Introduction
The term hypoxia describes a reduction in the normal
level of tissue oxygen tension, which may be acute or chronic.
Hypoxia is the most notable and prevailing characteristic of the
microenvironment of solid tumors (1). Consequently, adaptations to hypoxia
are necessary for the survival of tumor cells in such
microenvironments. Hypoxia-inducible factor-1 (HIF-1) is a
transcriptional complex that is activated in response to hypoxia
and other growth factors, and has a significant role in tumor
progression, invasion and metastasis (2,3). A
pool of studies has provided evidence indicating that the
overexpression of HIF-1α subunit is associated with a poor
prognostic outcome as well as resistance to chemotherapy and
radiation (4–7). It is also well established that
HIF-1α regulates multiple proangiogenic factors, including vascular
endothelial growth factor (VEGF). Hypoxia-induced VEGF may activate
the MAPK-related VEGF receptor signaling pathway to elicit
anti-apoptotic effects in a self-activating manner and induce
angiogenesis primarily through its interaction with two tyrosine
kinase receptors expressed in vascular endothelial cells, VEGFR1
(Flt1) and VEGFR2 (Flk1/KDR) (8).
Furthermore, there is emerging evidence demonstrating that VEGF may
also form an autocrine loop with HIF-1α, contributing to tumor cell
survival as well as drug resistance during hypoxia in
neuroblastoma, osteosarcoma, rhabdomyosarcoma and breast cancer
cells (9–11). The anticancer effect of the
pharmacological VEGF antagonism beyond interference with
angiogenesis is therefore a significant area of cancer
research.
Hypoxia and DNA damage occur simultaneously when
therapies that cause DNA damage are applied to tumors bearing
hypoxic regions. Cells may be eliminated following DNA damage
through various forms of programmed death, including apoptosis,
autophagy, mitotic catastrophe and necrosis. Among these, a common
paradigm is that chemotherapeutic agents stimulate cells to undergo
apoptosis, implying that impairment of apoptotic pathways may be
involved in the development of chemoresistance (12).
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer and a leading cause of cancer-related
mortality worldwide. As a highly vascularized tumor with poor
prognosis, elevated levels of VEGF have been identified and a
greater expression of VEGF has been associated with shorter
survival in HCC patients (13–15).
Therefore, inhibition of VEGF is an attractive target in HCC
treatment (16). Significant
clinical and biologic research into bevacizumab, a humanized
monoclonal antibody that binds VEGF prior to its attachment to its
natural receptors, has been undertaken with regard to HCC, since
systemic therapy with cytotoxic agents has demonstrated a notable
benefit (17–19).
Given the potential roles of chemotherapeutic agents
and VEGF antagonism in cancer therapeutics, one rational strategy
would be to combine anti-VEGF treatment with DNA damage agents such
as etoposide. We used the SMMC-7721 hepatoma cell line in the
present study, which is an identified chemoresistant phenotype when
subjected to hypoxia, with elevated serum VEGF levels contributing
to tumor growth and metastasis. This study investigates the role of
this combination strategy and provides evidence that pre-exposure
of SMMC-7721 cells to hypoxia weakens etoposide-induced
cytotoxicity. Notably, the anti-VEGF strategy facilitated DNA
damage ability of etoposide followed by cell cycle delay and
apoptosis induction, which served to overcome hypoxia-driven
etoposide resistance.
Materials and methods
Reagents and antibodies
Etoposide was purchased from Sigma-Aldrich (St.
Louis, MO, USA); a stock solution of 100 mM was prepared with
dimethyl sulfoxide and stored at −20°C. The stock solution was
further diluted with the appropriate medium immediately prior to
use. The antibodies to HIF-1α and Flt1 (VEGFR1) were purchased from
Calbiochem (La Jolla, CA, USA) and R&D systems (Minneapolis,
MN, USA), respectively. The antibodies to VEGF, p53, Bcl-2, Bax,
procaspase 3, cyclin A, cyclin B1, cdc2, AKT, ERK, α-tubulin and
β-actin, as well as HRP-labeled secondary anti-goat, anti-mouse and
anti-rabbit antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The antibodies to γH2AX,
p-AKT (Ser473), p-ERK, p-IκB-α (Ser32) and GAPDH were obtained from
Cell Signaling Technology, Inc. (Beverly, MA, USA). Recombinant
human VEGF165 (rhVEGF165) and a neutralizing
antibody against VEGF (anti-mouse monoclonal antibody) were
purchased from R&D Systems.
Cell culture and establishment of hypoxia
culture conditions
Human HCC SMMC-7721 cells (Shanghai Institute of
Biological Sciences, Shanghai, China) were cultured in RPMI-1640
(Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with
10% fetal bovine serum containing 100 U/ml penicillin and 100 μg/ml
streptomycin (Sigma-Aldrich). The cells were kept in a humidified
atmosphere of 5% CO2 and 20% O2 at 37°C.
Environmental hypoxic conditions (0.6% O2) were achieved
in an airtight humidified chamber continuously flushed with a mixed
gas where N2 was used to compensate for the reduced
O2 level.
Cytotoxicity assay
Cell proliferation was determined by a standard
3-[4,5-dimethylthia-zol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells were seeded in 96-well plates, and hypoxic cells were
allowed to attach for one day before exposure to etoposide at
concentrations specified or vehicle control alone. Plates were
assayed 48 and 72 h after the initiation of the designated
treatment. All experiments were repeated three times.
Flow cytometric determination of cell
cycle
To determine the phase distribution of the DNA
content, propidium iodide (PI) staining was performed. Following
the designated treatment of actively proliferating cultures, cells
were collected and washed twice with phosphate-buffered saline
(PBS) buffer and fixed in 70% ice-cold ethanol overnight. The cell
pellet was re-suspended in PBS plus 0.5 mg/ml RNaseA at 37°C for 30
min prior to staining with 5 mg/ml PI (Sigma-Aldrich) at room
temperature in the dark for another 30 min. The analysis was
performed with a FACScan flow cytometer (BD Biosciences, San Jose,
CA, USA).
Flow cytometric determination of
apoptosis
Cells were collected and washed twice with ice-cold
PBS buffer following the designated treatment of actively
proliferating cultures. Subsequently, cells were stained with
annexin V-fluorescein and PI according to the descriptions in the
commercial apoptosis detection kit (BD Pharmingen, San Diego, CA,
USA). Analysis was performed with a FACScan flow cytometer (BD
Biosciences).
Protein expression
Proteins were extracted in radioimmunoprecipitation
assay buffer (150 mM NaCl, 50 mM Tris, 2 mM EGTA, 2 mM EDTA, 25 mm
NaF, 25 mm glycerophosphate, 0.2% Triton X-100, 0.3% NONIDET P-40
and 0.1 mM PMSF). Total protein concentrations of whole cell
lysates were determined using the Bio-Rad reagent (Bio-Rad,
Hercules, CA, USA) bicinchoninic acid method. Equal amounts of 40
μg total protein were loaded per lane. Proteins were fractionated
on 8–12% Tris-Glycine pre-cast gels (Novex, San Diego, CA, USA),
transferred to Immobilon-P transfer membrane (Millipore, Billerica,
MA, USA), and probed with primary antibodies and then HRP-labeled
secondary antibodies. Proteins were visualized using ECL western
blotting detection reagents (Amersham Biosciences, Piscataway, NJ,
USA).
Results
Hypoxia impairs etoposide-induced
cytotoxicity
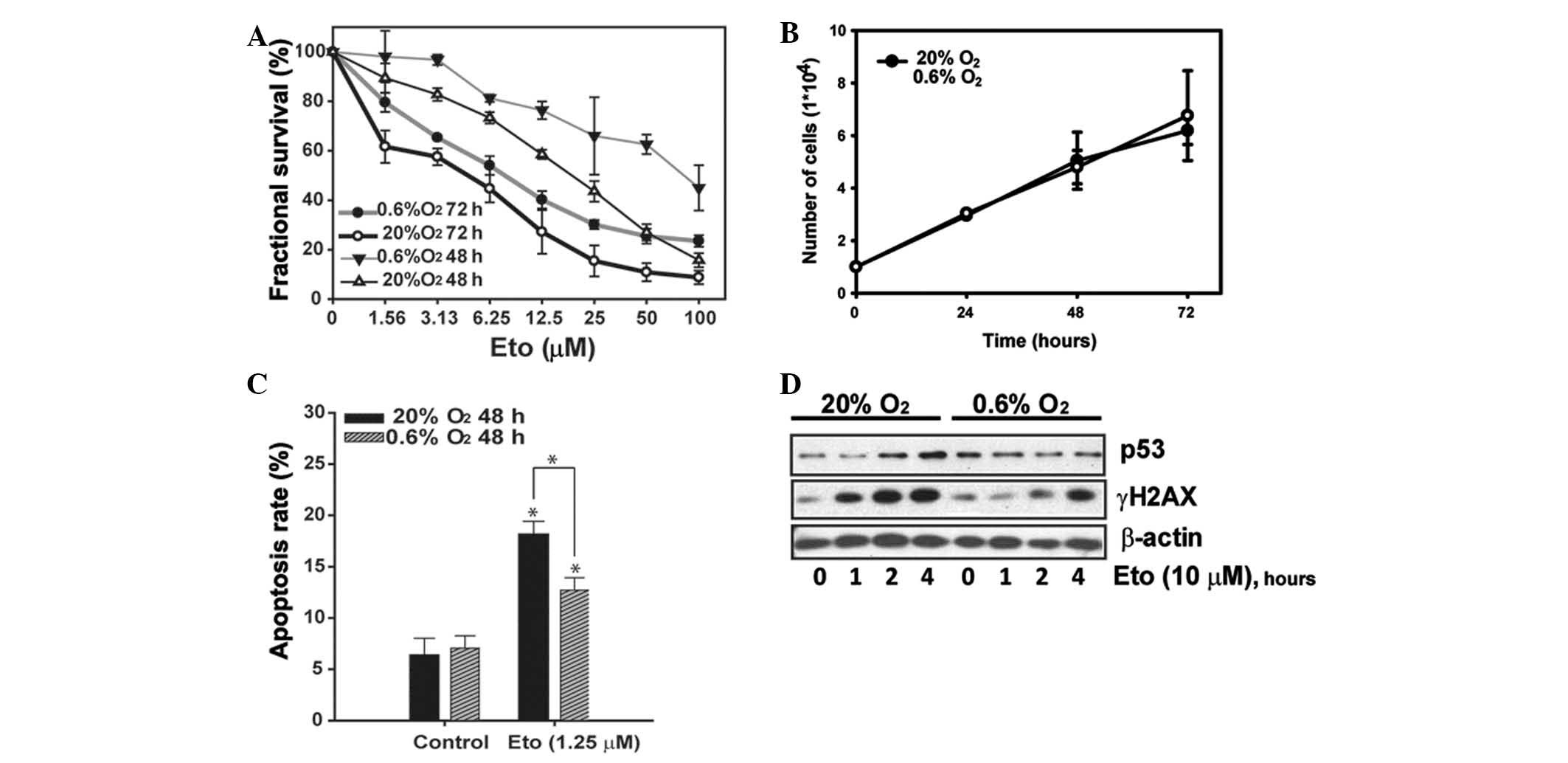
To examine the effect of hypoxia, SMMC-7721 cells
were exposed to normoxia or hypoxia (0.6% O2),
respectively, in the presence of serum to avoid glucose deprivation
or cell cycle inhibition. It was noted that etoposide exerted
significant concentration- and time-dependent cytotoxicity in a
normoxic atmosphere, as shown in Fig.
1A. Conversely, an impaired anti-proliferative effect was
observed under the hypoxic environment. To exclude the possibility
that the diminished cytotoxicity of etoposide was due to a general
anti-proliferative effect of hypoxic conditions that render more
slowly cycling cells, cell growth curves were analyzed under
various oxygen conditions, as shown in Fig. 1B, to demonstrate that hypoxia alone
did not lower cell proliferation within 72 h. However, the
etoposide-induced apoptosis was significantly reduced (Fig. 1C).
Etoposide is known to stabilize the cleavable
complex formed between topoisomerase II and DNA, and subsequently
form strand breakage. The topoisomerase II inhibitor causes
replication stress and generates the dominant damage-induced
phosphorylation of the minor histone H2A (γH2AX), which has become
one of the most widely used measures of DNA damage and is observed
within a short-term exposure to etoposide (20). We treated SMMC-7721 cells for 0–4 h
with a relatively high concentration (10 μM) and then measured the
γH2AX level. The western blotting results shown in Fig. 1D reveal that a significant increase
of γH2AX was observed after 1 h of etoposide treatment, which
reached a platform at 4 h. Since p53 was reported to be involved in
apoptosis in response to DNA damage, p53 accumulation was also
analyzed (21). A significant
accumulation of p53 was detected following a 2 h challenge of
etoposide. However, far less significant expression of γH2AX and
p53 was observed in hypoxia (Fig.
1D).
Anti-VEGF intervention improves
etoposide-induced apoptosis in hypoxia
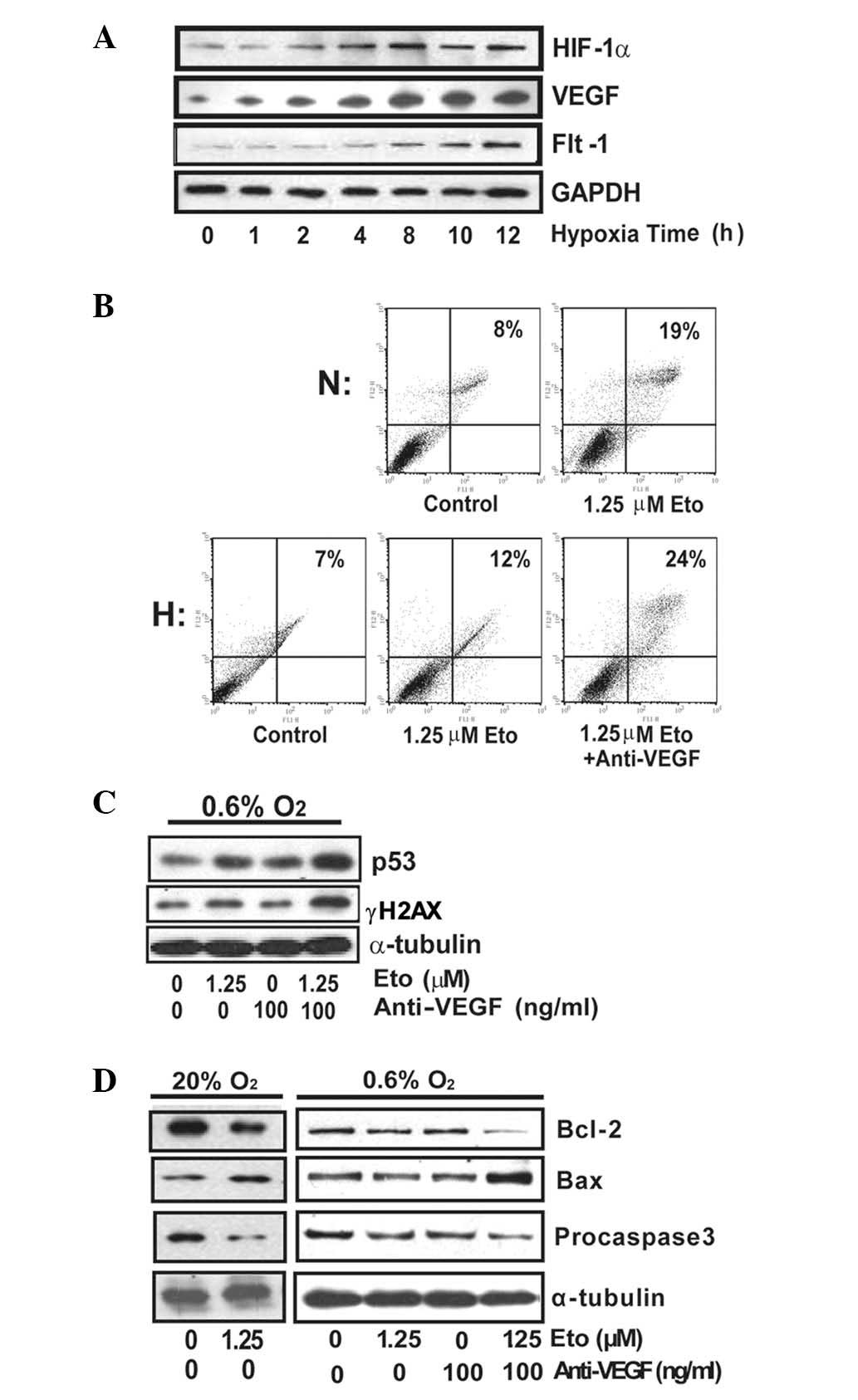
By culturing SMMC-7721 cells under hypoxic
conditions, increased levels of HIF-1α and VEGF were observed
(Fig. 2A). To investigate the
effect of VEGF in hypoxia-induced chemoresistance to etoposide, we
exposed SMMC-7721 cells to etoposide with or without anti-VEGF
under hypoxia and further detected the apoptosis. As expected, VEGF
interference increased the apoptotic fraction (Fig. 2B).
To demonstrate how this combination strategy might
affect etoposide function in DNA damage under hypoxia, SMMC-7721
cells were treated with or without anti-VEGF upon etoposide
administration (10 μM). Combined treatment of anti-VEGF and
etoposide significantly upregulated γH2AX and p53 levels, to a
level similar to that observed with a single treatment of etoposide
in normoxia (Fig. 2C).
Mitochondria play a pivotal role in the regulation
of apoptosis by regulating the balance between anti-apoptotic
family members including Bcl-2 and pro-apoptotic family members
including Bax. To determine whether the downstream activators of
apoptosis were induced, we analyzed the expression of Bcl-2, Bax
and procaspase 3. A significant increase in Bax as well as
significant decreases in Bcl-2 and procaspase 3 were observed upon
etoposide treatment in normoxic conditions (Fig. 2D). However, the expression of these
proteins was not significantly affected by etoposide alone in
hypoxia, while the anti-VEGF combination helped to further
downregulate Bcl-2 and procaspase 3, which was consistent with
apoptosis data (Fig. 2B). Taken
together, the anti-VEGF combination strategy may affect
mitochondrial-related apoptosis and improve etoposide-induced
SMMC-7721 apoptosis under hypoxia.
Anti-VEGF facilitates etoposide-induced
cell cycle arrest under hypoxia
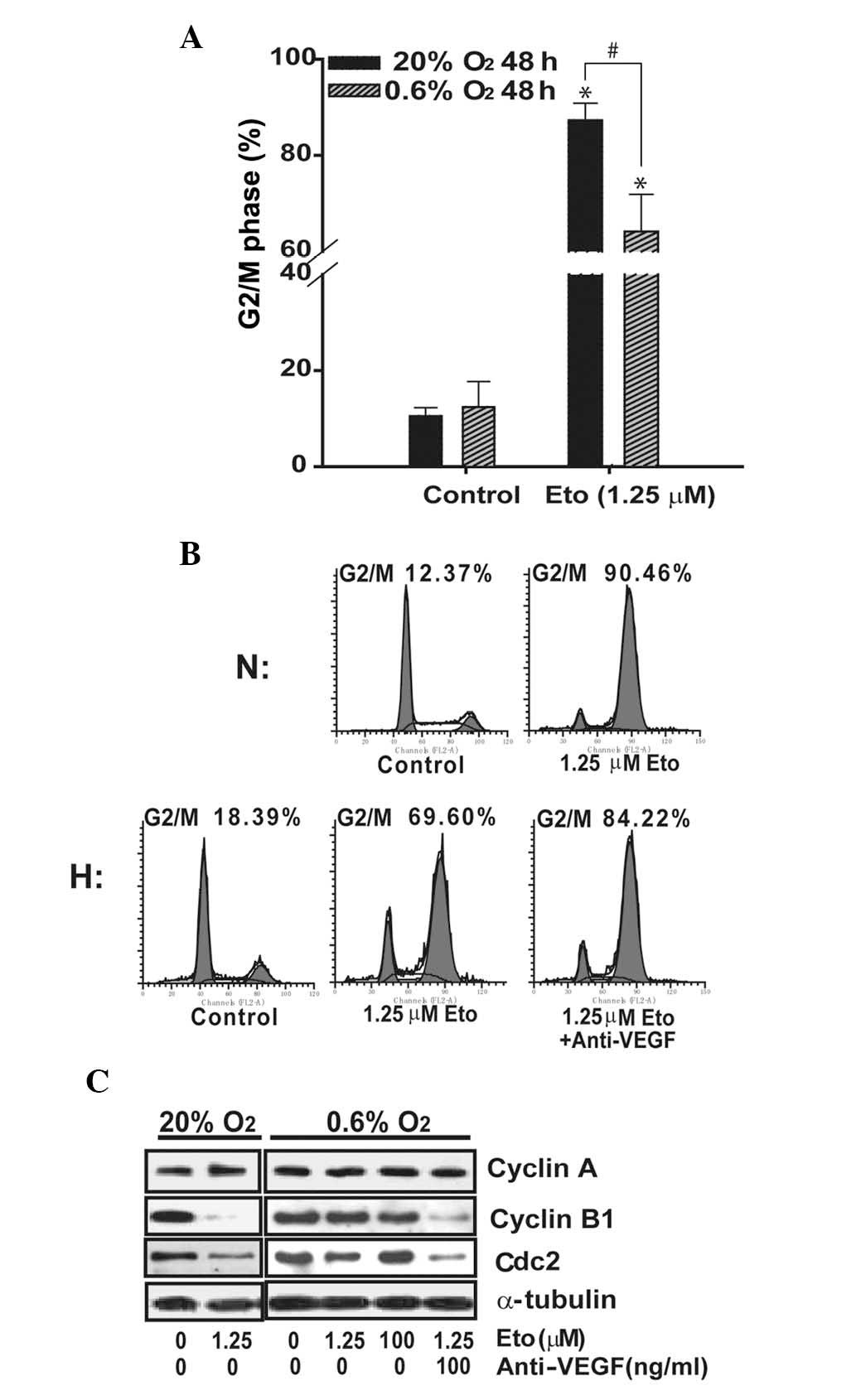
Hypoxia is also reported to induce cell cycle arrest
in certain tumors, leading to chemo- and/or radio-resistance
(22). SMMC-7721 cells were
treated with various concentrations of etoposide for 48 h before
analyses of cell cycle phase distribution were performed. Hypoxia
alone did not appear to induce cell cycle arrest since there were
no notable differences between the hypoxic and normoxic cells.
Etoposide exhibited a concentration-dependent effect in G2/M arrest
(data not shown here). As shown in Fig. 3A, 48-h treatment of single
etoposide (1.25 μM) triggered the arrest of 87.3±3.5% cells
(control, 10.5±1.7%) in the G2/M phase under normoxic conditions.
In contrast, a rate of only 64.1±7.7% (control, 12.4±5.3%) was
observed under hypoxia. However, an increased proportion of G2/M
phase arrested cells was observed when cells were treated with both
anti-VEGF and etoposide, suggesting that VEGF reduced etoposide in
cell cycle arrest (Fig. 3B).
Next, we assessed the effects of etoposide alone or
combined with anti-VEGF on cell cycle regulating proteins. In
normoxia, challenge with etoposide alone resulted in a significant
decrease in cyclin B1 and Cdc2 (Fig.
3C). However, in hypoxia, etoposide only caused a slight
decrease in cyclin B1 and Cdc2. Significant downregulation of
cyclin B1 and Cdc2 was observed when the cells were co-treated with
etoposide and anti-VEGF, which was similar to that observed under
normoxia.
Anti-VEGF combination strategy is
correlated with the AKT, ERK and nuclear factor-κB (NF-κB)
pathway
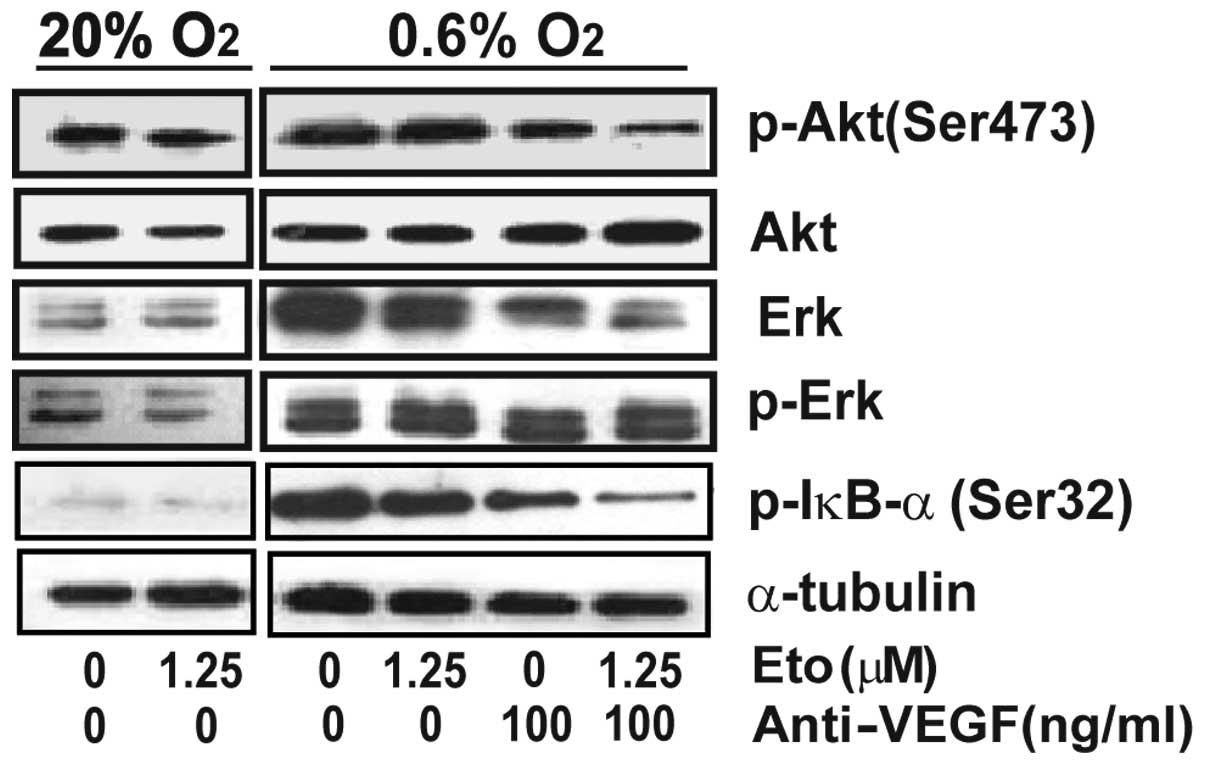
Current studies suggest that the AKT and ERK
pathways are the most relevant survival pathways in tumor cells
(23). We thus assessed whether or
not AKT and ERK play a role in hypoxia-induced insensitivity to
etoposide in SMMC-7721 cells. As shown in Fig. 4, hypoxia triggered phosphorylation
of AKT and ERK. Administration of etoposide alone resulted in a
slight decrease in the phosphorylation of AKT and ERK. However,
anti-VEGF alone or the combination treatment completely eliminated
the phosphorylation of these proteins, suggesting that the AKT and
ERK pathways are involved in anti-VEGF-restored sensitivity to
etoposide.
The transcription factor NF-κB has been implicated
in resistance to chemotherapeutic treatment. Recent evidence has
demonstrated significant cross-talk and the inter-independence of
NF-κB and HIF-1α signaling (24,25).
NF-κB is a transcriptional activator of HIF-1α and, conversely,
HIF-1α accumulation is shown to promote NF-κB. Since
phosphorylation of IκB-α at Ser32 is essential for the release of
active NF-κB, phosphorylation at this site is a significant marker
of NF-κB activation (26,27). Here, we demonstrate that
phosphorylation of IκB-α at Ser32 was induced by hypoxia which was
eliminated by VEGF inhibition, suggesting the involvement of
NF-κB.
Discussion
Hypoxia-induced chemoresistance is an aspect of
tumor biology that has received increasing attention over the last
decades, with the characterization of HIFs being of particular
significance (29,30). Genetic approaches and
small-molecule inhibitors targeting HIF-1 have proven effective in
decreasing hypoxia-induced resistance to chemotherapeutics in
tumors (31,32). Previous studies suggest that
VEGF/Flt1 may also form an autocrine loop with HIF-1α, which
contributes to cell survival and drug resistance during hypoxia
(9–11). Although KDR is generally considered
to be the major mediator of the mitogenic and angiogenic effects of
VEGF (33), emerging studies
demonstrate that Flt1 is present and functional in a diverse group
of tumors. In addition, Flt1 is the only receptor associated with
VEGF that contains a binding site for HIF-1α, which further
supports the recruitment of the VEGF/Flt1 system in HIF-1α-driven
tumor progression. Bevacizumab has been shown to improve treatment
outcomes in selected patients with advanced colorectal, breast and
non-small cell lung cancer. However, it is also being evaluated
among patients with earlier-stage cancer, and among patients with
other types of cancer. The results of a phase II clinical trial
(18,19) revealed that the combination of two
targeted therapies, bevacizumab and erlotinib, exhibited anticancer
activity in patients with advanced HCC and is therefore worthy of
further study.
The DNA damaging reagent etoposide has been
demonstrated to induce apoptosis in a variety of tumor cell lines
harboring either wild-type or mutant p53. Although the signaling
pathways mediating etoposide-induced apoptosis are unclear, one
pathway may involve p53 since DNA damage induced by etoposide has
been shown to activate p53. It is also known that p53 protein is a
potent negative regulator of HIF-1α and VEGF in hypoxia. p53
represses VEGF during hypoxia by binding the transcription factor
SP1 and inhibiting its ability to bind the VEGF promoter (34). HIF-1α activity may also be
inhibited by p53, which directly binds HIF-1α and targets protein
for degradation and thereby downregulated VEGF transcription
(35,36). In addition, the ability of p53 to
inhibit the HIF system is mediated by its physical interaction with
HIF-1α and does not even require its transcriptional activity.
Using γH2AX as an indicator of DNA damage, we observed that hypoxia
weakened etoposide-induced DNA damage. Notably, the anti-VEGF
combination strategy enhanced γH2AX and p53 expression.
Subsequently, cell cycle and apoptosis data suggested that the
anti-VEGF combination with etoposide could also restore etoposide
function to a level comparable with that observed in normoxia.
Previous studies have shown indirect links between
HIF-1α and NF-κB transcription pathways (37). The inactive NF-κB is primarily
localized in the cytoplasm, and its activation involves its release
from IκB-α and translocation to the nucleus. This phosphorylation
induces the degradation of IκB-α by the ubiquitin-proteasome system
and the release of NF-κB for nuclear translocation. Preventing
NF-κB activation exerts a tumor-suppressive effect by promoting
apoptosis of transformed cells which would otherwise have given
rise to cancer. In addition, NF-κB is a transcriptional activator
of HIF-1α, and basal NF-κB activity is required for HIF-1α
accumulation in normoxia and during hypoxia. Previous studies have
demonstrated significant cross-talk and the inter-independence of
NF-κB and HIF-1α signaling, indicating that NF-κB is a
transcriptional activator of HIF-1α and, conversely, HIF-1α
accumulation is shown to promote NF-κB (24,25).
Furthermore, in vitro and in vivo studies have
already shown that targeted inhibition of NF-κB sensitizes tumor
cells to chemotherapy and radiation (38).
In this study, we combined etoposide, a commonly
used chemotherapeutic agent, with monoclonal VEGF antibody to
evaluate its role in hypoxia-induced resistance in hepatoma
SMMC-7721 cells. The results revealed that hypoxia impaired
etoposide function in DNA damage and resultant cell death and
resulted in drug resistance. In addition, interference of VEGF
inhibited hypoxia induction of HIF-1α. The anti-VEGF combination
treatment enhanced etoposide efficacy by the attenuation of DNA
damage followed by limited cell cycle delay and/or apoptosis and
reversed drug resistance in SMMC-7721 cells.
Acknowledgements
This study was supported by National Natural Science
Funds (no. 81273535 and no. 81272611) and Hangzhou Core Scientific
research innovation project (no. 20112313A01). It also received
support from the National Natural Science Foundation of China
(81072657 and 91029745), Zhejiang Provincial Natural Science
Foundation of China (Z2090053) and the Program for New Century
Excellent Talents of the Ministry of Education of China.
Abbreviations:
|
HIF
|
hypoxia-inducible factor
|
|
VEGF
|
vascular endothelial growth factor
|
|
HCC
|
hepatocellular carcinoma
|
|
rhVEGF165
|
recombinant human vascular endothelial
growth factor (165)
|
|
NF-κB
|
nuclear factor κB.
|
References
|
1
|
Höckel M and Vaupel P: Tumor hypoxia:
definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carroll VA and Ashcroft M: Role of
hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the
regulation of HIF target genes in response to hypoxia, insulin-like
growth factor-I, or loss of von Hippel-Lindau function:
implications for targeting the HIF pathway. Cancer Res.
66:6264–6270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semenza GL, Shimoda LA and Prabhakar NR:
Regulation of gene expression by HIF-1. Novartis Found Symp.
272:33–36. 2006.
|
|
4
|
Schnitzer SE, Weigert A, Zhou J, et al:
Hypoxia enhances sphingosine kinase 2 activity and provokes
sphingosine-1-phosphate-mediated chemoresistance in A549 lung
cancer cells. Mol Cancer Res. 7:393–401. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang B, Fan L, Fang L, et al:
Hypoxia-mediated fenretinide (4-HPR) resistance in childhood acute
lymphoblastic leukemia cells. Cancer Chemother Pharmacol.
58:540–546. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Flamant L, Notte A, Ninane N, et al:
Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast
cancer cells under hypoxia. Mol Cancer. 9:1912010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sermeus A, Cosse JP, Crespin M, et al:
Hypoxia induces protection against etoposide-induced apoptosis:
molecular profiling of changes in gene expression and transcription
factor activity. Mol Cancer. 7:27–31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang D, Weng Q, Zhang L, et al: VEGF and
Bcl-2 interact via MAPKs signaling pathway in the response to
hypoxia in neuroblastoma. Cell Mol Neurobiol. 29:391–401. 2009.
View Article : Google Scholar
|
|
9
|
Das B, Yeger H, Tsuchida R, et al: A
hypoxia-driven vascular endothelial growth factor/Flt1 autocrine
loop interacts with hypoxia-inducible factor-1alpha through
mitogen-activated protein kinase/extracellular signal-regulated
kinase 1/2 pathway in neuroblastoma. Cancer Res. 65:7267–7275.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsuchida R, Das B, Yeger H, et al:
Cisplatin treatment increases survival and expansion of a highly
tumorigenic side-population fraction by upregulating VEGF/Flt1
autocrine signaling. Oncogene. 27:3923–3934. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee TH, Seng S, Sekine M, et al: Vascular
endothelial growth factor mediates intracrine survival in human
breast carcinoma cells through internally expressed VEGFR1/FLT1.
PLoS Med. 4:186–191. 2007. View Article : Google Scholar
|
|
12
|
Erler JT, Cawthorne CJ, Williams KJ, et
al: Hypoxia-mediated down-regulation of Bid and Bax in tumors
occurs via hypoxia-inducible factor 1-dependent and -independent
mechanisms and contributes to drug resistance. Mol Cell Biol.
24:2875–2889. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeng KS, Sheen IS, Wang YC, et al:
Prognostic significance of preoperative circulating vascular
endothelial growth factor messenger RNA expression in resectable
hepatocellular carcinoma: a prospective study. World J
Gastroenterol. 10:643–648. 2004.PubMed/NCBI
|
|
14
|
Jinno K, Tanimizu M, Hyodo I, et al:
Circulating vascular endothelial growth factor (VEGF) is a possible
tumor marker for metastasis in human hepatocellular carcinoma. J
Gastroenterol. 33:376–382. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwak BK, Shim HJ, Park ES, et al:
Hepatocellular carcinoma: correlation between vascular endothelial
growth factor level and degree of enhancement by multiphase
contrast-enhanced computed tomography. Invest Radiol. 36:487–492.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pang R and Poon RT: Angiogenesis and
antiangiogenic therapy in hepatocellular carcinoma. Cancer Lett.
242:151–167. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun W, Sohal D, Haller DG, et al: Phase 2
trial of bevacizumab, capecitabine, and oxaliplatin in treatment of
advanced hepatocellular carcinoma. Cancer. 117:3187–3192. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thomas MB, Morris JS, Chadha R, et al:
Phase II trial of the combination of bevacizumab and erlotinib in
patients who have advanced hepatocellular carcinoma. J Clin Oncol.
27:843–850. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaseb AO, Garrett-Mayer E, Morris JS, et
al: Efficacy of bevacizumab plus erlotinib for advanced
hepatocellular carcinoma and predictors of outcome: final results
of a phase II trial. Oncology. 82:67–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smart DJ, Halicka HD, Schmuck G, et al:
Assessment of DNA double-strand breaks and gammaH2AX induced by the
topoisomerase II poisons etoposide and mitoxantrone. Mutat Res.
641:43–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brady CA, Jiang D, Mello SS, et al:
Distinct p53 transcriptional programs dictate acute DNA-damage
responses and tumor suppression. Cell. 145:571–583. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Richard S, Geneviève CP, Lisa JF, et al:
hypoxia-inducible factor-1 activity associated with decreased
senescence and requires hypoxia-inducible factor-1 activity. Mol
Cancer Ther. 7:1961–1973. 2008. View Article : Google Scholar
|
|
23
|
Pommier Y, Sordet O, Antony S, et al:
Apoptosis defects and chemotherapy resistance: molecular
interaction maps and networks. Oncogene. 23:2934–2949. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jung Y, Isaacs JS, Lee S, et al:
Hypoxia-inducible factor induction by tumour necrosis factor in
normoxic cells requires receptor-interacting protein-dependent
nuclear factor kappa B activation. Biochem J. 370:1011–1017. 2003.
View Article : Google Scholar
|
|
25
|
Rius J, Guma M, Schachtrup C, et al:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perkins ND: Post-translational
modifications regulating the activity and function of the nuclear
factor kappa B pathway. Oncogene. 25:6717–6730. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Perkins ND and Gilmore TD: Good cop, bad
cop: the different faces of NF-kappaB. Cell Death Differ.
13:759–772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cummins EP, Berra E, Comerford KM, et al:
Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta,
giving insight into hypoxia-induced NFkappaB activity. Proc Natl
Acad Sci USA. 103:18154–18159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Unruh A, Ressel A, Mohamed HG, et al: The
hypoxia-inducible factor-1 alpha is a negative factor for tumor
therapy. Oncogene. 22:3213–3220. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Le QT, Denko NC and Giaccia AJ: Hypoxic
gene expression and metastasis. Cancer Metastasis Rev. 23:293–310.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sullivan R and Graham CH: Hypoxia prevents
etoposide-induced DNA damage in cancer cells through a mechanism
involving hypoxia-inducible factor 1. Mol Cancer Ther. 8:1702–1713.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rohwer N, Dame C, Haugstetter A, et al:
Hypoxia-inducible factor 1alpha determines gastric cancer
chemosensitivity via modulation of p53 and NF-kappaB. PLoS One.
5:120382010. View Article : Google Scholar
|
|
33
|
Ferrara N: Vascular endothelial growth
factor: basic science and clinical progress. Endocrine Rev.
25:581–611. 2004. View Article : Google Scholar
|
|
34
|
Pal S, Datta K and Mukhopadhyay D: Central
role of p53 on regulation of vascular permeability factor/vascular
endothelial growth factor (VPF/VEGF) expression in mammary
carcinoma. Cancer Res. 61:6952–6957. 2001.PubMed/NCBI
|
|
35
|
Ravi R, Mookerjee B, Bhujwalla ZM, et al:
Regulation of tumor angiogenesis by p53-induced degradation of
hypoxia-inducible factor 1alpha. Genes Dev. 14:34–44.
2000.PubMed/NCBI
|
|
36
|
Yang J, Ahmed A, Poon E, et al:
Small-molecule activation of p53 blocks hypoxia-inducible factor
1alpha and vascular endothelial growth factor expression in vivo
and leads to tumor cell apoptosis in normoxia and hypoxia. Mol Cell
Biol. 29:2243–2253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Walmsley SR, Print C, Farahi N, et al:
Hypoxia-induced neutrophil survival is mediated by
HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 201:105–115.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang CY, Cusack JC, Liu R, et al: Control
of inducible chemoresistance: enhanced anti-tumor therapy through
increased apoptosis by inhibition of NF-κB. Nat Med. 5:412–417.
1999. View Article : Google Scholar : PubMed/NCBI
|