Introduction
Chronic kidney disease (CKD) is a growing health
problem worldwide (1), which
manifests as a progressive loss of renal function. Numerous
factors, including oxidative stress, systemic inflammation,
hypertension, and dyslipidemia, contribute to the onset and
progression of CKD (2). Lipid
abnormalities in patients with CKD usually consist of reduced
high-density lipoprotein (HDL) concentrations and elevated plasma
triglyceride, low-density lipoprotein (LDL), and oxidized lipids
and lipoproteins (3). HDL
facilitates cholesterol efflux from peripheral tissues and delivers
cholesterol to the liver for excretion, therefore it has a key role
in reverse cholesterol transport (4). HDL confers protection from
cardiovascular disease due to its high anti-oxidant and
anti-inflammatory activities. LDL has converse roles to HDL in the
regulation of cholesterol transport. Oxidized LDL is capable of
inducing inflammatory responses and is therefore implicated in the
progression of numerous inflammatory disorders, such as
atherosclerosis (5) and CKD
(3).
The endoplasmic reticulum (ER) is a central
organelle of eukaryotic cells and has principle functions in lipid
synthesis and protein folding, maturation, and transport.
Disturbances to the normal functions of the ER (e.g. due to redox
dysregulation, aberrant calcium regulation, and glucose
deprivation) may result in the accumulation of unfolded or
misfolded proteins in the ER, a state known as ER stress (6). Three major ER stress response
transducers have previously been identified: Activating
transcription factor-6, inositol-requiring enzyme-1α (IRE1α), and
protein kinase RNA-like endoplasmic reticulum kinase (7). IRE1α is activated by homodimerization
and autophosphorylation under ER stress. It functions as an
endoribonuclease which splices X-box-binding protein-1 (XBP-1)
mRNA, yielding a potent transcriptional activator. Splicing of
XBP-1 has previously been shown to initiate the transcription of
genes involved in protein folding, transport and ER-associated
protein degradation (7).
ER stress initially serves as an adaptive response
but may lead to cell death, if severe or prolonged ER dysfunction
occurs. Previous research has demonstrated the importance of ER
stress in the pathophysiology of both acute and chronic kidney
diseases (8). Chiang et al
(9) reported that overwhelming ER
stress may induce renal cell apoptosis and subsequent fibrosis.
However, numerous studies have recently supported a protective role
of the induction of ER stress in cell damage (10–12).
Li et al (10) reported
that ER stress preconditioning mitigated retinal endothelial
inflammation, which was mediated through activation of the
XBP-1-mediated unfolded protein response (UPR) and inhibition of
NF-κB signaling. In a rat model, preconditioning with ER stress
ameliorated mesangioproliferative glomerulonephritis (12). These previous findings suggest that
transiently targeting ER stress may have therapeutic benefits in
the treatment of inflammatory diseases.
Inflammation is an important mechanism leading to
glomerular damage in CKD (13).
Native and modified LDL have previously been shown to increase the
production of numerous inflammatory mediators, including
interleukin-6 (IL-6), CD40, and macrophage migration inhibitory
factor, in human mesangial cells (HMCs) (14). The present study aimed to
investigate whether ER stress preconditioning could attenuate
LDL-induced inflammatory responses in HMCs, and to explore the
associated molecular mechanisms.
Materials and methods
Chemical reagents and antibodies
LDL was purchased from ProSpec (East Brunswick, NJ,
USA) and tunicamycin from Sigma-Aldrich (St. Louis, MO, USA). Mouse
anti-human monoclonal antibodies anti-XBP1 and anti-β-actin were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA),
anti-phospho-NF-κB p65 (Ser536), and anti-phospho-IκB kinase
(IKK)α/β (Ser176/180) were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA), and anti-phospho-IRE1α and anti-IRE1α were
purchased from Novus Biologicals (Littleton, CO., USA).
Cell culture and drug treatment
Primary HMCs were obtained from the Key Laboratory
of Infectious Diseases of Chongqing Medical University (Chongqing,
China). The cells were cultured in Dulbecco’s Modified Eagle’s
Medium, supplemented with 10% fetal bovine serum, 100 μg/ml
streptomycin, and 100 U/ml penicillin (Invitrogen Life
Technologies, Carlsbad, CA, USA), at 37°C in an atmosphere
containing 5% CO2. The experiments were performed with
cells taken from passages 3–8.
The HMCs were treated with LDL (200 nm) for various
durations (1–24 h), and then subjected to gene expression analysis.
To assess the effects of ER stress preconditioning on the
LDL-induced inflammatory responses, the cells were exposed for 8 h
to a low dose (0.1 μg/ml) of tunicamycin, an ER stress inducer,
followed by treatment with LDL.
Plasmids, small interfering RNA (siRNA)
oligonucleotides, and transfection
The full-length human XBP1 cDNA was amplified and
subcloned into an expression vector pEGFP-C1 (ClonTech, Palo Alto,
CA, USA), which encoded a C-terminal green fluorescent protein tag.
Specific siRNAs targeting human IRE1α and XBP1 were synthesized by
Invitrogen Life Technologies, with the following sense sequences:
IRE1α, 5′-GUCCCACUUUGUGUCCAAUTT-3′; and XBP1,
5′-CCAAGCUGGAAGCCAUUAATT-3′. A scrambled siRNA sequence,
5′-UUCUCCGAACGUGUCACGUTT-3′, was used as a negative control. The
final concentration of each siRNA was 2 μM. The HMCs were seeded in
6-well plates, at a density of 5×104 cells/well, 24 h
prior to transfection. The siRNA molecules were transfected into
the cells using Lipofectamine® 2000 reagent, according
to the manufacturer’s instructions (Invitrogen Life Technologies).
The transfection efficiency was determined by western blot analysis
of the corresponding protein levels, 24 h following siRNA
transfection.
Quantitative polymerase chain reaction
(qPCR)
Total cellular RNA was extracted using the RNeasy
kit, according to the manufacturer’s instructions (Qiagen, Hilden,
Germany). Reverse transcription was performed using the AMV First
Strand cDNA Synthesis kit (Bio Basic Inc., Amhurst, NY, USA). The
qPCR was conducted using an Applied Biosystems StepOnePlus
Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and
the SYBR® green PCR master mix (Applied Biosystems).
GAPDH was amplified in a parallel reaction, and was used as an
internal quantitative control. The cycle threshold (Ct) was
calculated for each gene tested. The relative mRNA expression
levels were normalized to those of GAPDH, using the
2−ΔΔCt method (15).
Primers are listed in Table I.
 | Table ISequences of the primers for RT-qPCR
analysis. |
Table I
Sequences of the primers for RT-qPCR
analysis.
| Gene | Sequence (5′-3′) |
|---|
| IL-6 | Forward:
TGCAATAACCACCCCTGACC
Reverse: ATTTGCCGAAGAGCCCTCAG |
| TNF-α | Forward:
CTTCTCGAACCCCGAGTGAC
Reverse: TGAGGTACAGGCCCTCTGAT |
| CD40 | Forward:
ACCCTTGGACAAGCTGTGAG
Reverse: GGCAAACAGGATCCCGAAGA |
| MCP-1 | Forward:
AGCAGCAAGTGTCCCAAAGA
Reverse: TTTGCTTGTCCAGGTGGTCC |
| XBP1 | Forward:
CAGTTGTCACCCCTCCAGAA
Reverse: GGCTGGTAAGGAACTGGGTC |
| IRE1α | Forward:
AAAACTACGCCTCCCCTGTG
Reverse: GTCAGATAGCGCAGGGTCTC |
| GAPDH | Forward:
CAATGACCCCTTCATTGACC
Reverse: GACAAGCTTCCCGTTCTCAG |
Western blot analysis
Following LDL treatment, the cells were lysed in
lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% NP-40, and 0.1%
SDS), supplemented with protease inhibitors (2 μg/ml aprotinin, 2
μg/ml leupeptin, and 1 mm phenylmethylsulfonyl fluoride). The
protein samples were separated by electrophoresis on polyacrylamide
gels containing 0.1% SDS, and then transferred to nitrocellulose
membranes (Pierce Biotechnology, Inc., Rockford, IL, USA). The
membranes were incubated with the individual primary antibodies
(goat anti-mouse NF-κB p65 antibody, sc-166748; Santa Cruz
Biotechnology, Inc; dilution, 1:1,000) overnight at 4°C, followed
by incubation for 1 h with the appropriate secondary antibodies
(Alexa Fluor® 488-labeled goat anti-rabbit
immunoglobulin G, #4412; Cell Signaling Technology, Inc.; dilution,
1:200), at room temperature. Immune complexes were visualized using
enzyme-linked chemiluminescence (GE Heakthcare Life Sciences,
Chalfont, UK). The band density was densitometrically analyzed
using the Quantity One software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), and normalized against the density of
β-actin.
Immunocytochemistry staining
The cells were washed with phosphate-buffered saline
(PBS; pH 7.4) and fixed in 4% formaldehyde in PBS for 15 min. The
cells were blocked by preincubation with 10% normal goat serum in
PBS for 1 h at room temperature, and were then incubated with
anti-NF-κB p65 antibodies, at 4°C overnight. Following further
washes with PBS, the cells were incubated with Alexa
Fluor® 488-labeled goat anti-rabbit immunoglobulin G
(1:200 dilution), at room temperature for 1 h. The cells were
counterstained with 4,6-diamidino-2-phenylindole, in order to
visualize the nuclei. The cells were visualized under a fluorescent
microscope (LeicaTCS-SP5, Leica, Mannheim, Germany).
Statistical analysis
The data are expressed as the means ± standard
deviation. The statistical differences among the numerous groups
were calculated using a one-way analysis of variance, followed by
the Tukey post hoc test. A P<0.05 was considered to
indicate a statistically significant difference. All statistical
tests were performed using SPSS version 13.0 software package for
Windows (SPSS, Inc., Chicago, IL, USA)
Results
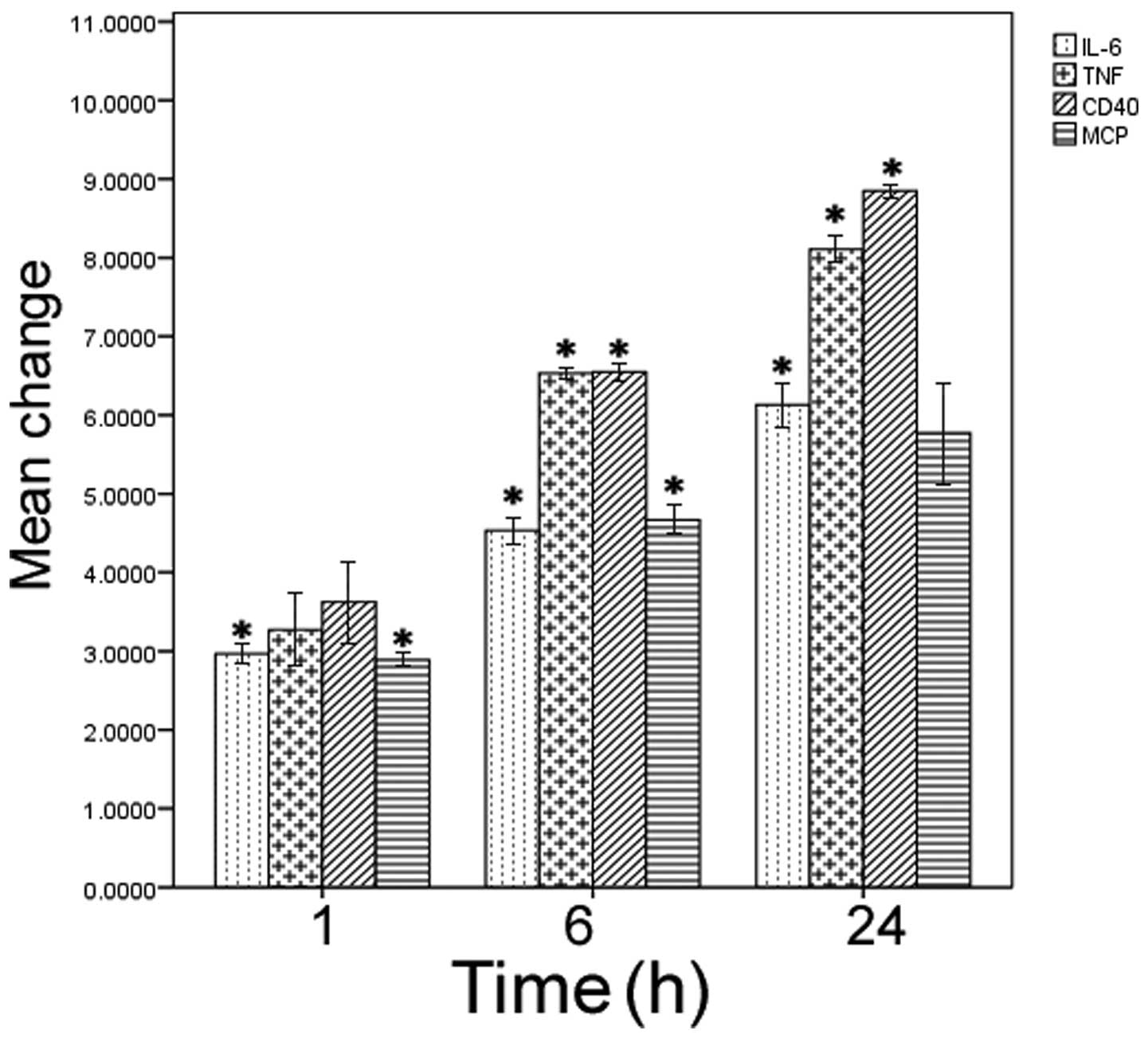
LDL treatment stimulates expression of
CD40 and proinflammatory cytokines in HMCs
HMCs were exposed to LDL for various durations, and
any changes to gene expression levels were determined by qPCR. LDL
treatment caused a significant increase in the relative mRNA
expression levels of IL-6, monocyte chemoattractant protein-1
(MCP-1), tumor necrosis factor-α (TNF-α), and CD40, as compared
with the untreated control cells (P<0.05; Fig. 1). Furthermore, this induction was
time-dependent, peaking at 24 h of culture.
Activation of the IRE1α/IKK/NF-κB pathway
is involved in LDL-induced proinflammatory cytokine expression
Treatment with LDL induced a time-dependent increase
in the phosphorylation of IRE1α, IKK, and NF-κB p65, as determined
by western blot analysis (Fig.
2A). This result is indicative of activation of the
IRE1α/IKK/NF-κB pathway. However, the total protein expression
levels of IRE1α, IKK, and NF-κB p65 remained unchanged (data not
shown). To further assess the activation of NF-κB signaling,
immunocytochemistry was used to examine the nuclear translocation
of NF-κB p65. Treatment with LDL markedly promoted NF-κB p65
nuclear translocation, in a time-dependent manner (Fig. 2B).
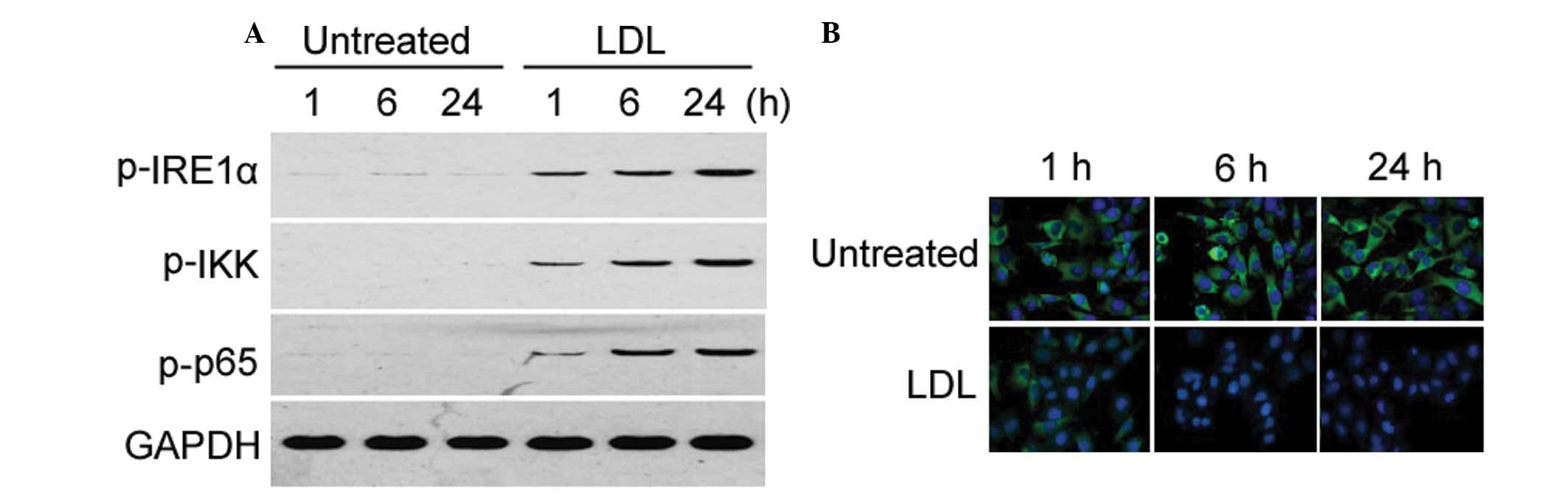 | Figure 2Treatment with low-density lipoprotein
(LDL) activates the inositol-requiring enzyme (IRE)1α/IκB kinase
(IKK)/nuclear factor (NF)-κB pathway in human mesangial cells. (A)
Western blot analysis of the phosphorylation of IRE1α, IKK, and
NF-κB p65 in HMCs, with or without LDL treatment. The Western blots
for LDL 1, 6, 24 h are presented again in Figs. 3B, 4B and 7B, and are used as a comparison for the
other experimental conditions. Representative blots of three
independent experiments with similar results are shown. (B)
Immunohistochemistry for NF-κB p65 subcellular expression in HMCs,
with or without LDL treatment. Green staining indicates the
localization of p65, and blue staining indicates the nucleus.
Magnification, ×200; h, hours; p, phosphorylated. |
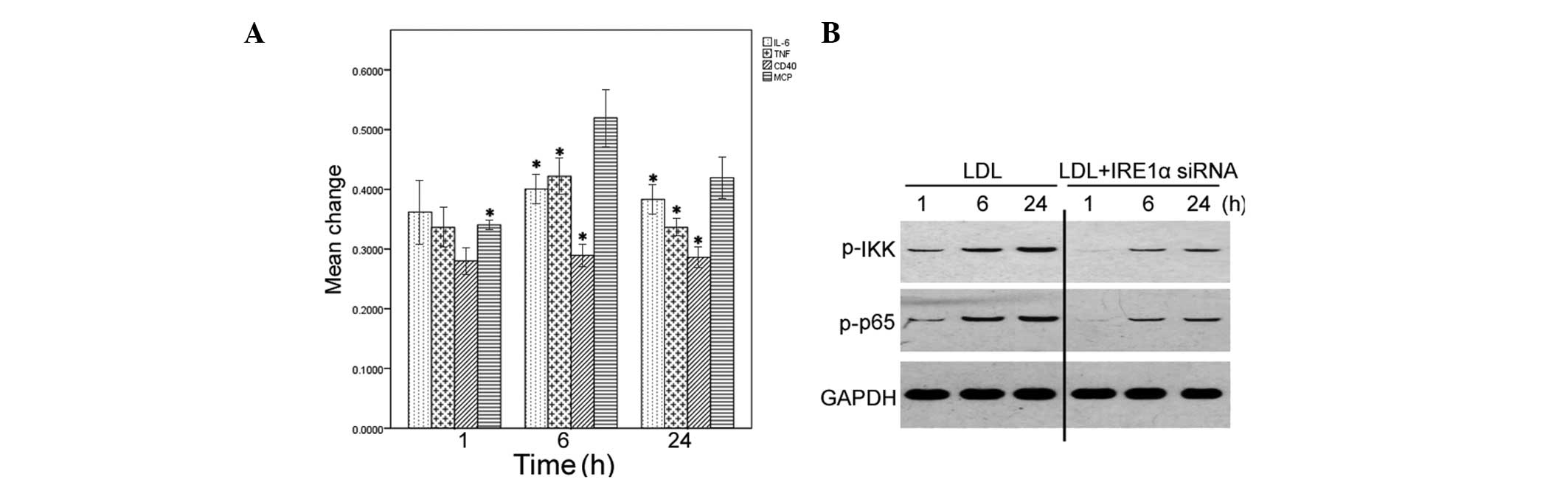
To confirm the essential role of IRE1α in
LDL-induced inflammatory responses, HMCs were transfected with
IRE1α specific siRNA, or scrambled control siRNA, 24 h before
treatment with LDL. The delivery of the IRE1α specific siRNA, but
not control siRNA, markedly reduced the mRNA expression levels of
endogenous IRE1α in HMCs, as compared with the non-transfected
cells (data not shown). Depletion of IRE1α expression significantly
blocked the induction of IL-6, MCP-1, TNF-α, and CD40 exerted by
LDL, as determined by qPCR (P<0.05; Fig. 3A). Furthermore, knockdown of IRE1α
expression markedly reduced the phosphorylation levels of IKK and
NF-κB p65 (Fig. 3B).
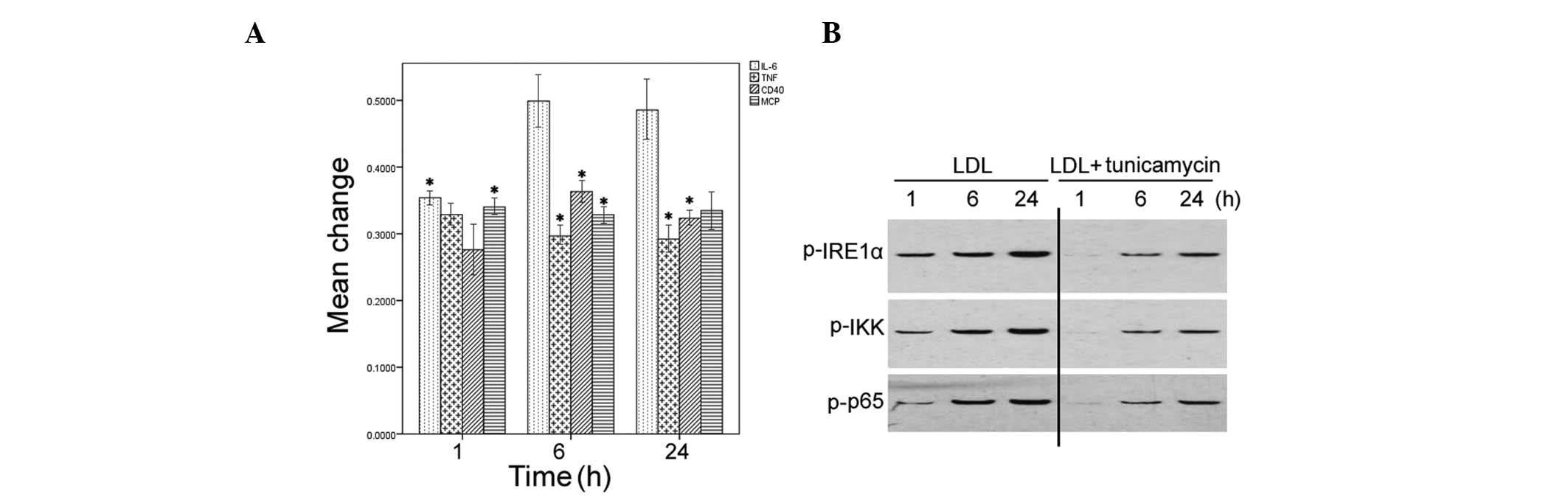
ER stress preconditioning attenuates the
LDL-induced inflammatory response
The effects of ER stress preconditioning were also
determined on LDL-induced inflammatory responses in HMCs.
Pretreatment with tunicamycin significantly attenuated the
induction of IL-6, MCP-1, TNF-α, and CD40 by LDL (P<0.05;
Fig. 4A). Furthermore, the
phosphorylation levels of IRE1α, IKK, and NF-κB p65 were markedly
reduced in the LDL-treated HMCs with tunicamycin pretreatment
(Fig. 4B).
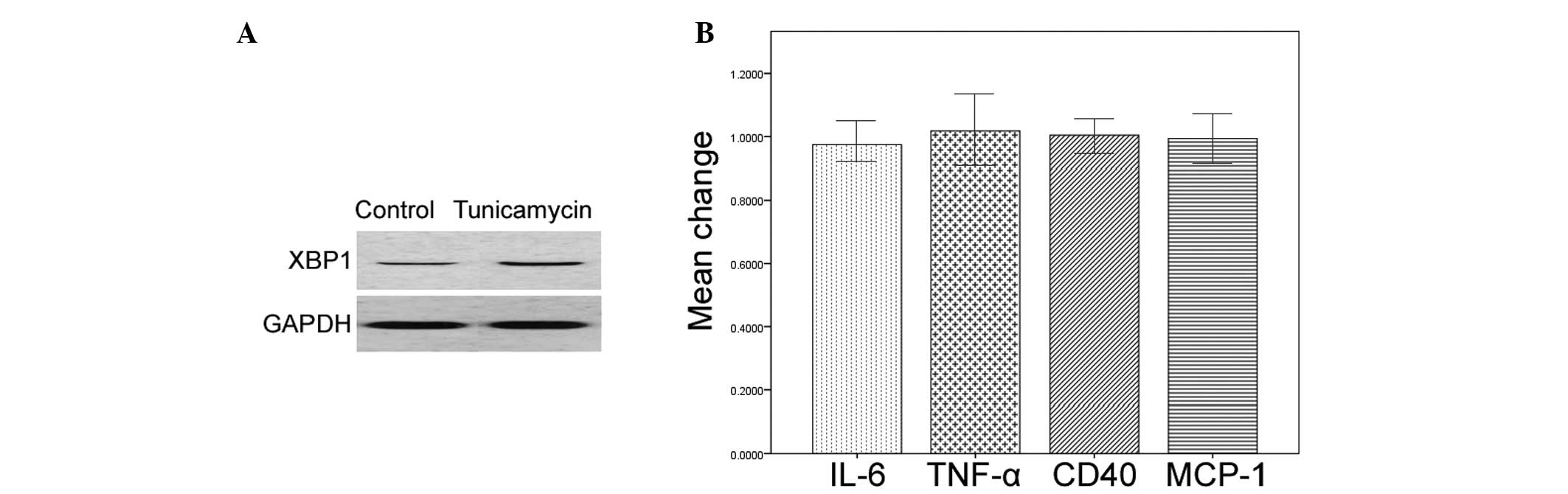
Tunicamycin pretreatment upregulates the
expression levels of XBP1 in HMCs
Treatment with tunicamycin alone markedly increased
the protein expression levels of XBP1 in the HMCs, as determined by
western blot analysis (Fig. 5A).
However, the mRNA expression levels of IL-6, MCP-1, TNF-α, and CD40
remained unchanged in the tunicamycin-treated cells (Fig. 5B).
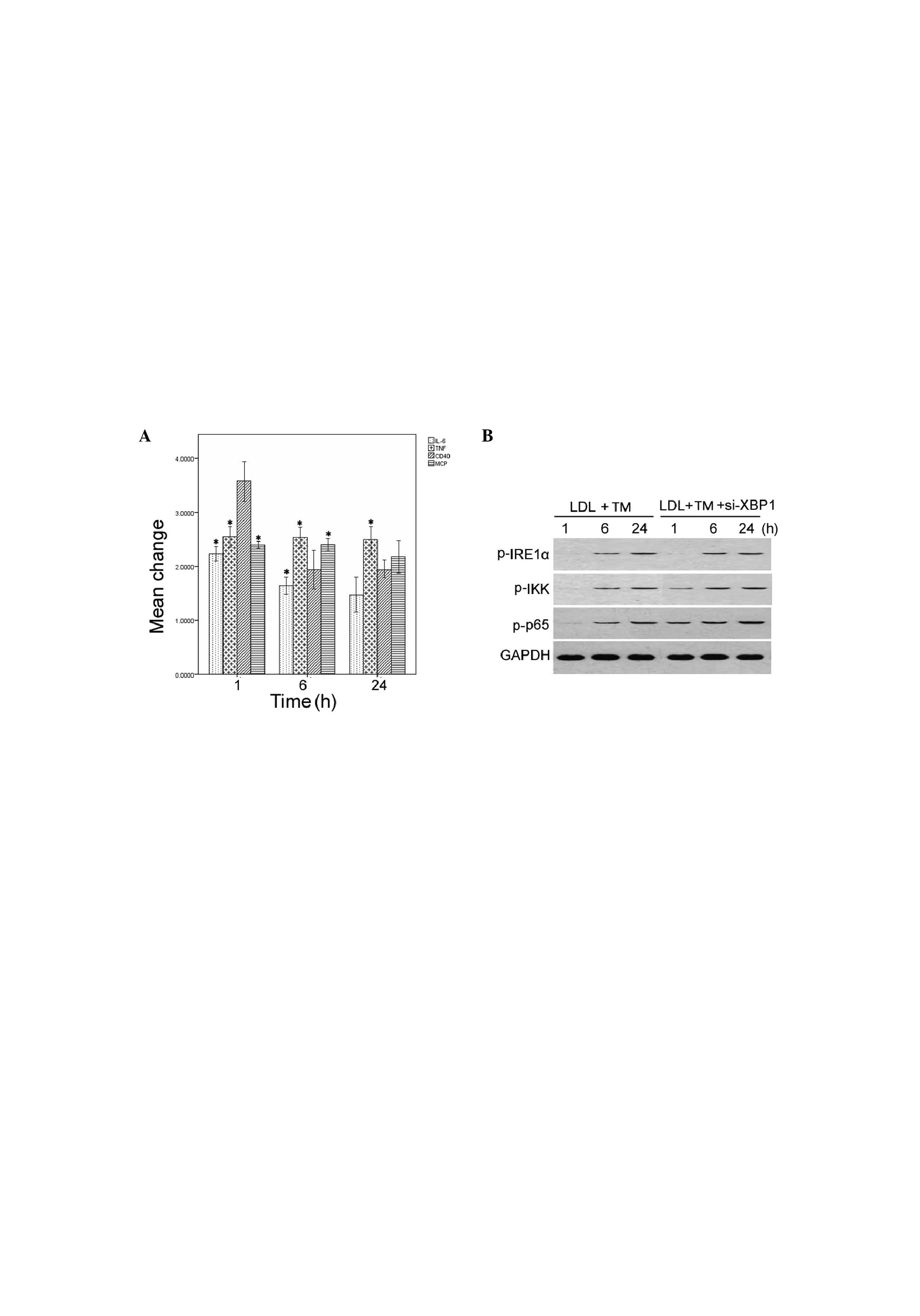
Depletion of XBP1 reverses the
anti-inflammatory effects of ER stress preconditioning on
LDL-treated HMCs
The role of XBP1 on the anti-inflammatory effects of
ER stress preconditioning in HMCs was then determined. Transfection
with specific XBP1 siRNA resulted in a marked reduction in
endogenous XBP1 protein expression levels in the HMCs, as compared
with the cells transfected with control siRNA (data not shown).
Notably, silencing XBP1 expression significantly restored the mRNA
expression of IL-6, MCP-1, TNF-α, and CD40, in the LDL-treated HMCs
with ER stress preconditioning (P<0.05; Fig. 6A). Furthermore, the phosphorylation
levels of IRE1α, IKK, and NF-κB p65 were markedly increased in the
XBP1-depleted HMCs with ER stress preconditioning (Fig. 6B).
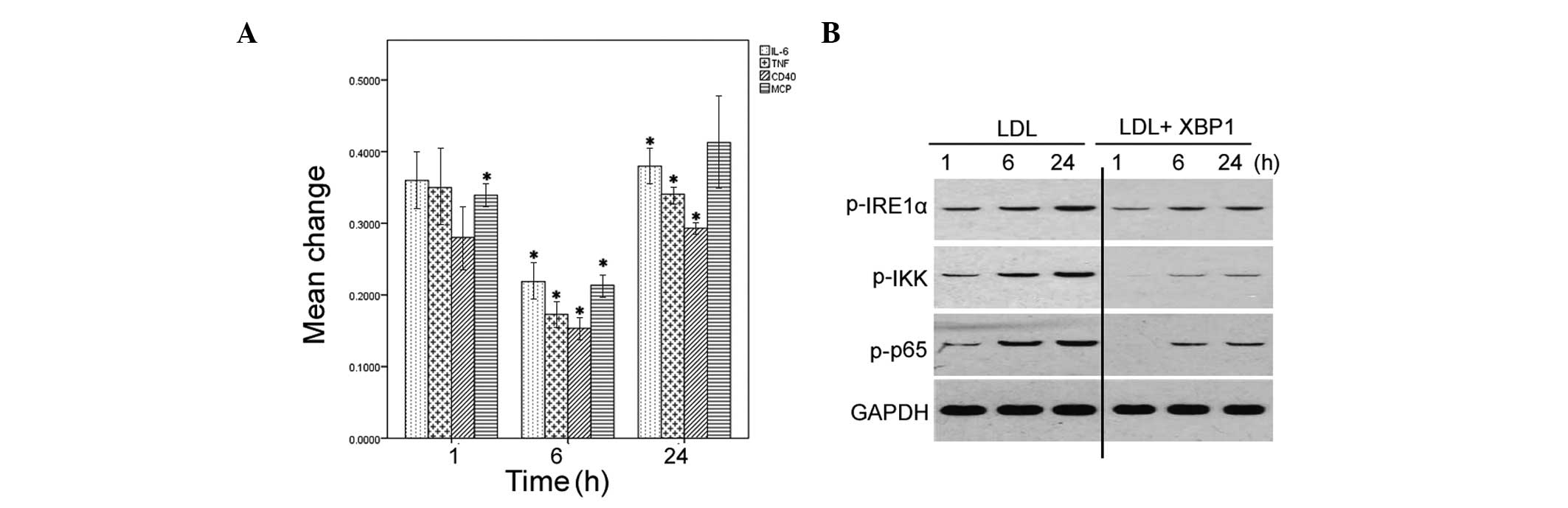
Overexpression of XBP1 antagonizes
LDL-induced inflammation
The present study also examined whether
overexpression of XBP1 could block LDL-induced inflammatory
responses in HMCs. The overexpression of XBP1 in the HMCs
transfected with the XBP1-expressing plasmid was confirmed by
western blot analysis (data not shown). Overexpression of XBP1
significantly reduced the mRNA expression levels of IL-6, MCP-1,
TNF-α, and CD40 in LDL-treated HMCs, as determined by qPCR
(Fig. 7A). Furthermore, the
phosphorylation levels of IRE1α, IKK, and NF-κB p65 in LDL-treated
cells were diminished in response to overexpression of XBP1
(Fig. 7B).
Discussion
Native and modified LDL has previously been shown to
possess potent proinflammatory activities in various biological
contexts. Meng et al (16)
reported that oxidized LDL triggers inflammatory responses in
cultured human mast cells by activating the Toll-like receptor
4-dependent signaling pathway. Oxidized LDL has also been shown to
stimulate proinflammatory responses in alternatively activated M2
macrophages (17). The present
study showed that LDL treatment promoted inflammatory responses in
HMCs, as evidenced by the increased expression levels of
proinflammatory cytokines IL-6, MCP-1, and TNF-α. These results are
concordant with previous studies (14,18).
Systemic inflammation is regarded as a key factor in the
pathogenesis of CKD (13). The
proinflammatory activity of LDL in HMCs may partially explain the
association between dyslipidemia and the progression of CKD
(2).
CD40 is expressed in a wide range of cells,
including macrophages, lymphocytes, endothelial cells, vascular
smooth muscle cells, and mesangial cells (19,20).
CD40/CD40 ligand interactions between infiltrating mononuclear
cells and resident renal cells, are associated with the
pathogenesis of immune-mediated glomerulonephritis (21). Increasing evidence has indicated
that activation of the CD40/CD40 ligand pathway is associated with
the initiation of inflammatory responses (19). Tanaka et al (22) reported that human platelets
stimulate mesangial cells to produce MCP-1, via the CD40/CD40
ligand pathway. The results of the present study demonstrated that
upregulation of CD40 expression occurs in LDL-induced inflammatory
responses in HMCs. In addition to initiation of inflammatory
responses, activation of CD40-dependent signaling has been found to
protect HMCs from oxidized LDL-induced apoptosis (23). These findings suggest that
LDL-mediated toxic effects on HMCs are associated with the
regulation of CD40 signaling.
There is known to be a close association between ER
stress and the inflammatory response (24). Li et al (25) showed that induction of ER stress
contributes to retinal inflammation in diabetic retinopathy
(24). The results of the present
study showed that, in parallel with induction of proinflammatory
cytokines, LDL treatment resulted in a time-dependent increase in
the phosphorylation of IRE1α, IKK, and NF-κB p65. IRE1α is a major
sensor of unfolded proteins in the ER, and is activated by
autophosphorylation (7). In
response to ER stress, activated IRE1α may bind to the IKK complex
and activate NF-κB (10,26). In the present study siRNA
technology was used to induce a specific depletion of IRE1α
expression. The loss of IRE1α expression antagonized the induction
of IL-6, MCP-1, TNF-α, and CD40 exerted by LDL, coupled with the
suppression of NF-κB activation. These findings indicate that ER
stress has a mediating role in LDL-induced inflammatory responses
in HMCs, which involves the activation of the IRE1α/IKK/NF-κB
pathway. Consistently, phospholipolyzed LDL has been found to
induce an inflammatory response in endothelial cells, by activating
ER stress signaling pathways (27).
Accumulating evidence indicates that ER stress
preconditioning confers protection against cellular injury in
various biological settings (12,28).
Hung et al (28) reported
that ER stress preconditioning attenuated hydrogen peroxide-induced
oxidative injury in LLC-PK1 renal epithelial cells. Li et al
(10) previously demonstrated that
preconditioning with ER stress mitigated retinal endothelial
inflammation. The present study showed that LDL-induced
inflammatory cytokine production was significantly diminished in ER
stress-primed HMCs, suggesting a protective role for ER stress
preconditioning. Notably, the results of the present study revealed
that pretreatment with the ER stress inducer tunicamycin, caused an
upregulation of XBP1, but not of the proinflammatory cytokines. In
addition, siRNA-mediated inhibition of XBP1 attenuated the
protective effects of tunicamycin pretreatment on LDL-induced
inflammation, which was coupled with an enhanced activation of the
IRE1α/IKK/NF-κB pathway. Numerous studies have shown that XBP1
ablation triggers feedback activation of its upstream enzyme IRE1α
(29,30). Furthermore, the present study
revealed that XBP1 overexpression inhibited LDL-induced activation
of the IRE1α/IKK/NF-κB pathway. These results collectively indicate
that ER stress preconditioning ameliorates LDL-induced inflammation
in HMCs, which is largely mediated through activation of XBP1
signaling and blockade of the IRE1α/IKK/NF-κB pathway.
In conclusion, activation of the IRE1α/IKK/NF-κB
pathway is required for LDL-induced inflammation in HMCs. ER stress
preconditioning resulted in the upregulation of XBP1 expression and
subsequent inhibition of the IRE1α/IKK/NF-κB pathway, which
consequently interfered with the LDL-induced inflammatory
responses. These findings warrant further investigation of the
therapeutic potential of ER stress preconditioning in inflammatory
disorders, such as CKD.
References
|
1
|
Wakasugi M, Kazama JJ and Narita I:
Differences in the local and national prevalences of chronic kidney
disease based on annual health check program data. Clin Exp
Nephrol. 16:749–754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nitta K: Clinical assessment and
management of dyslipidemia in patients with chronic kidney disease.
Clin Exp Nephrol. 16:522–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaziri ND and Norris K: Lipid disorders
and their relevance to outcomes in chronic kidney disease. Blood
Purif. 31:189–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Podrez EA: Anti-oxidant properties of
high-density lipoprotein and atherosclerosis. Clin Exp Pharmacol
Physiol. 37:719–725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm.
2013:1527862013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Inagi R: Endoplasmic reticulum stress as a
progression factor for kidney injury. Curr Opin Pharmacol.
10:156–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dickhout JG and Krepinsky JC: Endoplasmic
reticulum stress and renal disease. Antioxid Redox Signal.
11:2341–2352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiang CK, Hsu SP, Wu CT, et al:
Endoplasmic reticulum stress implicated in the development of renal
fibrosis. Mol Med. 17:1295–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Wang JJ and Zhang SX:
Preconditioning with endoplasmic reticulum stress mitigates retinal
endothelial inflammation via activation of X-box binding protein 1.
J Biol Chem. 286:4912–4921. 2011. View Article : Google Scholar :
|
|
11
|
Aminzadeh MA, Sato T and Vaziri ND:
Participation of endoplasmic reticulum stress in the pathogenesis
of spontaneous glomerulosclerosis - role of intra-renal angiotensin
system. Transl Res. 160:309–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inagi R, Kumagai T, Nishi H, et al:
Preconditioning with endoplasmic reticulum stress ameliorates
mesangioproliferative glomerulonephritis. J Am Soc Nephrol.
19:915–922. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cottone S, Lorito MC, Riccobene R, et al:
Oxidative stress, inflammation and cardiovascular disease in
chronic renal failure. J Nephrol. 21:175–179. 2008.PubMed/NCBI
|
|
14
|
Santini E, Lupi R, Baldi S, et al: Effects
of different LDL particles on inflammatory molecules in human
mesangial cells. Diabetologia. 51:2117–2125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak MJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Meng Z, Yan C, Deng Q, et al: Oxidized
low-density lipoprotein induces inflammatory responses in cultured
human mast cells via Toll-like receptor 4. Cell Physiol Biochem.
31:842–853. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Tits LJ, Stienstra R, van Lent PL, et
al: Oxidized LDL enhances pro-inflammatory responses of
alternatively activated M2 macrophages: a crucial role for
Krüppel-like factor 2. Atherosclerosis. 214:345–349. 2011.
View Article : Google Scholar
|
|
18
|
Massy ZA, Kim Y, Guijarro C, et al:
Low-density lipoprotein-induced expression of interleukin-6, a
marker of human mesangial cell inflammation: effects of oxidation
and modulation by lovastatin. Biochem Biophys Res Commun.
267:536–540. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Antoniades C, Bakogiannis C, Tousoulis D,
Antonopoulos AS and Stefanadis C: The CD40/CD40 ligand system:
linking inflammation with atherothrombosis. J Am Coll Cardiol.
54:669–677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yellin MJ, D’Agati V, Parkinson G, et al:
Immunohistologic analysis of renal CD40 and CD40L expression in
lupus nephritis and other glomerulonephritides. Arthritis Rheum.
40:124–134. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuroiwa T, Schlimgen R, Illei GG, McInnes
IB and Boumpas DT: Distinct T cell/renal tubular epithelial cell
interactions define differential chemokine production: implications
for tubulointerstitial injury in chronic glomerulonephritides. J
Immunol. 164:3323–3329. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka T, Kuroiwa T, Ikeuchi H, et al:
Human platelets stimulate mesangial cells to produce monocyte
chemoattractant protein-1 via the CD40/CD40 ligand pathway and may
amplify glomerular injury. J Am Soc Nephrol. 13:2488–2496. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deambrosis I, Scalabrino E, Deregibus MC,
Camussi G and Bussolati B: CD40-dependent activation of
phosphatidylinositol 3-kinase/AKT pathway inhibits apoptosis of
human cultured mesangial cells induced by oxidized LDL. Int J
Immunopathol Pharmacol. 18:327–337. 2005.PubMed/NCBI
|
|
24
|
Su J, Zhou L, Kong X, et al: Endoplasmic
reticulum is at the crossroads of autophagy, inflammation, and
apoptosis signaling pathways and participates in the pathogenesis
of diabetes mellitus. J Diabetes Res. 2013:1934612013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Wang JJ, Yu Q, Wang M and Zhang SX:
Endoplasmic reticulum stress is implicated in retinal inflammation
and diabetic retinopathy. FEBS Lett. 583:1521–1527. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hayakawa K, Hiramatsu N, Okamura M, et al:
Acquisition of anergy to proinflammatory cytokines in nonimmune
cells through endoplasmic reticulum stress response: a mechanism
for subsidence of inflammation. J Immunol. 182:1182–1191. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gora S, Maouche S, Atout R, et al:
Phospholipolyzed LDL induces an inflammatory response in
endothelial cells through endoplasmic reticulum stress signaling.
FASEB J. 24:3284–3297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hung CC, Ichimura T, Stevens JL and
Bonventre JV: Protection of renal epithelial cells against
oxidative injury by endoplasmic reticulum stress preconditioning is
mediated by ERK1/2 activation. J Biol Chem. 278:29317–29326. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
So JS, Hur KY, Tarrio M, et al: Silencing
of lipid metabolism genes through IRE1α-mediated mRNA decay lowers
plasma lipids in mice. Cell Metab. 16:487–499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee AH, Heidtman K, Hotamisligil GS and
Glimcher LH: Dual and opposing roles of the unfolded protein
response regulated by IRE1alpha and XBP1 in proinsulin processing
and insulin secretion. Proc Natl Acad Sci USA. 108:8885–8890. 2011.
View Article : Google Scholar : PubMed/NCBI
|