cat. no. D1306; Thermo Fisher Scientific
Inc.). Finally, the fluorescence intensity in each cell was
detected using the IX71 fluorescent microscope (Olympus, Hamburg,
Germany).
Western blot analysis
SH-SY5Y cells were harvested from culture dishes
(100 mm in diameter) following 24-h treatment with distilled water
(vehicle) or selenite dissolved in distilled water subsequent to
TNF-α treatment. These cell pellets were solubilized in PRO-PREP
Protein Extraction Solution (Intron Biotechnology, Inc., Seongnam,
Korea) containing 1.0 mM phenylmethylsulfonyl fluoride, 1.0 mM
ethylenediamine tetraacetic acid, 1 μM Pepstatin A, 1 μM Leupeptin
and 1 μM Aprotinin. The supernatant was collected from these
lysates subsequent to centrifugation for 10 min at 10,000 × g at
4°C. The protein concentrations were then determined using a BCA
Protein Assay Kit (Thermo Fisher Scientific Inc.). Total protein
(30 μg) isolated from neuroblastoma cell lysate was separated by
polyacrylamide gel electrophoresis on a 8–12% sodium dodecyl
sulphate gel for 2 h, and then transferred for 2 h at 40 V onto
nitrocellulose membranes. Each membrane was incubated overnight at
4°C with each of the primary antibodies anti-tau (cat. no. sc-5587;
Santa Cruz Biotechnology, Inc.), anti-p-tau (Ser404, cat. no.
sc-12952; Santa Cruz Biotechnology, Inc.), anti-glycogen synthase
kinase 3β (GSK-3β), anti-p-GSK-3β, anti-Akt, anti-p-Akt (cat. nos.
9325, 9336, 9272 and 4058 respectively; all from Cell Signaling
Technology, Inc., Boston, MA, USA), and anti-β-actin (cat. no.
A5316; Sigma-Aldrich). The membranes were then washed with washing
buffer (as in the immunofluorescence assay above) and incubated
with horseradish peroxidase-conjugated goat anti-rabbit IgG
(dilution, 1:1,000; Zymed Laboratories Inc.) at room temperature
for 1 h. The membrane blots were developed using an enhanced
chemiluminescence (ECL) Reagent Plus kit (Amersham Pharmacia
Biotech, Piscataway, NJ, USA).
Statistical analysis
Non-treated and TNF-α-treated groups were compared
by a one-way analysis of variance using the software SPSS for
Windows (release 10.10, standard version; IBM, Armonk, NY, USA).
Post hoc tests were performed to identify significant differences
between the vehicle and selenite-treated groups. All values were
expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Selenite treatment-mediated recovery in
cell death induced by TNF-α
TNF-α treatment induces pathological cell death
(necrosis) and physiological gene-directed cell death (apoptosis)
in various cells (14). We
therefore examined whether selenite treatment can prevent neuronal
cell death induced by TNF-α treatment. For this purpose, cell
viability was measured in neuroblastoma cells treated with
different concentrations of selenite using the MTT assay and DAPI
staining analysis. TNF-α-treated cells showed a low level of
viability compared to non-treated cells. Cell viability was
significantly increased in cells cotreated with TNF-α and high
concentrations of selenite, but not when lower concentrations of
selenite were used (Fig. 1A).
Similar results were observed with DAPI staining analysis.
Specifically, a higher number of apoptotic cells with fragmented or
compacted nuclei was detected in TNF-α+vehicle-treated cells
relative to the non-treated group, whereas a reduced number of
apoptotic cells was observed in the TNF-α+selenite-treated cells
relative to TNF-α+vehicle-treated cells (Fig. 1B and C). However, there was not a
significant number of apoptotic cells identified in SH-SY5Y normal
cells (Data not shown). Taken together, our findings indicate that
selenite treatment may inhibit the cell death induced by TNF-α
treatment in neuroblastoma cells.
Effects of selenite treatment on tau
hyperphosphorylation
To determine whether selenite treatment can alter
tau hyperphosphorylation, the levels of total tau and
phosphorylated-tau at Ser404 were measured in the
TNF-α+selenite-treated group. Multiple tau isoforms were observed
in the 46–80 kDa range of molecular weight (Fig. 2A). However, it is very difficult to
correspond a specific band as six isoforms, as tau is subject to a
variety of post-translational modifications including
phosphorylation, polyubiquitination and glycation (15). The total level of tau
phosphorylation was significantly higher in the
TNF-α+vehicle-treated compared to the non-treated group. Following
selenite treatment, the level of p-tau was significantly decreased,
while that of unphosphorylated tau was markedly increased. Notably,
the band intensity of the tau isoform 46–50 kDa decreased
three-fold in the TNF-α+selenite-treated relative to the
TNF-α+vehicle-treated group, while the band intensity of tau
isoform with 65–70 kDa was enhanced in the same group. The level of
β-actin, used as an endogenous control, remained constant in all
groups (Fig. 2A and B).
To determine whether hyperphosphorylation of the tau
protein induced by TNF-α is involved in cell death, the cells were
stained with the anti-p-tau antibody, and fluorescence intensity
was observed under a fluorescence microscope. The fluorescence
intensity of the p-tau protein in the TNF-α+vehicle-treated group
was higher than that observed in non-treated cells, while a lower
level of p-tau intensity was observed in TNF-α+selenite-treated
cells compared to TNF-α+vehicle-treated cells, confirmed by
repeated experiments. Furthermore, a high number of cells
containing fragmented or compacted nuclei was detected in
TNF-α+vehicle-treated cells exhibiting a high intensity of p-tau
when compared to the non-treated group. Following TNF-α+selenite
treatment, the number of apoptotic cells with fragmented or
compacted nuclei was reduced (Fig.
2C). Overall, these results suggest that the inhibition of tau
hyperphosphorylation observed after selenite treatment may
associate with the inhibition of apoptotic cell death in
neuroblastoma cells.
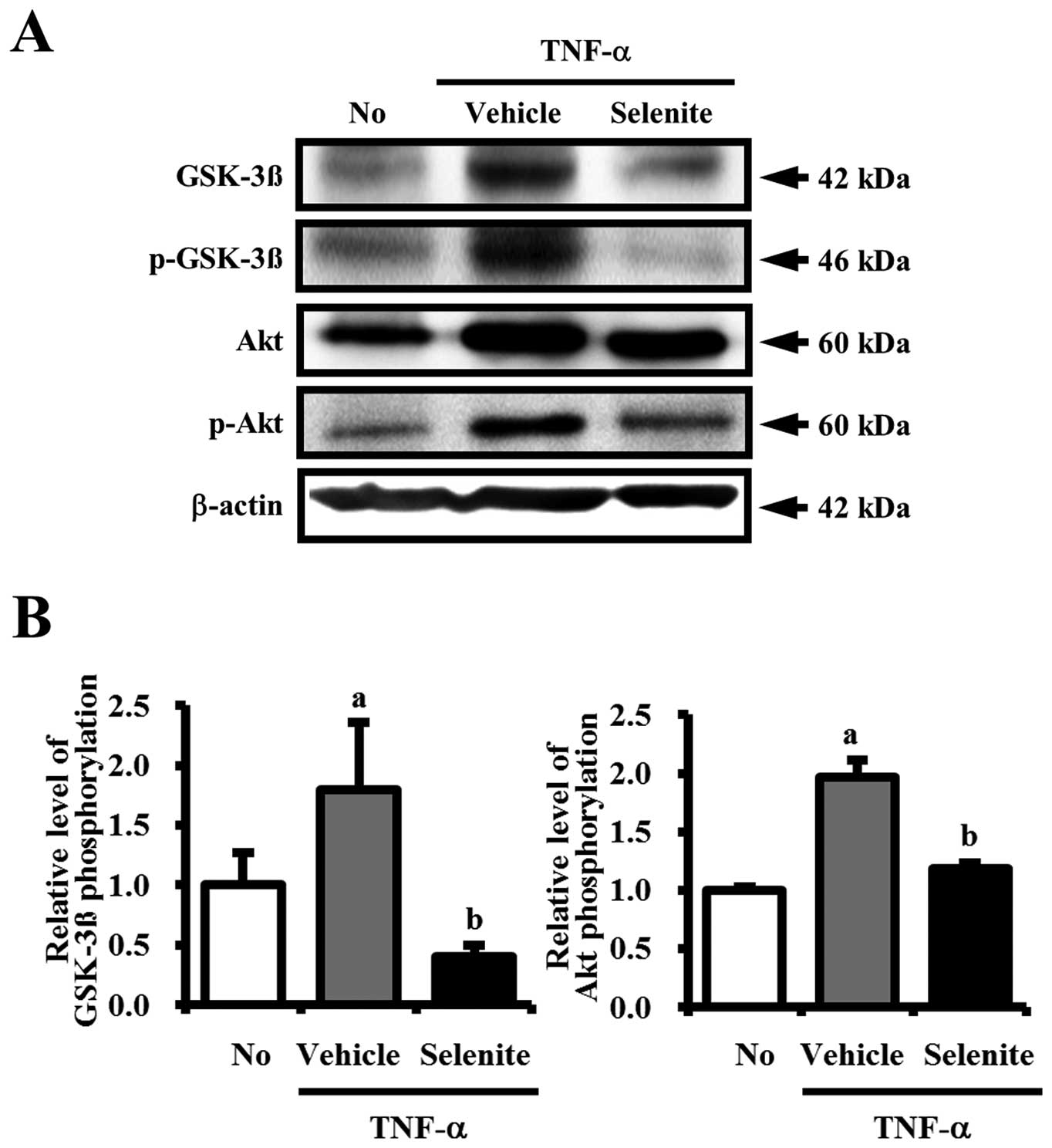
Effects of selenite treatment on GSK-3β
and Akt phosphorylation
The tau protein is a phosphoprotein that potentially
has 80 serine/threonine and 5 tyrosine phosphorylation sites
(15). Phosphorylation of tau was
shown to be regulated by kinases, such as GSK-3β and Akt (15,16).
To determine whether selenite treatment is accompanied by
downregulation of the activity of these kinases, the
phosphorylation level of GSK-3β and Akt was measured. The
phosphorylation level of GSK-3β was higher in the
TNF-α+vehicle-treated group compared to the non-treated group,
although the total level of GSK-3β also increased in response to
TNF-α treatment. However, the level of p-GSK-3β was markedly
decreased in the TNF-α+selenite-treated compared to the
TNF-α+vehicle-treated group. Alterations in the phosphorylation
level of Akt were very similar to those of GSK-3β. Specifically,
the phosphorylation level of Akt was reduced down to a level
similar of that of the non-treated group upon TNF-α+selenite
treatment (Fig. 3A and B). Taken
together, these results indicate that selenite treatment may
inhibit tau phosphorylation through inhibition of GSK-3β and Akt
phosphorylation.
Discussion
The selenium content of healthy adult humans can
vary widely, ranging from 3 to 20 mg, based on the influence of the
natural environment on the selenium content of soils, crops and
human tissues (17). Furthermore,
selenium is widely distributed in several tissues of the human
body. Approximately 30% of tissue selenium is found in the liver,
15% in the kidney, 30% in muscles and 10% in the blood serum
(17). In addition, the effects of
different selenium concentrations on tau phosphorylation have been
shown to slightly differ among studies. In SH-SY5Y neuroblastoma
cells overexpressing human tau (P301L mutation), the
phosphorylation level of tau at Ser422 was markedly reduced in the
10–1,000 μM selenate-treated group, while it was not altered in
response to treatment with <10 μM selenite (11). However, analysis of the activity of
the serine-threonine phosphatase PP2A revealed a significant effect
on tau phosphorylation in fresh complete growth medium containing
100 μM sodium selenate (12). In
this study, the cell viability and tau hyperphosphorylation were
significantly altered in response to treatment with 5 μM selenite.
These results greatly differ from those of previous studies, in
which tau phosphorylation was affected by high concentrations of
selenate. We argue that this difference is due to variations in
treatment conditions, e.g. on the duration of the treatment, and in
the structure of the tested compounds.
Although selenium is an essential trace element, it
is toxic to humans if consumed in excess. A follow-up study of five
Chinese patients suggested that selenosis can be induced by
exceeding the tolerable upper intake level of 400–800 μg/day
(18). In addition, serum selenium
concentrations are categorized by the determination of caused
symptoms in the patients as follows: i) 400–30,000 μg/l, acute
toxicity; ii) 500–1,400 μg/l, chronic toxicity; and iii) <1,400
μg/l, no toxicity (19). In
neuroblastoma cells overexpressing the human tau mutant (P301L)
protein, two forms of selenium were found to exert different
cytotoxic effects. Specifically, selenate cytotoxicity observed
above a dose of 1 mM, while selenite was toxic only above a dose of
10 μM. However, selenite showed no cytotoxicity below a dose of 10
μM (11). In our study, selenite
did not induce any specific toxicity at <5 μM, although cell
viability was not significantly decreased at 2.5 μM (data not
shown). The results of the present study are very similar with
those of a previous study although above data was not measured
toxicity at a dose of 5 μM.
Sodium selenate has been reported to reduce tau
phosphorylation both in vitro and in vivo. For
example, selenate-treated cells showed a dose-dependent reduction
in tau phosphorylation at multiple phosphorylation sites, including
pS422, 12E8 and PHF-1 (11). In
addition, the same study showed that two independent tau transgenic
mice models with NFT pathology exhibit a significant reduction in
tau phosphorylation and complete abrogation of NFT formation after
chronic oral treatment with sodium selenate (11). Acute treatment with sodium selenate
rapidly reduced tau phosphorylation in either neuroblastoma cells
or healthy aged mice, through regulation of PP2A, while it induced
improved spatial learning and memory and decreased tau
phosphorylation in TAU441 transgenic mice (12). The present study is the first to
demonstrate the effects of sodium selenite on tau phosphorylation
induced by TNF-α treatment. Specifically, cells treated with TNF-α
and selenite recovered to the level of non-treated cells in terms
of cell viability, tau hyperphosphorylation and activity of tau
kinases. These results are in agreement with those of previous
studies (12), which suggested
that sodium selenate may mitigate tau phosphorylation and
specifically activate PP2A in neuroblastoma cells and transgenic
mice.
Taken together, our results showed that sodium
selenite is associated with mitigation of TNF-α-induced
pathogenesis, by inhibiting cell death, tau hyperphosphorylation
and activation of kinases in neuroblastoma cells. Our findings
suggest that sodium selenite may play a crucial role in the
regulation of tau phosphorylation via modulation of the activity of
tau kinases, and it thus may serve as a new therapeutic agent in AD
treatment by modulating tau phosphorylation.
Acknowledgements
This study was supported by the 2013 Specialization
Project Research Grant funded by the Pusan National University.
References
|
1
|
Hanisch UK: Microglia as a source and
target of cytokines. Glia. 40:140–155. 2002.PubMed/NCBI
|
|
2
|
Klintworth H, Garden G and Xia Z: Rotenone
and paraquat do not directly activate microglia or induce
inflammatory cytokine release. Neurosci Lett. 462:1–5. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Montgomery SL and Bowers WJ: Tumor
necrosis factor-alpha and the roles it plays in homeostatic and
degenerative processes within the central nervous system. J
Neuroimmune Pharmacol. 7:42–59. 2012. View Article : Google Scholar
|
|
4
|
Rubio-Perez JM and Morillas-Ruiz JM: A
review: inflammatory process in Alzheimer’s disease, role of
cytokines. ScientificWorldJournal. 2012:7563572012.
|
|
5
|
Breder CD, Tsujimoto M, Terano Y, Scott DW
and Saper CB: Distribution and characterization of tumor necrosis
factor-α-like immunoreactivity in the murine central nervous
system. J Comp Neurol. 337:543–567. 1993.
|
|
6
|
Swardfager W, Lanctot K, Rothenburg L,
Wong A, Cappell J and Herrmann N: A meta-analysis of cytokines in
Alzheimer’s disease. Biol Psychiatry. 68:930–941. 2010.
|
|
7
|
Dowlati Y, Herrmann N, Swardfager W, Liu
H, Sham L, Reim EK and Lanctot KL: A meta-analysis of cytokines in
major depression. Biol Psychiatry. 67:446–457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Locksley RM, Killeen N and Lenardo MJ: The
TNF and TNF receptor superfamilies: integrating mammalian biology.
Cell. 104:487–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brynskov J, Foegh P, Pedersen G, Ellervik
C, Kirkegaard T, Bingham A and Saermark T: Tumour necrosis factor
alpha converting enzyme (TACE) activity in the colonic mucosa of
patients with inflammatory bowel disease. Gut. 51:37–43. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilber CG: Toxicology of selenium: a
review. Clin Toxicol. 17:171–230. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Eersel J, Ke YD, Liu X, Delerue F,
Kril JJ, Gotz J and Ittner LM: Sodium selenate mitigates tau
pathology, neurodegeneration, and functional deficits in
Alzheimer’s disease models. Proc Natl Acad Sci USA.
107:13888–13893. 2010.PubMed/NCBI
|
|
12
|
Corcoran NM, Martin D, Hutter-Paier B,
Windisch M, Nguyen T, Nheu L, Sundstrom LE, Costello AJ and Hovens
CM: Sodium selenate specifically activates PP2A phosphatase,
dephosphorylates tau and reverses memory deficits in an Alzheimer’s
disease model. J Clin Neurosci. 17:1025–1033. 2010.PubMed/NCBI
|
|
13
|
Haratake M, Yoshida S, Mandai M, Fuchigami
T and Nakayama M: Elevated amyloid-β plaque deposition in dietary
selenium-deficient Tg2576 transgenic mice. Metallomics. 5:479–483.
2013.
|
|
14
|
Sarraf CE: Tumor necrosis factor and cell
death in tumors (Review). Int J Oncol. 5:1333–1339. 1994.PubMed/NCBI
|
|
15
|
Wang JZ, Xia YY, Grundke-Iqbal I and Iqbal
K: Abnormal hyperphosphorylation of tau: sites, regulation, and
molecular mechanism of neurofibrillary degeneration. J Alzheimers
Dis. 33(Suppl 1): S123–139. 2013.PubMed/NCBI
|
|
16
|
Chung ES, Bok E, Sohn S, Lee YD, Baik HH
and Jin BK: GT1b-induced neurotoxicity is mediated by the
Akt/GSK-3/tau signaling pathway but not caspase-3 in mesencephalic
dopaminergic neurons. BMC Neurosci. 11:742010. View Article : Google Scholar
|
|
17
|
Food and Agriculture Organization of the
United Nations (FAO)/World Health Organization (WHO). Human vitamin
and mineral requirements: report of a Joint FAO/WHO expert
consultation Bangkok, Thailand. FAO; Rome, Italy: pp. 235–255.
2002
|
|
18
|
Yang G and Zhou R: Further observations on
the human maximum safe dietary selenium intake in a seleniferous
area of China. J Trace Elem Electrolytes Health Dis. 8:159–165.
1994.PubMed/NCBI
|
|
19
|
Nuttall KL: Evaluating selenium poisoning.
Ann Clin Lab Sci. 36:409–420. 2006.PubMed/NCBI
|