Introduction
Autophagy is a cellular self-catabolic process
responsible for the degradation of long-lived proteins and
organelles. During autophagy, cytoplasmic constituents are
sequestered into autophagosomes and degraded via the lysosomal
pathway (1). Autophagy is able to
promote cell survival in response to stress. However, progressive
autophagy also induces cell death (2). Therefore, autophagy not only has a
crucial role in the regulation of normal development, but
dysregulated or defective autophagy is associated with disease.
MicroRNAs (miRNAs) are a group of endogenous,
non-coding, single-strand, small RNAs of 22–25 nucleotides, which
regulate gene expression at the post-transcriptional level,
predominantly through base pairing with the 3′-untranslated region
(3′-UTR) of target mRNAs, which results in mRNA degradation or
translational repression (3). It
has been revealed that miRNAs regulate >30% of genes, which are
associated with almost all major cellular processes, including cell
proliferation, differentiation, apoptosis and migration, as well as
immune responses (4). Myocardial
tissue injury induced by ischemia and hypoxia is a major factor
underlying the development of fatal diseases. The main cause of
myocardial ischemic injury is myocardial cell apoptosis induced by
myocardial hypoxia (5).
Furthermore, subsequent reoxygenation may further aggravate the
damage (6). Multiple miRNAs have
been shown to function as protectors against hypoxia/reoxygenation
(H/R)-induced myocardial injury (7–9).
However, the specific molecular mechanisms underlying this effect
have remained elusive.
Accumulating evidence has indicated that certain
miRNAs function in the induction or inhibition of autophagy in
cells under stress, including miRNA-101 (miR-101) (10). miR-101 has been demonstrated to
participate in the regulation of various cancers (11,12).
For example, miR-101 was demonstrated to be a potent inhibitor of
autophagy in breast cancer cells and the inhibition of miR-101
sensitized breast cancer cells to drug-mediated cell death
(10). In the present study, it
was demonstrated that H/R led to cellular apoptosis as well as
miR-101 downregulation in H9c2 cardiomyocytes. Further
investigation suggested that the inhibition of miR-101 attenuated
H/R-induced apoptosis, at least in part, via the induction of
autophagy. The effect of miR-101 on H/R-induced cardiomyocyte
apoptosis may be via direct targeting of Ras-related protein Rab-5A
(RAB5A), which has previously been demonstrated to be involved in
autophagy.
Materials and methods
Cell culture
H9c2 cardiomyocytes were obtained from the American
Type Culture Collection (Rockville, MD, USA). Cells were cultured
in DMEM supplemented with 10% fetal bovine serum (FBS) (Both from
Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in a
humidified incubator containing 5% CO2.
H/R treatment of H9c2 cardiomyocytes
Cells were pre-incubated with a selective autophagy
inhibitor 3-MA (10 mM; Sigma-Aldrich, St. Louis, MO, USA) prior to
hypoxia/reoxygenation treatment. Following culture in serum-free
DMEM at 37°C in 5% CO2 for 12 h, H9c2 cardiomyocytes
were cultured at 37°C in 1% O2/94% N2/5%
CO2 for 4 h. Subsequently, H9c2 cardiomyocytes were
cultured in DMEM containing 10% FBS and incubated at 37°C in 5%
CO2 for 3 h.
Reverse transcription polymerase chain
reaction (RT-PCR) assay
Total RNA was extracted using TRIzol (Invitrogen
Life Technologies, Carlsbad, CA, USA). For the analysis of mRNA
expression, the TaqMan Reverse Transcription kit (Thermo Fisher
Scientific, Waltham, MA, USA) was used to convert RNA into
complementary (c)DNA, and PCR was subsequently performed using the
Power SYBR Green kit (Thermo Fisher Scientific) on a Bio-Rad
MiniOption thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). β-actin was used as an endogenous control. The primers for
RAB5A were as follows: Forward, 5′-CATTGGGGCTGCCTTTCTA-3′ and
reverse, 5′-TCCTCTGGCTGAGTTTGCG-3′. The primers for stathmin 1
(STMN1) were as follows: Forward, 5′-GCGAGAGAAGGACAAGCACG-3′ and
reverse, 5′-TTGGATATTTAGGAAGGGGT-3′. The primers for β-actin were
as follows: Forward, 5′-AGGCCCCTCTGAACCCTAAG-3′ and reverse,
5′-CCAGAGGCATACAGGGACAAC-3′. The primers for U6 were as follows:
Forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′. For the analysis of miRNA expression,
an ABI miRNA reverse transcription kit (Applied Biosystems, Life
Technologies, Foster City, CA, USA) was used to convert RNA into
cDNA, according to the manufacturer’s instructions. Subsequently,
PCR was performed using an miRNA Q-PCR Detection kit (GeneCopoeia,
Rockville, MD, USA) on a Bio-Rad MiniOption thermocycler (Bio-Rad
Laboratories, Inc.). The U6 gene (GeneCopopia, Rockville, MD, USA)
was used as an endogenous control. PCR cycling conditions were as
follows: 95°C for 1 min, followed by 40 cycles of 95°C for 15 sec,
58°C for 15 sec and 72°C for 20 sec. All experiments were performed
in triplicate. For mRNA and miRNA, the relative expression was
analyzed using the 2−ΔΔCt method.
Western blot analysis
Cells were solubilized in cold
radioimmunoprecipitation assay lysis buffer (Fermentas, Thermo
Fisher Scientific, Pittsburgh, PA, USA). Proteins were separated by
12% SDS-PAGE, and transferred onto a polyvinylidene difluoride
(PVDF) membrane (EMD Millipore, Billerica, MA, USA), which was then
incubated with Tris-Buffered Saline and Tween 20 (Sigma-Aldrich)
containing 5% skimmed milk (BD Biosciences, Franklin Lakes, NJ,
USA) at 4°C for overnight. Subsequently, the membrane was incubated
with rabbit monoclonal anti-RAB5A (1:1,000; Abcam, Hong Kong,
China) and mouse monoclonal anti-β-actin (1:1,000; Abzoom, Dallas,
TX, USA) primary antibodies at room temperature for 4 h. Membranes
were also incubated at room temperature with rabbit monoclonal
anti-mouse/rat/human Beclin1 antibody (1/500; Abcam) for 4 h,
rabbit polyclonal anti-mouse/rat/human IL-1β antibody (1/200; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) for 4 h, rabbit
monoclonal anti-mouse/rat/human Bcl2 antibody (1/500; Abcam) for 3
h, rabbit polyclonal anti-mouse/rat/human LC3B antibody (1/1000;
Abcam) for 4 h, rabbit polyclonal anti-mouse/rat/human/quail
caspase-3 antibody (1/400; Abcam) for 4 h and mouse monoclonal
anti-mouse/rat/human/rabbit/pig β-actin antibody (1/2000; Abzoom,
Dallas, TX, USA) for 2 h. Following washing three times with
phosphate-buffered saline with Tween 20 (PBST), the membrane was
incubated with the goat anti-mouse or goat anti-rabbit secondary
antibodies (1:5,000; Abcam), respectively, at room temperature for
1 h. Following washing three times with PBST, an enhanced
chemiluminscence kit (Pierce Chemical, Rockford, IL, USA) was used
to perform chemiluminescent detection. Relative protein expression
was analyzed with Image-Pro plus software 6.0 (Media Cybernetics,
Inc., Rockville, MD, USA), represented as the density ratio versus
GAPDH.
Transfection
For the miR-101 functional analysis, cells were
transfected with the pre-miR-101, pre-con, anti-101 or anti-con
(Genecopoeia, Rockville, MD, USA) were transfected into H9c2
cardiomyocytes using Lipofectamine 2000 (Invitrogen Life
Technologies) according to the manufacturer’s instructions.
Dual luciferase reporter assay
A mutant 3′-UTR of RAB5A was generated using the
Quick-Change Site-Directed Mutagenesis kit (Stratagene, La Jolla,
CA, USA). For the luciferase assay, 100,000 cells were cultured to
~70% confluence in 24-well plates and co-transfected with
psiCHECK™2-RAB5A-3′-UTR or psiCHECK™2-mut RAB5A-3′-UTR vectors
(Promega Corp., Madison, WI, USA) plus 50 nM miR-101 or 100 mM
miR-101 inhibitor. Cells were incubated with Lipofectamine 2000/DNA
complex for 5 h and the medium was subsequently replaced with DMEM
containing 10% FBS. Forty-eight hours after transfection, a dual
luciferase reporter gene assay kit (Promega Corp.) was used to
determine the luciferase activities in each group using an LD400
luminometer (Promega Corp.). Renilla luciferase activity was
normalized to firefly luciferase activity.
Apoptosis analysis
Flow cytometry was used to determine cell apoptosis
with an Annexin V-fluorescein isothiocyanate (FITC) Apoptosis
Detection kit (BD Biosciences). At 24 h post-transfection, cells
were harvested and washed twice with cold PBS. Subsequently,
106 cells were resuspended in 200 μl binding buffer with
10 μl Annexin-V-FITC and 5 μl propidium iodide, and incubated in
the dark for 30 min. Finally, 300 μl binding buffer was added and
the cells were assessed using flow cytometric analysis (Moflo XDP;
Beckman Coulter, Brea, CA, USA).
Statistical methods
Values are expressed as the mean ± standard
deviation of three independent experiments. SPSS 18 software
(International Business Machines, Armonk, NY, USA) was used to
perform statistical analyses. Statistical analysis of differences
between values was performed by one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference between values.
Results
H/R induces apoptosis and miR-101
downregulation in H9c2 cardiomyocytes
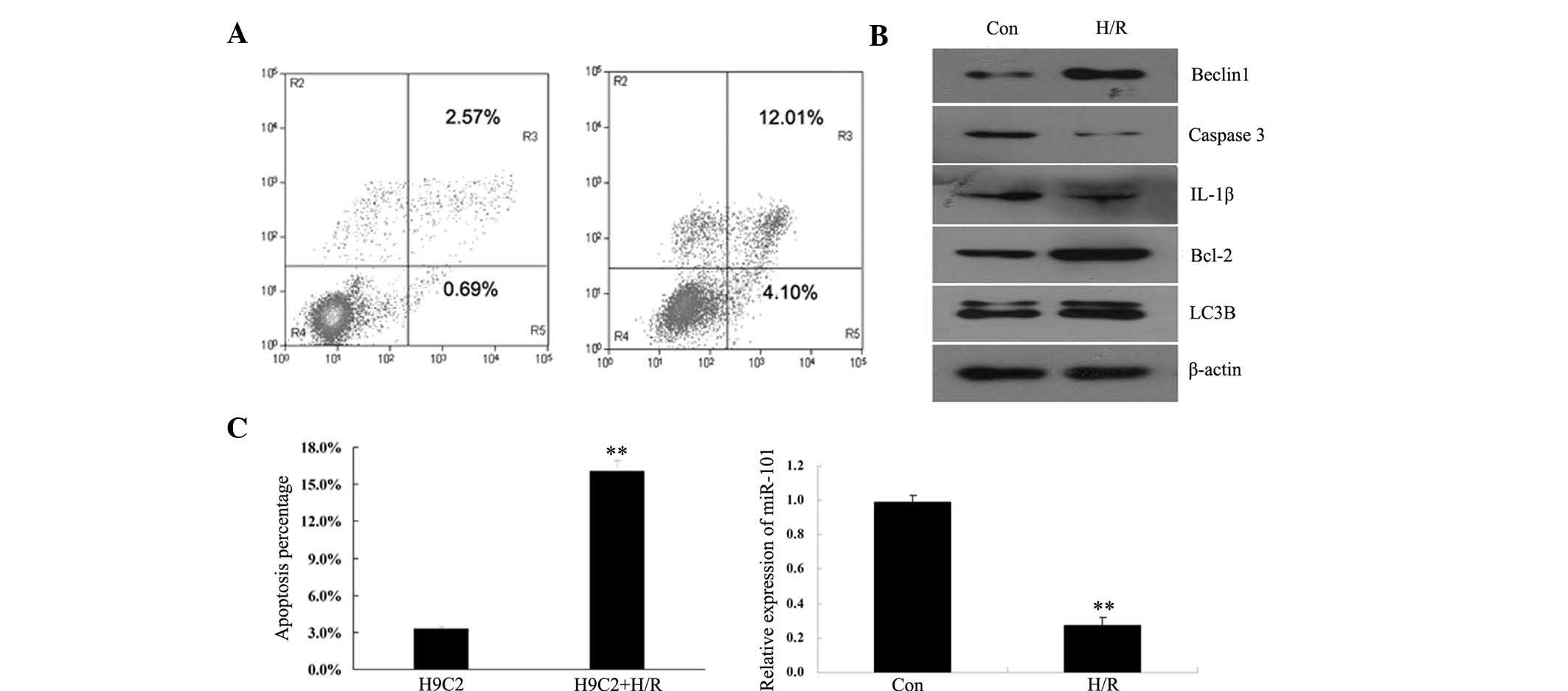
Following H/R treatment, the apoptotic rate of H9c2
cardiomyocytes was determined using flow cytometry. The results
indicated that the level of apoptosis was significantly increased
following H/R treatment of H9c2 cardiomyocytes (Fig. 1A), indicating that H/R induced
apoptosis in H9c2 cardiomyocytes. The expression levels of certain
apoptosis-associated proteins, including caspase-3, B-cell
lymphoma-2 (Bcl-2), interleukin 1β (IL-1β), as well as two
autophagy markers, Beclin1 and 1A/1B-light chain 3B (LC3B), were
also examined. As indicated in Fig.
1B, the protein expression levels of pro-apoptotic caspase-3
and IL-1β were significantly increased, while the expression of
anti-apoptotic Bcl-2 was markedly decreased following H/R.
Furthermore, the expression levels of autophagy-associated Beclin1
and LC3B were upregulated in H9c2 cardiomyocytes following H/R
treatment. The expression of miR-101 in H/R treated H9c2
cardiomyocytes was determined by RT-PCR. The results indicated that
miR-101 expression was markedly downregulated following H/R
treatment of H9c2 cardiomyocytes (Fig.
1C), suggesting that H/R may have had an inhibitory effect on
the expression of miR-101 in H9c2 cardiomyocytes.
RAB5A is a direct target of miR-101 in
H9c2 cardiomyocytes
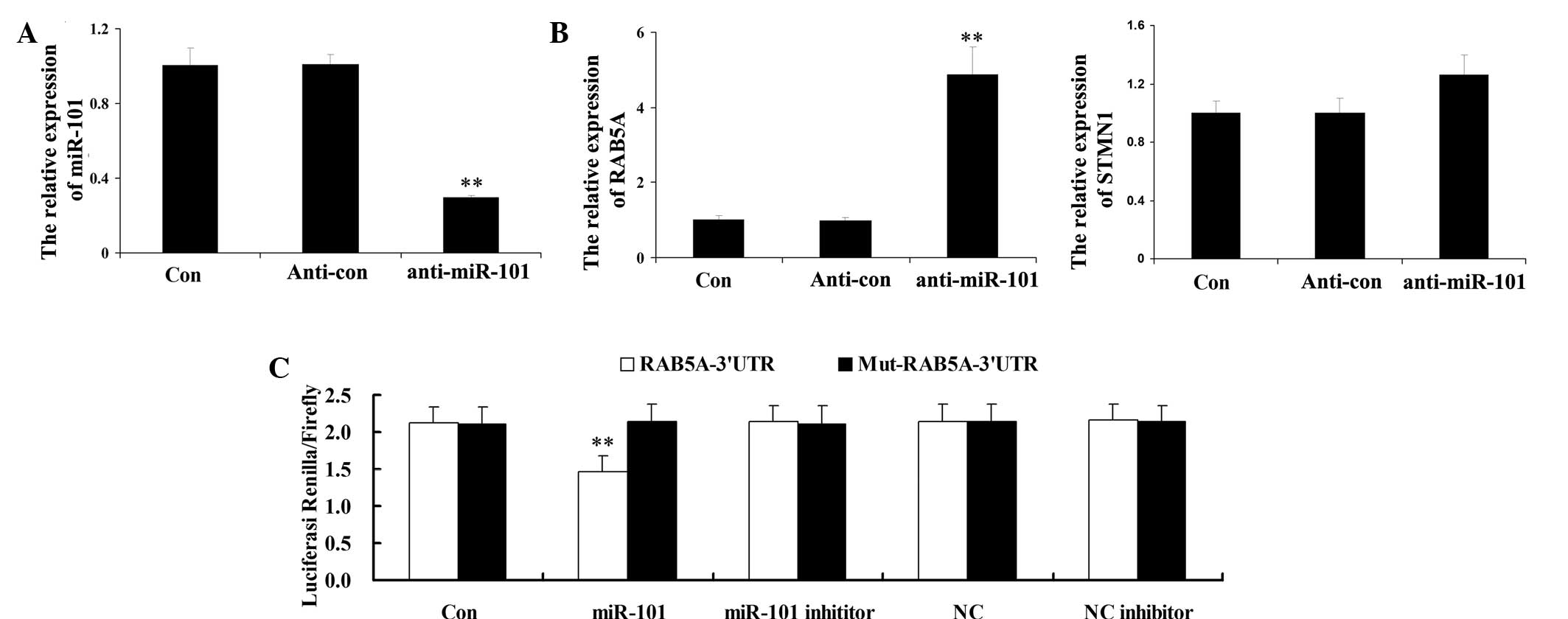
To investigate the potential targets of miR-101,
H9c2 cardiomyocytes were transfected with anti-miR-101. Following
transfection, the expression levels of miR-101 in H9c2
cardiomyocytes were evaluated. As shown in Fig. 2A, the expression levels of miR-101
were significantly reduced following transfection with
anti-miR-101, indicating that the transfection efficiency was
satisfactory. Subsequently, the expression levels of RAB5A and
STMN1 were determined by RT-PCR. As indicated in Fig. 2B, the mRNA expression levels of
RAB5A were significantly upregulated following the knockdown of
miR-101; however, the other potential target investigated, STMN1,
demonstrated no significant difference in expression to that of the
control group. These results suggested that RAB5A may be a direct
target of miR-101.
Based on these results, a luciferase reporter assay
was performed to determine whether RAB5A was a direct target of
miR-101 in H9c2 cardiomyocytes. As indicated in Fig. 2C, H9c2 cardiomyocytes
co-transfected with miR-101 mimic and the wild-type 3′-UTR of RAB5A
demonstrated a significant decrease in luciferase activity compared
to that of the control groups. However, H9c2 cardiomyocytes
co-transfected with miR-101 and mutant 3′-UTR RAB5A demonstrated no
significant difference in luciferase activity. These data indicated
that RAB5A may be a direct target of miR-101 in H9c2
cardiomyocytes.
miR-101 influences the apoptotic rate of
H/R-treated H9c2 cardiomyocytes
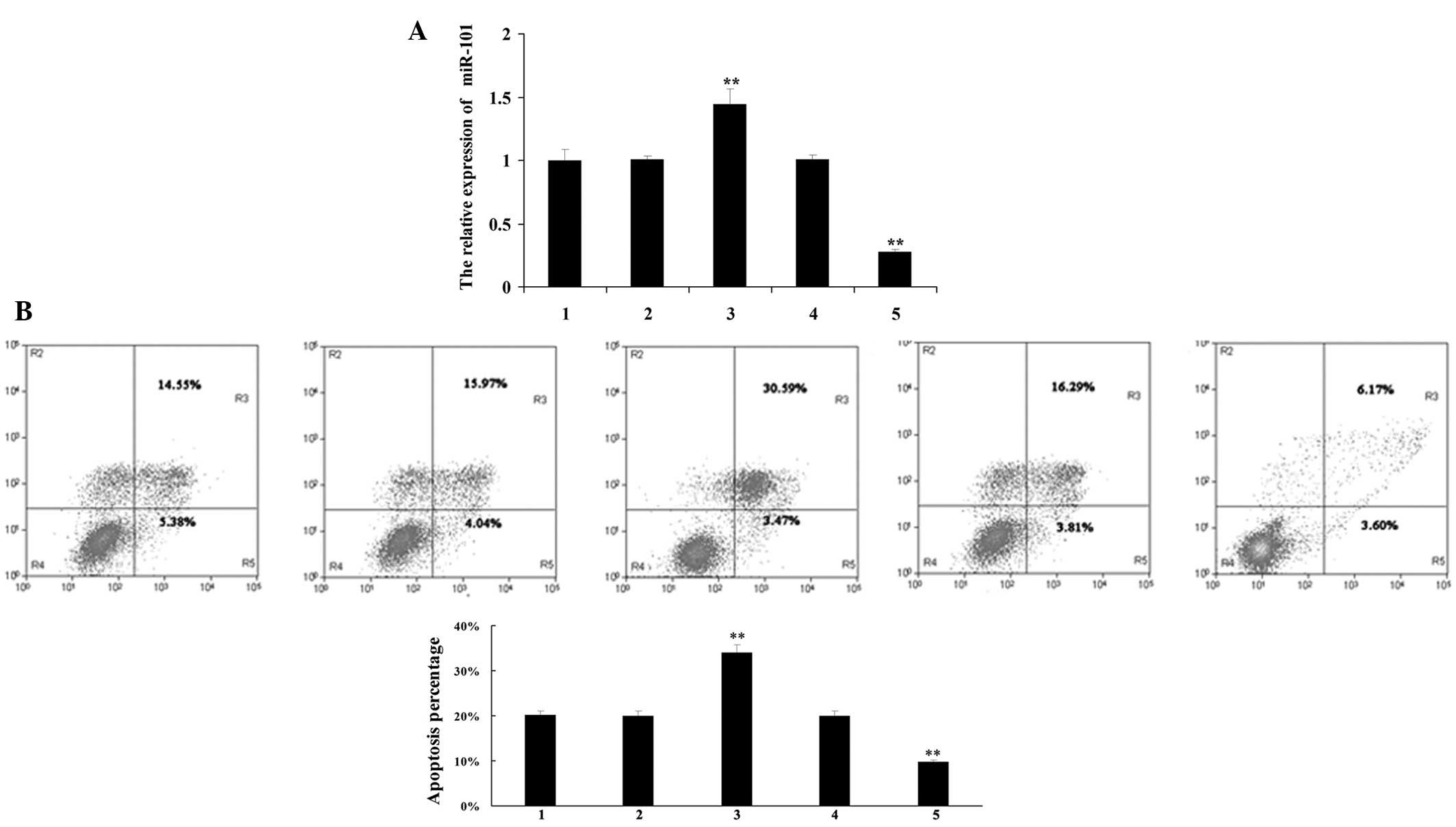
To further investigate the role of miR-101 in
H/R-treated H9c2 cardiomyocytes, H9c2 cardiomyocytes were
transfected with pre-miR-101 and anti-miR-101, respectively. Once
the transfection efficiency had been confirmed (Fig. 3A), H9c2 cardiomyocytes were treated
with H/R and the apoptotic rate in each group was determined by
flow cytometry. miR-101 upregulation significantly promoted
H/R-induced apoptosis, whereas miR-101 downregulation markedly
inhibited H/R-induced apoptosis in H9c2 cardiomyocytes (Fig. 3B), which suggested that miR-101 may
promote H/R-induced apoptosis in H9c2 cardiomyocytes.
miR-101 influences the expression of
autophagy- and apoptosis-associated factors in H/R-treated H9c2
cardiomyocytes
RT-PCR was performed in order to determine the
expression levels of certain key autophagy- and
apoptosis-associated factors, including IL-1β, caspase-3, Bcl-2 and
RAB5A. The expression levels of IL-1β and caspase-3 were found to
be increased following upregulation of miR-101, and decreased
following downregulation of miR-101. However, the expression levels
of Bcl-2 and RAB5A were reduced following upregulation of miR-101
and increased following downregulation of miR-101 (Fig. 4A).
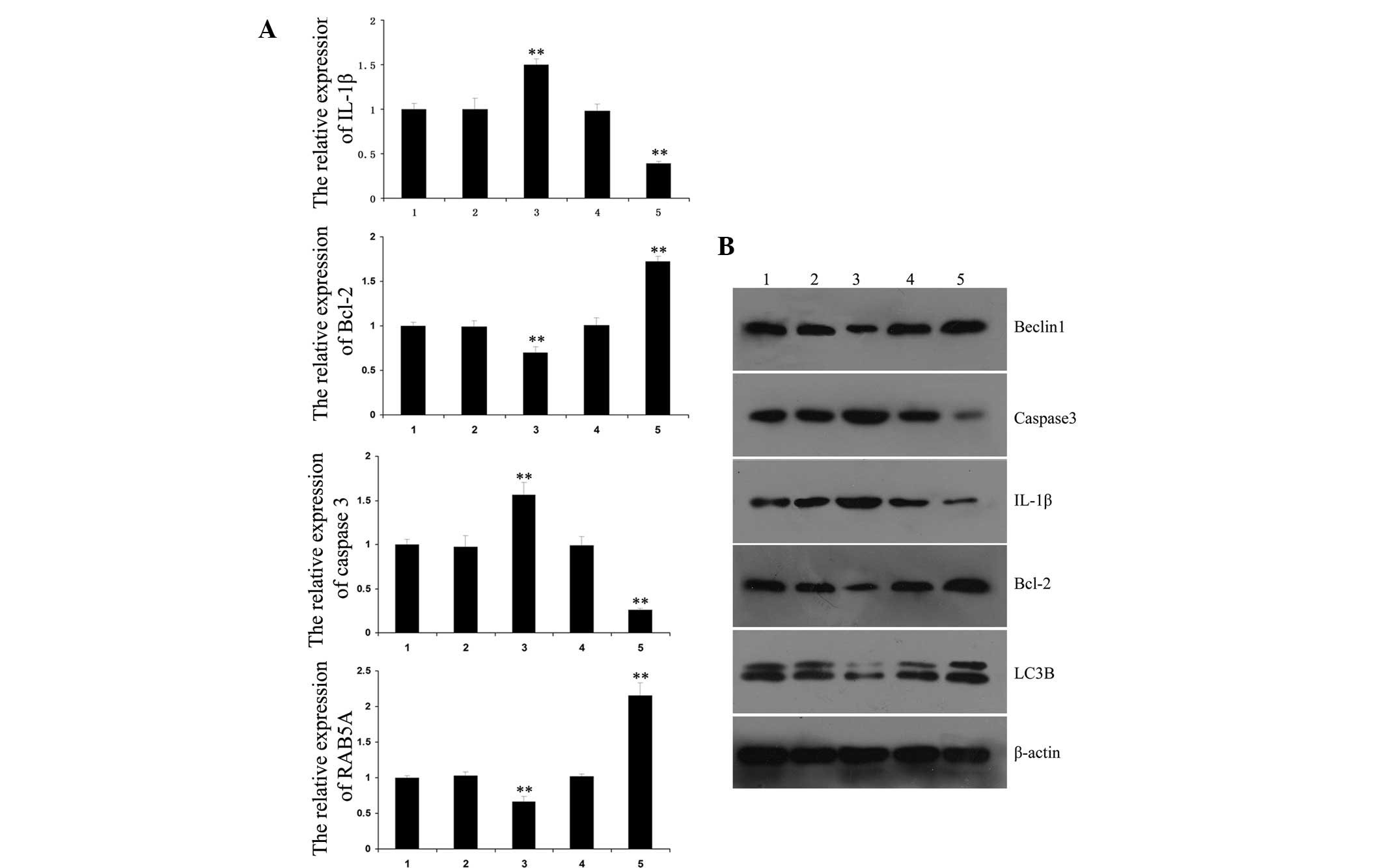 | Figure 4Effects of miR-101 on the expression
of autophagy- and apoptosis-associated factors in H/R-treated H9c2
cardiomyocytes. (A) Polymerase chain reaction analysis was
performed to determine the messenger RNA expression levels of
autophagy- and apoptosis-associated factors in H/R-treated H9c2
cardiomyocytes following transfection with pre-miR-101 or
anti-miR-101. **P<0.01 vs. 1. (B) Western blot
analysis was performed to determine the protein expression levels
of autophagy- and apoptosis-associated factors in H/R-treated H9c2
cardiomyocytes following transfection with pre-miR-101 or
anti-miR-101. 1, H9c2 cardiomyocytes without any treatment; 2, H9c2
cardiomyocytes transfected with control miRNAs; 3, H9c2
cardiomyocytes transfected with pre-miR-101; 4, H9c2 cardiomyocytes
transfected with control anti-miRNAs; 5, H9c2 cardiomyocytes
transfected with anti-miR-101; Values are presented as the mean
±standard deviation of a representative experiment performed in
triplicate. H/R, hypoxia/reoxygenation; miR-101, microRNA-101;
IL-1β, interleukin 1β; Bcl-2, B-cell lymphoma-2; LC3B, 1A/1B-light
chain 3B. |
Subsequently, the protein expression levels of
IL-1β, caspase-3, Bcl-2, Beclin1 and LC3B were evaluated. The
results indicated that the expression levels of miR-101 in each
group were negatively correlated with autophagy-associated Beclin1
and LC3B, as well as anti-apoptotic Bcl-2, but positively
correlated with pro-apoptotic IL-1β and caspase-3 (Fig. 4B).
Inhibition of miR-101 attenuates
H/R-induced apoptosis via an upregulation of autophagy
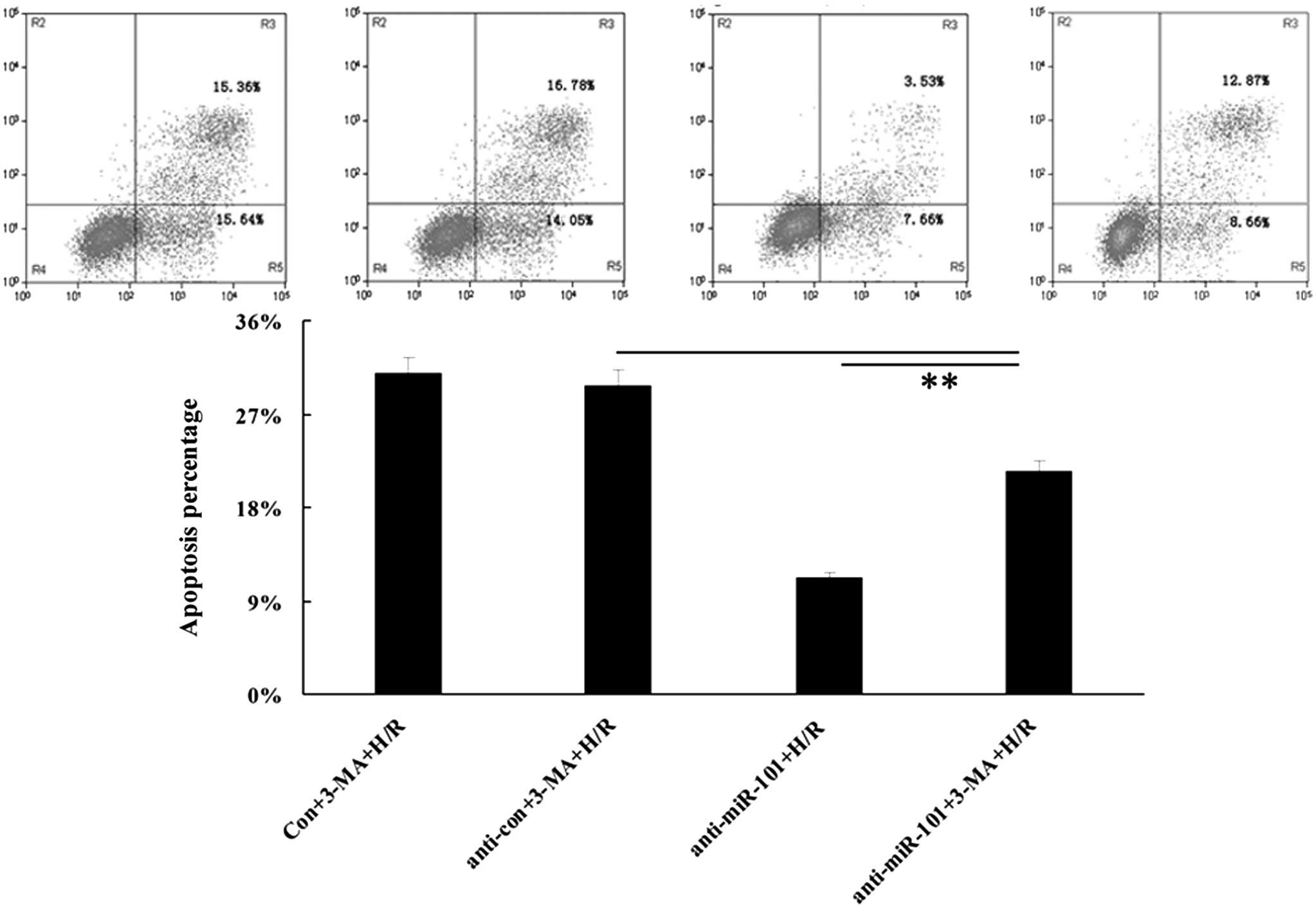
As autophagy is able to protect cells from
H/R-induced apoptosis to a certain extent, it was hypothesized that
the promoting effect of miR-101 on H/R-induced apoptosis may be a
result of its inhibitory effect on autophagy. In order to evaluate
the validity of this hypothesis, autophagy inhibitor
3-methyladenine (3-MA) was added into the medium following
transfection of H9c2 cardiomyocytes with anti-miR-101. Under H/R
conditions, cellular apoptosis in each group was determined. As
shown in Fig. 5, the apoptotic
rate in the anti-miR-101+H/R group was lower than that in the
anti-miR-101+3-MA+H/R group, suggesting that inhibition of miR-101
attenuated H/R-induced apoptosis, at least partially, via the
upregulation of autophagy.
Discussion
Under normal conditions, autophagy occurs at low
levels; however, this process is upregulated in response to stress,
including hypoxia, mitochondrial dysfunction and nutrient
deprivation (13,14). It has been reported that autophagy
promotes the survival of stressed cells via the recycling of
proteins to generate the fatty acids and free amino acids required
to maintain energy production, and the removal of damaged proteins
and organelles preventing their accumulation (1,15).
Accordingly, upregulation of autophagy within certain limits has
protective and beneficial effects on cells subjected to stress.
However, prolonged hypoxia and subsequent reperfusion may lead to
excessive autophagy, which results in cardiomyocyte death by
excessive self-digestion of organelles and proteins (10). Such abundant cardiomyocyte loss may
result in contractile dysfunction and heart failure (16). The manipulation of autophagy may
therefore become a promising therapeutic strategy for the
prevention or treatment of numerous types of heart disease. In the
present study, it was demonstrated that H/R induced apoptosis in
H9c2 cardiomyocytes, concurrently with the downregulation of
miR-101. Further investigation identified a novel target of
miR-101, RAB5A, which had previously been reported to be associated
with autophagy (10,16,17).
Subsequently, the role of miR-101 in the regulation of autophagy
under H/R was further investigated and the results suggested that
inhibition of miR-101 attenuated H/R-induced apoptosis, at least
partially, via the induction of autophagy.
In the present study, it was also demonstrated that
alongside cellular apoptosis accompanied by the inhibition of
miR-101, H/R induced the upregulation of autophagy-associated
Beclin 1 and LC3B, indicating that the level of autophagy was
upregulated. Enhanced autophagy was previously identified in
cardiac cells following H/R, as demonstrated by an increase in the
number of autophagic vesicles (18–20).
These results suggested that H/R resulted in enhanced apoptosis and
autophagy. As low levels of autophagy during ischemia and early
reperfusion protect against cell death, it was suggested that
spontaneous upregulation of autophagy may prevent excessive
apoptosis of cardiomyocytes in response to H/R. Numerous studies
have also demonstrated that autophagy is a beneficial response to
ischemia/reperfusion (I/R) (21–24).
For example, increased autophagy was demonstrated to correlate with
the functional recovery of the myocardium following I/R, and
cardiac myocytes exhibiting enhanced levels of autophagy were found
to be negative for apoptosis (25,26).
miR-101 has been shown to participate in the
regulation of autophagy. Frankel et al (10) for the first time, to the best of
our knowledge, identified miR-101 as a potent inhibitor of basal-,
etoposide- and rapamycin-induced autophagy in breast cancer cells.
They also found that STMN1, RAB5A and ATG4D were three targets of
miR-101 that were involved in miR-101-mediated autophagy.
Furthermore, they revealed that STMN1 overexpression partially
rescued cells from miR-101-mediated inhibition of autophagy, and
that inhibition of miR-101 sensitized breast cancer cells to
drug-mediated cell death (10). Xu
et al (16) reported
similar results, indicating that miR-101 had an inhibitory effect
on autophagy, and enhanced cisplatin-induced apoptosis in
hepatocellular carcinoma HepG2 cells, potentially via the
inhibition of autophagy by targeting RAB5A, STMN1 and ATG4D. In the
present study, it was also revealed that RAB5A expression levels
were significantly upregulated following inhibition of miR-101 in
H9c2 cardiomyocytes. However, no significant changes in STMN1
expression levels were detected, which suggested that the
regulatory association between miR-101 and STMN1 may be
cell-specific. In addition, it was confirmed that miR-101 directly
targeted RAB5A via binding to the 3′-UTR of RAB5A mRNA in H9c2
cardiomyocytes. Their targeting association has also previously
been revealed in breast cancer MCF7 cells and hepatocellular
carcinoma HepG2 cells (10,16).
Bcl-2 has been shown to function as a negative
regulator of cellular apoptosis (27) and increasing evidence has
implicated Bcl-2 in the regulation of autophagy (28,29).
Bcl-2 was found to bind to Beclin1 and disrupt autophagy, and
overexpression of Bcl-2 in the heart inhibited autophagy in
response to starvation. Furthermore, a Beclin1 mutant, which lacked
the Bcl-2 binding domain, induced excessive autophagy following
overexpression (30,31). These observations suggested that
Bcl-2 may function as a negative regulator of autophagy via the
inhibition of Beclin1. In the present study, it was demonstrated
that following upregulation of miR-101, expression levels of Bcl-2
were reduced and the apoptotic rate was upregulated, indicating
that miR-101 may promote H/R-induced cell apoptosis via
downregulation of Bcl-2. However, despite Bcl-2 functioning as a
negative regulator of Beclin1 and the reduced expression levels of
Bcl-2 following overexpression of miR-101, the expression of
Beclin1 was also decreased. It was therefore suggested that miR-101
may also have a suppressive effect on Beclin1 expression, through
which it may inhibit H/R-induced autophagy. To further verify the
hypothesis that autophagy was involved in the miR-101-mediated
apoptosis in cardiomyocytes treated by H/R, autophagy inhibitor
3-MA was used. The apoptotic rate was found to be reduced following
inhibition of miR-101 in H/R-treated cardiomyocytes; however, this
reduction was partially attenuated following the addition of 3-MA,
indicating that inhibition of autophagy attenuated the suppressive
effect of miR-101 downregulation on H/R-induced apoptosis in
cardiomyocytes.
In conclusion, the present study, for the first
time, to the best of our knowledge, suggested that the inhibition
of miR-101 attenuated H/R-induced apoptosis, at least in part, via
the induction of autophagy, and that miR-101 may function as a
potential therapeutic agent for the treatment or prevention of
heart disease.
References
|
1
|
Oku M, Takano Y and Sakai Y: The emerging
role of autophagy in peroxisome dynamics and lipid metabolism of
phyllosphere microorganisms. Front Plant Sci. 5:812014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Petersen M, Hofius D and Andersen SU:
Signaling unmasked: Autophagy and catalase promote programmed cell
death. Autophagy. 10:2004.
|
|
3
|
Tessitore A, Cicciarelli G, Del Vecchio F,
et al: MicroRNAs in the DNA Damage/Repair Network and Cancer. Int J
Genomics. 2014:8202482014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen LJ, Lim SH, Yeh YT, Lien SC and Chiu
JJ: Roles of microRNAs in atherosclerosis and restenosis. J Biomed
Sci. 19:792012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu T, Li D and Jiang D: Targeting cell
signaling and apoptotic pathways by luteolin: cardioprotective role
in rat cardiomyocytes following ischemia/reperfusion. Nutrients.
4:2008–2019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morita K: Surgical reoxygenation injury of
the myocardium in cyanotic patients: clinical relevance and
therapeutic strategies by normoxic management during
cardiopulmonary bypass. Gen Thorac Cardiovasc Surg. 60:549–556.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu LF, Liang Z, Lv ZR, et al:
MicroRNA-15a/b are up-regulated in response to myocardial
ischemia/reperfusion injury. J Geriatr Cardiol. 9:28–32. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qin Y, Yu Y, Dong H, Bian X, Guo X and
Dong S: MicroRNA 21 inhibits left ventricular remodeling in the
early phase of rat model with ischemia-reperfusion injury by
suppressing cell apoptosis. Int J Med Sci. 9:413–423. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu L, Zhang G, Liang Z, et al:
MicroRNA-15b enhances hypoxia/reoxygenation-induced apoptosis of
cardiomyocytes via a mitochondrial apoptotic pathway. Apoptosis.
19:19–29. 2014. View Article : Google Scholar
|
|
10
|
Frankel LB, Wen J, Lees M, et al:
microRNA-101 is a potent inhibitor of autophagy. EMBO J.
30:4628–4641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin X, Guan H, Li H, et al: miR-101
inhibits cell proliferation by targeting Rac1 in papillary thyroid
carcinoma. Biomed Rep. 2:122–126. 2014.PubMed/NCBI
|
|
12
|
Yin J, Wang M, Jin C and Qi Q: miR-101
sensitizes A549 NSCLC cell line to CDDP by activating caspase
3-dependent apoptosis. Oncol Lett. 7:461–465. 2014.PubMed/NCBI
|
|
13
|
Gong JS and Kim GJ: he role of autophagy
in the placenta as a regulator of cell death. Clin Exp Reprod Med.
41:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hou C, Zhu M, Sun M and Lin Y: MicroRNA
let-7i induced autophagy to protect T cell from apoptosis by
targeting IGF1R. Biochem Biophys Res Commun. 453:728–734. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gustafsson AB and Gottlieb RA: Recycle or
die: the role of autophagy in cardioprotection. J Mol Cell Cardiol.
44:654–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Y, An Y, Wang Y, et al: miR-101
inhibits autophagy and enhances cisplatin-induced apoptosis in
hepatocellular carcinoma cells. Oncol Rep. 29:2019–2024.
2013.PubMed/NCBI
|
|
17
|
Li Y, Zhao Y, Hu J, et al: A novel
ER-localized transmembrane protein, EMC6, interacts with RAB5A and
regulates cell autophagy. Autophagy. 9:150–163. 2013. View Article : Google Scholar :
|
|
18
|
Sybers HD, Ingwall J and DeLuca M:
Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct
Metab. 12:453–463. 1976.PubMed/NCBI
|
|
19
|
Zhang ZL, Fan Y and Liu ML: Ginsenoside
Rg1 inhibits autophagy in H9c2 cardiomyocytes exposed to
hypoxia/reoxygenation. Mol Cell Biochem. 365:243–250. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao X, Chen A, Yang P, et al: Alpha-lipoic
acid protects cardiomyocytes against hypoxia/reoxygenation injury
by inhibiting autophagy. Biochem Biophys Res Commun. 441:935–940.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang JH, Behrns KE, Leeuwenburgh C and Kim
JS: Critical role of autophage in ischemia/reperfusion injury to
aged livers. Autophagy. 8:140–141. 2012. View Article : Google Scholar :
|
|
22
|
Han Z, Cao J, Song D, et al: Autophagy is
involved in the cardioprotection effect of remote limb ischemic
postconditioning on myocardial ischemia/reperfusion injury in
normal mice, but not diabetic mice. PLoS One. 9:e868382014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang JW, Cho H and Lee SM: Melatonin
inhibits mTOR-dependent autophagy during liver
ischemia/reperfusion. Cell Physiol Biochem. 33:23–36. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y and Ren J: Targeting autophagy for
the therapeutic application of histone deacetylase inhibitors in
ischemia/reperfusion heart injury. Circulation. 129:1088–1091.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeng M, Wei X, Wu Z, et al: NF-κB-mediated
induction of autophagy in cardiac ischemia/reperfusion injury.
Biochem Biophys Res Commun. 436:180–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: The interplay between pro-death and pro-survival signaling
pathways in myocardial ischemia/reperfusion injury: apoptosis meets
autophagy. Cardiovasc Drugs Ther. 20:445–462. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu G, Wang T, Song J and Zhou Z: Effects
of apoptosis-related proteins caspase-3, Bax and Bcl-2 on cerebral
ischemia rats. Biomed Rep. 1:861–867. 2013.
|
|
28
|
Ni Z, Wang B, Dai X, et al: HCC cells with
high levels of Bcl-2 are resistant to ABT-737 via activation of the
ROS-JNK-autophagy pathway. Free Radic Biol Med. 70:194–203. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heath-Engel HM, Chang NC and Shore GC: The
endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2
protein family. Oncogene. 27:6419–6433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang XH, Kleeman LK, Jiang HH, et al:
Protection against fatal Sindbis virus encephalitis by beclin, a
novel Bcl-2-interacting protein. J Virol. 72:8586–8596.
1998.PubMed/NCBI
|
|
31
|
Pattingre S, Tassa A, Qu X, et al: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|