Introduction
Mild induced hypothermia (MIH) is considered to
improve survival rate and provide a potential neuroprotective
effect in acute neurological injury, with favorable neurological
outcomes. Several studies have identified MIH as a potential
therapeutic strategy for the treatment of traumatic brain injury
(TBI) (1,2). The first study of MIH as a treatment
option for patients with TBI was reported in 1945 (3). MIH, as a non-pharmacological measure,
exerts a complex range of effects in the pathophysiological process
of TBI. More specifically, MIH treatment may not only improve
histopathological and behavioral outcomes, but also has the ability
to reduce intracranial pressure in animal models and clinical
trials of TBI (4,5). However, the molecular and cellular
mechanisms underlying hypothermic protection remain to be
elucidated. The neuroprotective mechanism of MIH is initially
attributed to a reduction in the cerebral metabolic rate of oxygen
(6). Several studies over the last
decade have demonstrated that MIH was able to significantly enhance
glucose utilization, inhibit free radical production and suppress
inflammatory responses and apoptotic pathways. Additionally,
previous studies have indicated that therapeutic MIH may improve
traumatic outcomes, including the modulation of
temperature-sensitive microRNA expression and suggested that MIH
has a beneficial effect on the gene expression profile of the
hippocampus following TBI in rats (7). On the basis of these findings, the
present study assessed the hypothesis that MIH treatment has the
ability to regulate the expression of connexin 43 (Cx43) and
glutamate transporter 1 (GLT-1) in the hippocampus following TBI in
rats.
Gap junctions, composed of proteins from the
connexin family, enable intercellular communication between cells.
It has been reported that gap junctions have important regulatory
effects in essential cellular processes, including electrical
coupling, metabolic transport, proliferation, differentiation and
apoptosis (8). Cx43 is the most
widely and highly expressed gap junction protein in several
different tissues (9). In the
central nervous system, Cx43 is mainly expressed in astrocytes and
forms the major component protein in astrocytic gap junctions. In
addition, astrocytes establish a glial network and communicate
through gap junctions in the brain, indicating a potential
neuroprotective role of astrocytic gap junctions. Mroue et
al found that alterations in the expression of Cx43 could
affect cellular fate (10) and
several studies have demonstrated that Cx43 had a physiological and
pathophysiological impact on a variety of systems (11,12).
In addition, results from our previous study demonstrated that TBI
stimulated the increased expression of Cx43 on the surface of
astrocytes in rats (13). Ohsumi
et al also investigated the expression and distribution of
astrocytic Cx43 gap junctions in the hippocampus and the cortex
following TBI, suggesting that Cx43 participates in the
pathophysiological processes of brain damage (14). Notably, other studies have revealed
that Cx43 is significantly enhanced in the selectively vulnerable
CA1-CA2 region of the hippocampus following brain injury (15).
In addition to being gap-junctional proteins,
connexins may exist in the form of unopposed halves of gap junction
channels, termed hemichannels (16). Cx43-hemichannels in astrocytic gap
junctions remain closed under normal physiological conditions but
they may open following brain injury. Astrocytes have been
demonstrated to release relatively large molecules, including
adenosine triphosphate and glutamate, by the opening of
Cx43-hemichannels, which was involved in the propagation of injury
in the brain (17). Glutamate, a
major excitatory neurotransmitter, causes excitotoxicity and
neuronal cell death at high concentrations. Glutamate transport is
the only mechanism involved in extracellular glutamate clearance
(18). GLT-1, as the major
glutamate transporter, is predominantly expressed in astrocytes.
GLT-1 provides the majority of glutamate clearance capacity by
transporting it into the intracellular space (19). Previous studies have demonstrated
that the mRNA and protein levels of GLT-1 are depressed in the
cortex following TBI, which contributes to an increase in
extracellular glutamate concentration following TBI (20). Therefore, stimulating an increase
in GLT-1 expression following brain injury may improve neuronal
damage and functional deficit.
These findings suggest that Cx43 and GLT-1 may be
potential targets for the treatment of TBI. In the present study,
the potential therapeutic effect of MIH was investigated in a rat
model of TBI and the possibility that treatment with MIH may reduce
the increase in Cx43 expression and reverse post-traumatic GLT-1
suppression in the hippocampus following TBI was assessed.
Materials and methods
Animals and TBI model
All procedures were performed in accordance with the
guidelines of the Chinese Council on Animal Protection and were
approved by Hebei United University Committee for the use of
animals in research (Tangshan, China). A total of 90 male
Sprague-Dawley (SD) rats (aged 12–16 weeks and weighing 350–375 g)
were purchased from Vital River Laboratory Animal Technology Co.,
Ltd. (Bejing, China). The rat model of TBI was induced by using a
modified weight-drop device, as described previously by Marmarou
et al (21). All animals
were anesthetized with pentobarbital sodium (Nembutal; 60 mg/kg). A
midline incision was made to expose the skull between the bregma
and lambda suture lines and a steel disc (diameter, 10 mm;
thickness, 3 mm) was adhered to the exposed skull using dental
acrylic. Animals were moved onto a foam mattress underneath a
weight-drop device, in which a 450 g weight falls freely through a
vertical tube from a height of 1.5 m onto the steel disc. Animals
in the sham group underwent the same surgical procedure without
weight-drop impact. Following treatment, the rats were individually
housed in separate cages. The rats were placed on warming pads for
24 h post-surgery, to aid them in maintaining a regular body
temperature.
Sham, TBI normothermia and MIH treatment
groups
The 90 male SD rats were randomly divided into three
groups (n=30): Sham group, TBI normothermia group (37°C) and
MIH-treated group (33°C). Mild hypothermia was induced, as
described previously (22). The
control of the temperature was initiated 30 min post-TBI.
Temperature probes were used to measure temporalis muscle and
rectal (core) temperature. Selective brain hypothermia was induced
30 min after injury, with a target hypothermic temperature achieved
40 min after TBI and maintained using cooled air and heating lamps
for 4 h. Animals were then rewarmed slowly over a 1.5 h period.
Rats were anesthetized and sacrificed at 6, 12, 24, 48 and 72 h
following TBI.
Evaluation of brain edema
Brain edema was evaluated by analysis of brain water
content, as described previously (23). Rat brains were separated and
weighed immediately with a chemical balance to obtain a wet weight
(WW). Following drying in a desiccating oven for 24 h at 100°C, dry
tissues were weighed again to obtain a constant dry weight (DW).
The percentage of water in the tissues was calculated according to
the following formula: % brain water = (WW − DW)/WW) × 100.
Morris water maze test
Spatial learning ability was assessed using a Morris
water maze, as described previously (24). The Morris water maze consists of a
black circular pool (diameter, 180 cm; depth, 45 cm) filled with
water to a depth of 30 cm (temperature, 26°C). The maze is divided
into four equivalent quadrants: north (N), west (W), south (S) and
east (E). A 2 cm submerged escape platform (12 cm diameter, 28 cm
height, made opaque with paint) was placed in the middle of one of
the quadrants, equidistant from the sidewall and the center of the
pool. Rats were trained to find the platform prior to TBI or sham
surgery. For each trial, the rat was randomly placed at a quadrant
start point (N, S, E or W) facing the wall of the pool. The rats
were permitted a maximum of 60 sec to escape to a platform, rats
which failed to escape within 90 sec were placed on the platform
for a maximum of 20 sec and returned to the cage for a new trial
(intertrial interval 20 sec). The escape latency performance was
recorded using a video camera and video tracking system (HVS
Imaging, Hampton, UK). The average escape latency of a total of
five trials was calculated. This test was conducted at 24, 48 and
72 h post TBI.
Immunohistochemical analysis
Immunohistochemical analysis was performed using an
SABC immunohistochemistry kit (Wuhan Boster Biological Engineering
Co., Ltd., Wuhan, China) according to the manufacturer’s
instructions. Paraffin embedded brain tissue sections (5 μm) were
heated for 30 min at 60°C, dewaxed and rehydrated, followed by
microwave antigen retrieval procedures. In brief, sections in
Citrate salt buffer (Wuhan Boster Biological Engineering Co., Ltd.,
Wuhan, China) were heated in a microwave oven for 5 min. Endogenous
peroxidase was inactivated with 3% H2O2 for
10 min at room temperature. The sections were incubated in 5%
bovine serum albumin (Gibco BRL, Carlsbad, CA, USA) solution for 20
min, to inhibit the nonspecific binding. Subsequently, sections
were incubated overnight at 4°C with rabbit anti-Cx43, GLT-1
polyclonal antibodies (Santa Cruz Biotechnology Inc., Santa Cruz,
CA, USA) diluted 1:50 and then with polyclonal horseradish
peroxidase-conjugated anti-rabbit immunoglobulin G (IgG; Wuhan
Boster Biological Engineering Co., Ltd., Wuhan, China) antibodies
for 30 min. Diaminobenzidine (Wuhan Boster Biological Engineering
Co., Ltd.) was used to reveal the immunohistochemical reaction.
Western blot analysis
Total proteins from the hippocampal CA1 regions were
rapidly isolated and extracted. The concentration of total protein
was measured using a bicinchoninic acid reagent (Solarbio Science
and Technology Co., Ltd., Beijing, China). Samples were subjected
to sodium dodecyl sulfate polyacrylamide gel electrophoresis. The
proteins separated on the gel were transferred onto polyvinylidene
difluoride membranes (Roche Diagnostics GmBH, Mannheim, Germany).
Blots were inhibited with 5% fat-free dry milk for 2 h at room
temperature and were incubated overnight at 4°C with rabbit
anti-Cx43, GLT-1 and β-actin polyclonal antibody (Santa Cruz
Biotechnology, Inc.; diluted 1:200). The blots were then incubated
with horseradish peroxidase conjugated anti-rabbit IgG (Cell
Signaling Technology, Inc., Danvers, MA, USA; diluted 1:5,000) for
2 h at room temperature. Following incubation, the immunoblot on
the membrane was visible following development with an enhanced
chemiluminescence detection system (ChemiDoc XRS; Bio Rad,
Hercules, CA, USA) and the densitometric signals were quantified
using an imaging program (Image Lab 4.1; Bio-Rad). The
immunoreactive bands of all proteins expressed were normalized to
the intensity of corresponding bands for β-actin. The results of
the western blot analysis were analyzed using National Institutes
of Health Image 1.41 software (Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) was used for statistical
analysis of the data. Statistical analysis was performed using
analysis of variance and subsequently by the Student-Newman-Keuls
post-hoc test or Student’s t-test (comparison of two means).
P<0.05 was considered to indicate a statistically significant
difference.
Results
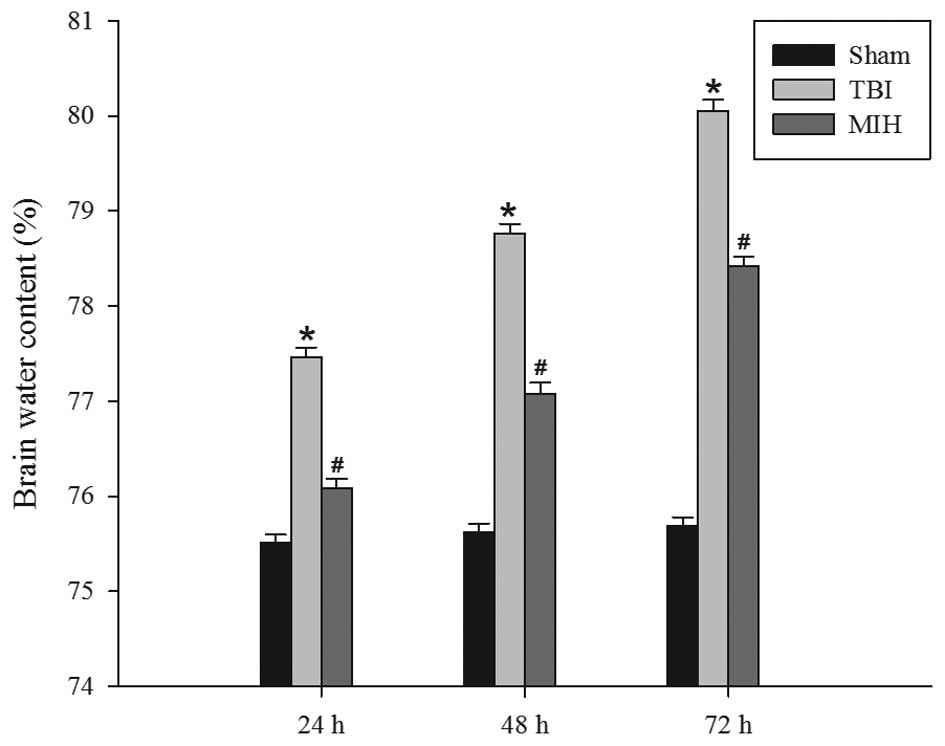
MIH treatment attenuates TBI-induced
brain edema
The wet-dry weight method was used to evaluate brain
edema. As shown in Fig. 1, TBI
induced a significant increase in brain edema at 24, 48 and 72 h
compared with the sham group. However, treatment with MIH post-TBI
significantly reduced brain edema compared with the TBI group at
the corresponding time points.
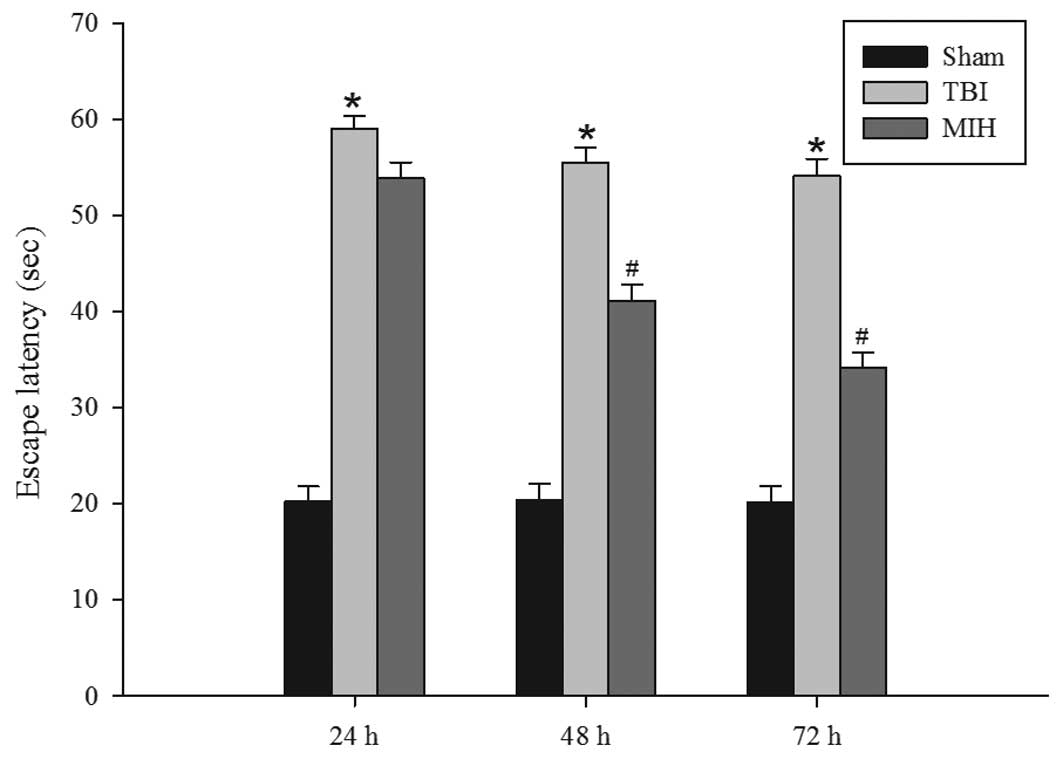
MIH treatment improves learning and
memory ability
Following the observation that therapeutic MIH
treatment attenuated brain edema post-TBI, the present study next
assessed whether MIH was able to improve spatial learning ability
using a Morris water maze. As shown in Fig. 2, TBI caused a significant deficit
in spatial learning compared with the sham group at 24, 48 and 72
h. However, MIH treatment post-TBI markedly reduced the escape
latency compared with the TBI group at 48 and 72 h.
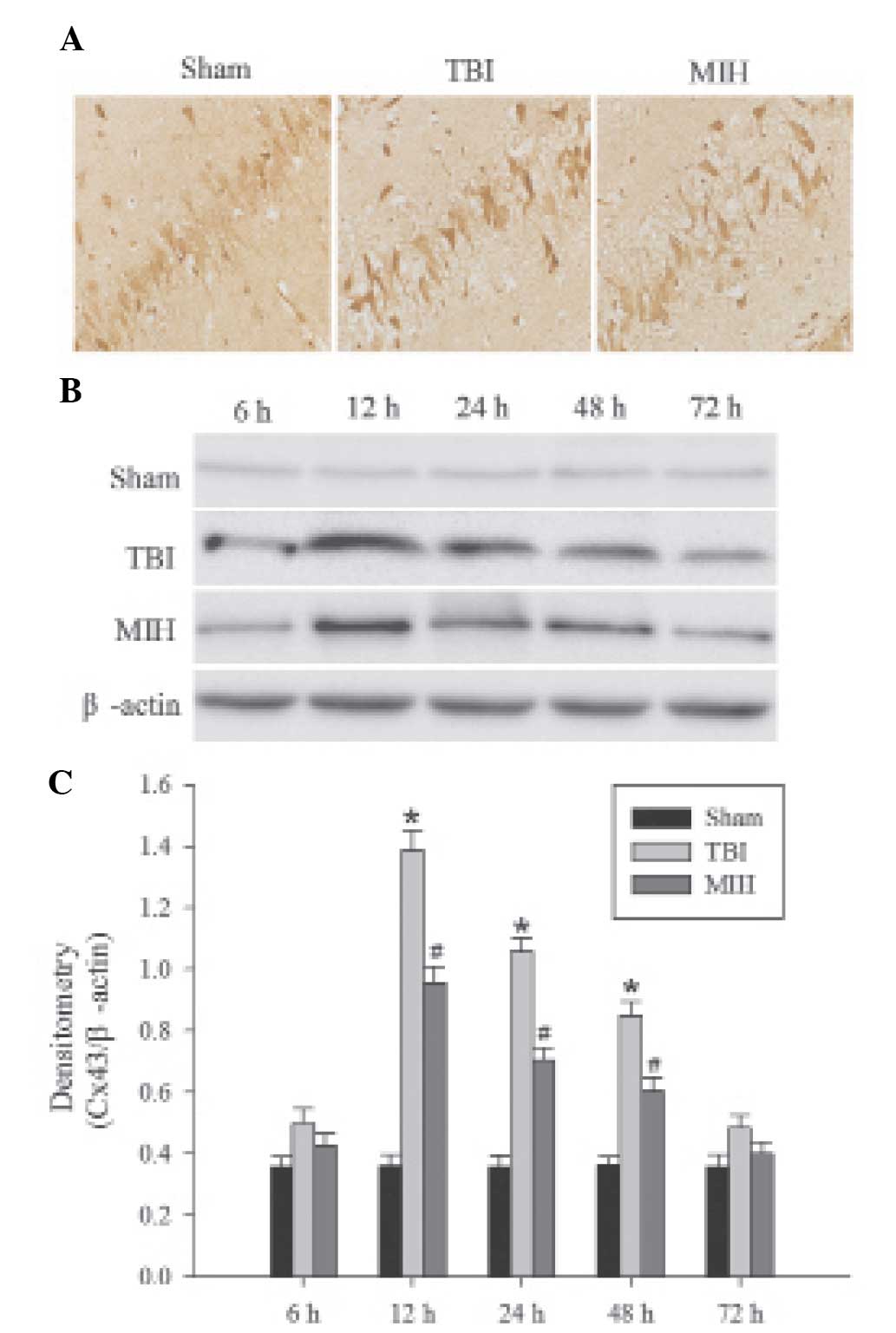
MIH treatment depresses Cx43 protein
expression
Immunohistochemical analysis was used to determine
the localization of Cx43 protein. As shown in Fig. 3A, the brown particles observed in
the cells indicated positive results. The majority of brown
particles appeared in the astrocytes of the hippocampus,
particularly in the membranes of astrocytes and were occasionally
expressed in the cytoplasm. Western blot analysis and
immunohistochemical analysis revealed the levels of Cx43 expression
in the different groups. As shown in Fig. 3B and C, the protein expression of
Cx43 in the hippocampus was present at a consistently low level in
the sham group, whereas Cx43 exhibited a significant increase at
different time points in the TBI group. The expression of Cx43
protein in the hippocampus was increased at 6 h post TBI, peaked at
12 h and then gradually decreased, but remained higher than the
expression levels in the sham group. Treatment with MIH following
TBI caused a significant reduction in the TBI-induced upregulation
of Cx43 expression compared with the TBI group at 12, 24 and 48
h.
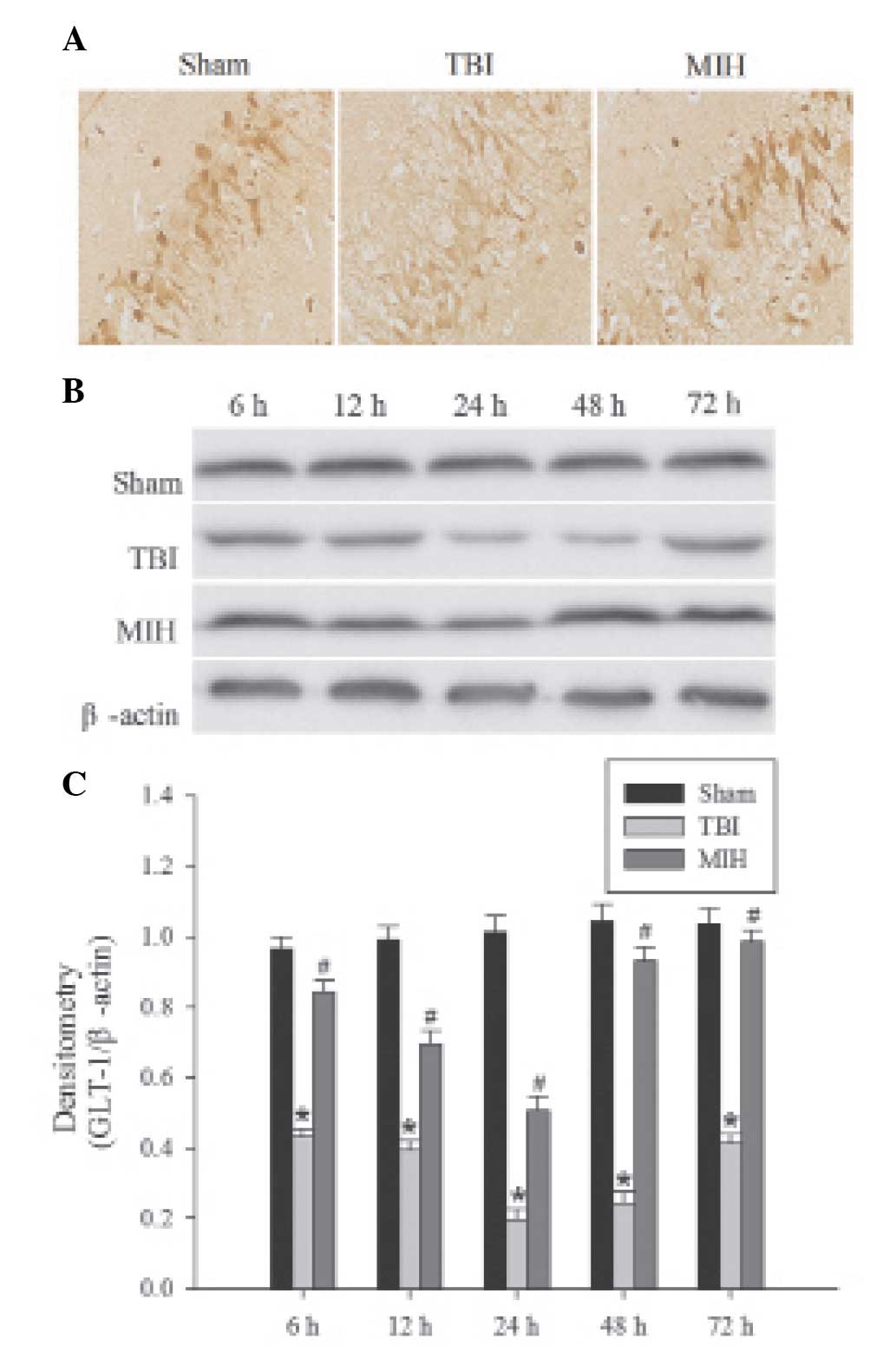
MIH treatment upregulates GLT-1 protein
expression
The localization of GLT-1 protein was determined by
immunohistochemical analysis. GLT-1, a membrane receptor, is
predominantly expressed in astrocytes (Fig. 4A). Brown particles were mainly
observed in the membranes of astrocytes in the hippocampus and, in
part, in the cytoplasm. The levels of GLT-1 were detected by
western blot analysis. In the sham group, GLT-1 protein in the
hippocampus demonstrated basal expression. As shown in Fig. 4B and C, the expression of GLT-1 was
suppressed at 6 h post TBI, persisted at a low level until 24 h
after TBI and then gradually increased, but remained lower than the
GLT-1 expression in the sham group. However, treatment with MIH
significantly inhibited the downregulation of GLT-1 protein
compared with the TBI groups at 6, 12, 24, 48 and 72 h.
Discussion
TBI, an extremely complex neurological condition,
leads to acute functional deficit in the brain. However, the
molecular events underlying TBI remain to be elucidated. Currently,
TBI is a serious health issue with additional social and
psychological burdens and the increasing prevalence and high
mortality rate of TBI poses a challenge in neurological research.
The management of TBI aims to attenuate the amount of secondary
brain injury occurring immediately following primary brain insult.
Notably, several studies have revealed that MIH treatment following
TBI contributes to a reduction in secondary cerebral damage and
exerts significant neuroprotective effects (25). In the present study, the
neuroprotective action of MIH was confirmed by reducing brain edema
and facilitating the recovery of cognitive function in a rat model
of TBI. The results suggested a potential role of MIH in limiting
secondary damage and promoting improved functional outcome
following TBI, which is consistent with the results of Marion and
Bullock (26) and Dietrich et
al (27). Furthermore,
immunohistochemical results demonstrated that CX43 was widely
located in the membrane of astrocytes and Cx43 was predominantly
overexpressed in the hippocampal CA1 following TBI. The results of
the present study clearly demonstrated that Cx43 immunoreactivity
was increased in experimental models of brain injury.
Cx43-hemichannels remain closed under normal physiological
conditions but can open and release glutamate following TBI.
Excessive extracellular glutamate following TBI contributes to
excitotoxic cell death and is important in the development of
secondary injuries (28). However,
the levels of glutamate may be effectively controlled by astrocytic
glutamate transporters, including GLT-1. The present study revealed
that GLT-1 proteins were predominantly localized in astrocytes and
were downregulated shortly following TBI. In conclusion, the
downregulation of GLT-1 further exacerbated extracellular glutamate
accumulation, which resulted in neuronal loss and associated
neurological deficits, which have been associated with TBI-induced
secondary brain injury in hippocampal tissues (18). Therefore, inhibition of
Cx43-hemichannels or upregulation of the expression of GLT-1 can
improve neural outcomes, representing novel therapeutic targets for
reducing the severity of TBI.
In the present study, the therapeutic potential of
MIH as a neuroprotective strategy for the treatment of TBI was
investigated. MIH treatment following TBI was found to be
beneficial for neurological recovery. In particular, MIH
significantly attenuated brain edema and improved learning and
memory ability following TBI in the rat model. However, the
underlying mechanisms are yet to be elucidated. The protective
effect of hypothermia may involve multiple pathways, including
preventing the initiation of apoptotic/necrotic processes, reducing
cellular/neuronal damage and inhibiting excitotoxicity (29). An important finding of the present
study was that MIH treatment downregulated levels of Cx43 and
upregulated the expression of GLT-1 in the hippocampus following
TBI. Based on the available evidence, the beneficial effects of MIH
in TBI treatment may be explained as follows: MIH reduced the
protein expression of Cx43 in the astrocytes and decreased the
release of extracellular glutamate from Cx43-hemichannels, or MIH
also reversed GLT-1 reduction in the astrocytes and further
enhanced the uptake of extracellular glutamate. Taken together, MIH
may have the ability to balance glutamate content between
intracellular and extracellular environments, therefore having a
neuroprotective role following TBI.
MIH treatment exerts a neuroprotective effect on TBI
by attenuating brain edema and improving neurological outcomes.
These protective effects may be associated with reversing the
increase in Cx43 and upregulation of GLT-1 levels following TBI.
The present study provided evidence in vivo that MIH has the
potential to become a valuable neuroprotective intervention in
numerous neurological events.
Acknowledgements
The present study was supported by a grant from the
Science and Technology Development Project of Tangshan City (no.
12140209A-31).
Abbreviations:
|
TBI
|
traumatic brain injury
|
|
MIH
|
mild induced hypothermia
|
|
Cx43
|
connexin 43
|
|
GLT-1
|
glutamate transporter 1
|
|
TH
|
therapeutic hypothermia
|
|
CNS
|
central nervous system
|
References
|
1
|
Cheng SX, Zhang S, Sun HT and Tu Y:
Effects of mild hypothermia treatment on rat hippocampal β-amyloid
expression following traumatic brain injury. Ther Hypothermia Temp
Manag. 3:132–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jia F, Mao Q, Liang YM and Jiang JY:
Effect of post-traumatic mild hypothermia on hippocampal cell death
after traumatic brain injury in rats. J Neurotrauma. 26:243–252.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fay T: Observations on generalized
refrigeration in cases of severe cerebral trauma. Assoc Res Nerv
Ment Dis Proc. 24:611–619. 1945.
|
|
4
|
Kramer C, Freeman WD, Larson JS, et al:
Therapeutic hypothermia for severe traumatic brain injury: a
critically appraised topic. Neurologist. 18:173–177. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andrews PJ, Sinclair LH, Harris B, et al:
Study of therapeutic hypothermia (32 to 35°C) for intracranial
pressure reduction after traumatic brain injury (the
Eurotherm3235Trial): outcome of the pilot phase of the trial.
Trials. 14:1–8. 2013. View Article : Google Scholar
|
|
6
|
Liu L and Yenari MA: Therapeutic
hypothermia: neuroprotective mechanisms. Front Biosci. 12:816–825.
2007. View Article : Google Scholar
|
|
7
|
Feng JF, Zhang KM, Jiang JY, Gao GY, Fu XA
and Liang YM: Effect of therapeutic mild hypothermia on the
genomics of the hippocampus after moderate traumatic brain injury
in rats. Neurosurgery. 67:730–742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laird DW: The gap junction proteome and
its relationship to disease. Trends Cell Biol. 20:92–101. 2010.
View Article : Google Scholar
|
|
9
|
Márquez-Rosado L, Solan JL, Dunn CA,
Norris RP and Lampe PD: Connexin43 phosphorylation in brain,
cardiac, endothelial and epithelial tissues. Biochi Biophys Acta.
1818:1985–1992. 2012. View Article : Google Scholar
|
|
10
|
Mroue R, El-Sabban M and Talhouk R:
Connexins and the gap in context. Integ Biol. 3:255–266. 2011.
View Article : Google Scholar
|
|
11
|
Ongstad EL, O’Quinn MP, Ghatnekar GS, Yost
MJ and Gourdie G: A connexin43 mimetic peptide promotes
regenerative healing and improves mechanical properties in skin and
heart. Adv Wound Care (New Rochelle). 2:55–62. 2013. View Article : Google Scholar
|
|
12
|
Yu M, Zhang C, Li L, Dong S, Zhang N and
Tong X: Cx43 reverses the resistance of A549 lung adenocarcinoma
cells to cisplatin by inhibiting EMT. Oncol Rep. 31:2751–2758.
2014.PubMed/NCBI
|
|
13
|
Sun LQ, Gao JL, Cui CM, et al: Astrocytic
p-connexin43 regulates neuronal autophagy in the hippocampus
following traumatic brain injury in rats. Mol Med Rep. 9:77–82.
2014.
|
|
14
|
Ohsumi A, Nawashiro H, Otani N, Ooigawa H,
Toyooka T and Shima K: Temporal and spatial profile of
phosphorylated connexin43 after traumatic brain injury in rats. J
Neurotrauma. 27:1255–1263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rami A, Volkmann T and Winckler J:
Effective reduction of neuronal death by inhibiting gap junctional
intercellular communication in a rodent model of global transient
cerebral ischemia. Exp Neurol. 170:297–304. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang S, Wang YQ, Xu CF, Li YN, Guo R and
Li L: Involvement of connexin43 in the infrasonic noise-induced
glutamate release by cultured astrocytes. Neurochem Res.
39:833–842. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Obrenovitch TP and Urenjak J: Is high
extracellular glutamate the key to excitotoxicity in traumatic
brain injury? J Neurotrauma. 14:677–698. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yi JH and Hazell AS: Excitotoxic
mechanisms and the role of astrocytic glutamate transporters in
traumatic brain injury. Neurochem Int. 48:394–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Ma A, Zhu W, et al: The role of
connexin 43 and hemichannels correlated with the astrocytic death
following ischemia/reperfusion insult. Cell Mol Neurobiol.
33:401–410. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goodrich GS, Kabakov AY, Hameed MQ, Dhamne
SC, Rosenberg PA and Rotenberg A: Ceftriaxone treatment after
traumatic brain injury restores expression of the glutamate
transporter, GLT-1, reduces regional gliosis and reduces
post-traumatic seizures in the rat. J Neurotrauma. 30:1434–1441.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats: Part I: Pathophysiology and biomechanics. J
Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Truettner JS, Alonso OF, Bramlett HM and
Dietrich WD: Therapeutic hypothermia alters microRNA responses to
traumatic brain injury in rats. J Cereb Blood Flow Metab.
31:1897–1907. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang J, Liu J, Zhou C, et al: Mmp-9
deficiency enhances collagenase-induced intracerebral hemorrhage
and brain injury in mutant mice. J Cereb Blood Flow Metab.
24:1133–1145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hui-guo L, Kui L, Yan-ning Z and Yong-jian
X: Apocynin attenuate spatial learning deficits and oxidative
responses to intermittent hypoxia. Sleep Med. 11:205–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tokutomi T, Morimoto K, Miyagi T,
Yamaguchi S, Ishikawa K and Shigemori M: Optimal temperature for
the management of severe traumatic brain injury: effect of
hypothermia on intracranial pressure, systemic and intracranial
hemodynamics, and metabolism. Neurosurgery. 52:102–112. 2003.
|
|
26
|
Marion D and Bullock MR: Current and
future role of therapeutic hypothermia. J Neurotrauma. 26:455–467.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dietrich WD, Atkins CM and Bramlett HM:
Protection in animal models of brain and spinal cord injury with
mild to moderate hypothermia. J Neurotrauma. 26:301–312. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Obrenovitch TP and Urenjak J: Is high
extracellular glutamate the key to excitotoxicity in traumatic
brain injury? J Neurotrauma. 14:677–698. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Broessner G, Fischer M, Schubert G,
Metzler B and Schmutzhard E: Update on therapeutic temperature
management. Critical Care. 16:A12012.
|