Introduction
Cigarette smoking is not only one of the most severe
public health issues but also one of the most supported etiological
factors contributing to the development of bronchogenic carcinoma
and chronic obstructive pulmonary disease (COPD) (1). Cigarette smoke (CS) is a complex
combination of chemicals, including high levels of oxidants.
Alveolar epithelial cells (AECs) appear to be a major target for
oxidant injury of the various cell types of the lung (2–4).
Injury of the alveolar epithelium by CS is hypothesized to be an
important process in the pathogenesis of smoking-associated
pulmonary diseases. Therefore, protection of AECs from injury by CS
appears to be crucial for the management of numerous lung diseases
associated with cigarette smoking.
The breach in epithelial integrity may constitute
one of the earliest elements of lung injury in response to CS
exposure (5–7). Breaches of the epithelial barrier may
induce wound-repair responses and a persistent increase in
epithelial permeability. This may lead to the subepithelial tissue
being directly exposed to CS. Oxidative stress and active
cytoskeletal rearrangement are critical for the development of
epithelial barrier disruption induced by CS (8,9).
Decreases in the levels of zona occludens (ZO)-1 and occludin are
associated with epithelial barrier dysfunction and increased
epithelial permeability (10,11).
Accumulation of occludin and ZO-1 at tight junctions (TJs) is
associated with protection of the epithelial barrier (12). Occludin is anchored to the
cytoskeleton by ZO-1 (13). In the
process of abnormal epithelial repair, the expression of ZO-1 and
occludin decreases (14), which
causes epithelial cells to lose their normal structure. ZO-1 and
occludin are two well-characterized proteins in TJs, which are
associated with epithelial barrier structure and integrity
(15). CS causes the
delocalization of ZO-1 and occludin from the cell-cell boundaries
and a subsequent loss of epithelial integrity (16).
Smoking not only affects epithelial cell structure,
but also various functions of epithelial cells, including synthesis
and secretion. Vascular endothelial growth factor (VEGF) is a
potent angiogenic protein that has been implicated in a number of
structural and functional alterations of vascular endothelial
cells. The survival of endothelial cells is largely achieved
through the action of VEGF, which is abundantly expressed in the
lung (17). VEGF has autocrine
effects on the growth and proliferation of pulmonary epithelial
cells in vitro (18). VEGF
is predominantly secreted by AECs and cigarette smoke extract (CSE)
reduces VEGF production in epithelial cells (19). Decreased levels of VEGF are known
to occur in smokers, in the lungs of patients with COPD and in rat
lungs in response to CS exposure (20,21).
As a result, inhibiting these decreases in VEGF induced by CSE may
prevent emphysema development.
Macrolides are a group of antibiotics that are
characterized by a macrocyclic lactone ring with various amino
sugars attached. In addition to their antimicrobial activity, a
number of these antibiotics also have immunomodulatory properties,
as demonstrated in multiple in vitro and in vivo
studies (22–24). The immunomodulatory effects are
associated with the lactone ring, which is only observed in the 14
(erythromycin, clarithromycin and roxithromycin) and the 15
(azithromycin, AZM) membered macrolides (25). AZM is different from other
macrolide antibacterial drugs in that it possesses unusual
pharmacokinetic properties. It accumulates at a high rate in cells
and tissues and has a plasma half-life of >40 h (26). In the airway epithelial cells and
neutrophils of cystic fibrosis patients, AZM has been demonstrated
to have antioxidant capacity (27–29).
However, there have been no studies, to the best of our knowledge,
investigating the possible role of AZM in the protection of human
AECs from oxidative injury induced by CSE. The present study
examined the effect of AZM on the regulation of CSE-induced injury
in the human alveolar epithelial cell line A549.
Materials and methods
Cell culture and drug treatment
Cell cultures of the A549 human type II alveolar
epithelial cell line (Cell Research Center, Institute of Basic
Medical Sciences, Chinese Academy of Medical Sciences, Beijing,
China) were grown in Dulbecco’s modified Eagle’s medium (DMEM)/F-12
culture medium (HyClone, Logan, UT, USA) containing 10%
heat-inactivated fetal calf serum (HyClone), 100 kU/l penicillin
and 100 mg/l streptomycin (Invitrogen Life Technologies, Carlsbad,
CA, USA). The cells were maintained at 37°C in a humidified
atmosphere at 5% CO2. The cell cultures were maintained
until they were ~70–80% confluent and subsequently incubated in
serum-free DMEM (SF-DMEM) for 16 h. AZM (Zithromax; Pfizer
Pharmaceuticals, Dublin, Ireland) and N-acetyl-L-cysteine (NAC)
were dissolved in sterilized phosphate-buffered saline (PBS;
Zhongshan Biotechnology, Beijing, China). Prior to incubation with
or without CSE, AZM and NAC were added to cells for 2 h.
Preparation of CSE
Fresh CSE was prepared for each experiment. Briefly,
one commercial filtered cigarette (Derby; China Tobacco Anhui
Industrial Co., Ltd, Hefei, China) was passed through 10 ml of
preheated DMEM using a peristaltic pump, with the pH adjusted to
7.4, and subsequently filtered through a 0.22 mm filter. The
absorbance of 320 nm measured using a Hitachi U-3900H (Hitachi
High-Technologies, Tokyo, Japan)revealed few differences between
different preparations of CSE. The solution was considered 100% CSE
and was diluted for each experiment.
Assay of A549 cell viability
The viability of the A549 cells was determined using
a colorimetric,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Sigma-Aldrich, St. Louis, MO, USA). Briefly, the cells were
cultured in 96-well tissue culture plates, grown to 70–80%
confluence and subsequently incubated for over 16 h in SF-DMEM F-12
medium. After 24 h CSE treatment, the cells were incubated with 0.5
mg/ml MTT in fresh medium for a further 4 h. The blue formazan was
dissolved by adding dimethyl sulfoxide (Sigma-Aldrich)and was
spectrophotometrically measured at a wavelength of 570 nm using a
Thermo Scientific Multiskan FC (Thermo Fisher Scientific, Waltham,
MA, USA).
Protein preparation and western blot
analysis
Following incubation, the cells were washed with
ice-cold PBS twice. Proteins were extracted from the A549 cells
using radioimmunoprecipitation assay buffer [50 mM Tris/HCl, pH
7.4, 150 mM NaCl, 1% (v/v) NP-40, 0.1% (w/v) SDS; Solarbio Science
and Technology Co., Ltd., Beijing, China] containing a protease
inhibitor cocktail (AEBSF, bestatin, E-64, leupeptin, pepstatin A
and 1,10-phenanthroline; catalogue number, P9599; 100:1, v:v;
Sigma-Aldrich). The cell lysates were subjected to centrifugation
at 12,000 × g at 4°C for 15 min and the supernatant was collected
as total protein. Protein quantitation was performed using the
bicinchoninic acid method according to the manufacturer’s
instructions (Pierce Biotechnology, Inc., Rockford, IL, USA). The
samples were separated on 10% SDS-polyacrylamide gels and
transferred onto a polyvinylidene difluoride membrane (Millipore,
Billerica, MA, USA), which was soaked in 5% milk in Tris-buffered
saline with Tween 20 (TBST; pH 7.6; Sigma-Aldrich) for 2 h at room
temperature. The membrane was incubated overnight with a rabbit
VEGF monoclonal antibody (1909-1; Epitomics, Burlingame, CA, USA)
at a dilution of 1:250, a rabbit ZO-1 polyclonal antibody (ab59720;
Abcam, Cambridge, UK) at a dilution of 1:100 or a rabbit polyclonal
antibody occludin (71-1500; Invitrogen Life Technologies) at a
dilution of 1:250 on a rotating platform at 4°C. Following washing
with TBST (pH 7.6), the membranes were incubated in horseradish
peroxidase-conjugated goat anti-rabbit and mouse IgG, at a 1:5,000
dilution (M21003; Abmart, Shanghai, China) for 2 h on a rotating
platform at room temperature (RT). The antibody complexes were
detected by chemiluminescence (Millipore) according to the
manufacturer’s instructions. Bands were quantified using a
PhosphorImager and ImageQuant software version 1.46 (Amersham
Biosciences, Amersham, UK) and normalized to GAPDH.
Immunofluorescence
Cells cultured on six-well chamber slides were
washed with PBS three times for 5 min per wash and fixed in 4%
paraformaldehyde for 30 min at RT. Following a further three washes
with PBS for 5 min per wash, the slides were incubated with 3%
bovine serum albumin (BSA; Sigma-Aldrich) in PBS for 1 h at RT.
Subsequently, cells were incubated with primary antibodies against
human ZO-1 and occludin, and diluted at 1:100 in PBS with 1% BSA.
Following incubation with the primary antibodies overnight at 4°C,
the cells were washed with PBS and incubated with Alexa Fluor
488-conjugated anti-rabbit IgG (ZF-0511; Zhongshan Biotechnology
Co., Ltd., Beijing, China; diluted at 1:50 in PBS with 1% BSA) for
1 h at RT. Following three washes in PBS, the slides were stained
with 10 μg/ml Hoechst 33258 for 10 min at RT. The slides were
washed again and mounted. Immunofluorescence images were captured
by fluorescence microscopy (Eclipse 80i; Nikon Corp., Tokyo,
Japan).
Fluorescence staining
Cells cultured on six-well chamber slides were
washed with PBS three times for 5 min per wash and subsequently
incubated with ROS Fluorescent Probe-dihydroethidium (DHE; Vigorous
Biotechnology Beijing Co., Ltd., Beijing, China) in SF-DMEM F-12
medium for 30 min at 37°C in darkness and fixed in 4%
paraformaldehyde for 30 min at RT. The slides were washed again and
mounted. Immunofluorescence images were captured by fluorescence
microscopy (Eclipse 80i; Nikon Corp.).
Quantification of intracellular ROS
Intracellular ROS was measured using ROS Fluorescent
Probe-DHE. Following a 24 h treatment of CSE or 200 μM
H2O2, the cells were washed with PBS and
subsequently incubated with 5 μM DHE in PBS for 30 min at 37°C in
darkness. The cells were harvested and washed with PBS. Following
centrifugation at 800 × g for 6 min, the cells were suspended in
PBS. Relative fluorescence intensities in the A549 cells were
analyzed with flow cytometry (FACS Calibur; BD Biosciences,
Franklin Lakes, NJ, USA).
ELISA
Following pretreatment with AZM and incubation with
CSE for 24 h or 10% CSE for 6, 12, 24 or 48 h, the cell culture
medium was centrifuged at 1006.2 × g for 20 min and subsequently
the supernatant was collected. Quantification of VEGF levels was
performed using human VEGF ELISA kits (Bio-rexd, Beijing, China)
according to the manufacturer’s instructions.
Statistical analysis
All data are expressed as the mean ± standard error
of the mean. The statistical significance of the differences was
evaluated by analysis of variance and subsequently by Tukey’s
multiple-comparison procedure. P<0.05 was considered to indicate
a statistically significant difference.
Results
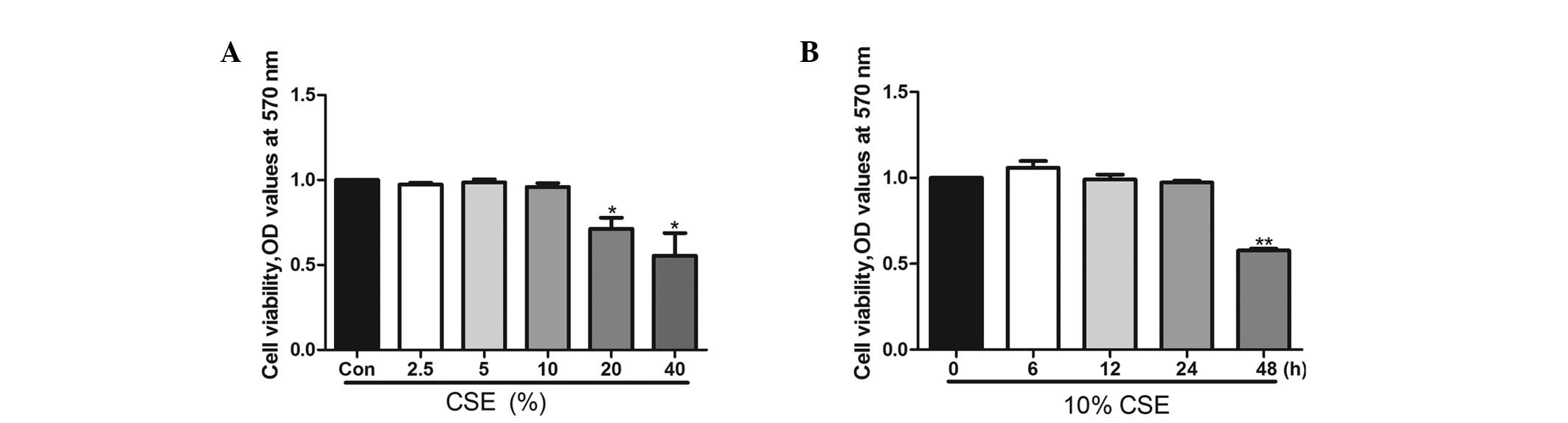
CSE affects A549 cell viability in a
dose- and time-dependent manner
A549 cells were exposed to 2.5, 5, 10, 20 and 40%
CSE for 24 h or to 10% CSE for 6, 12, 24 and 48 h. A549 cell
viability was decreased when the cells were treated with 20% CSE
for 24 h (Fig. 1A) or with 10% CSE
for 48 h (Fig. 1B). Following 24 h
of incubation, CSE at concentrations of 10% did not affect cell
viability. In the subsequent experiments, CSE was used at a
concentration of 10% for 24 h.
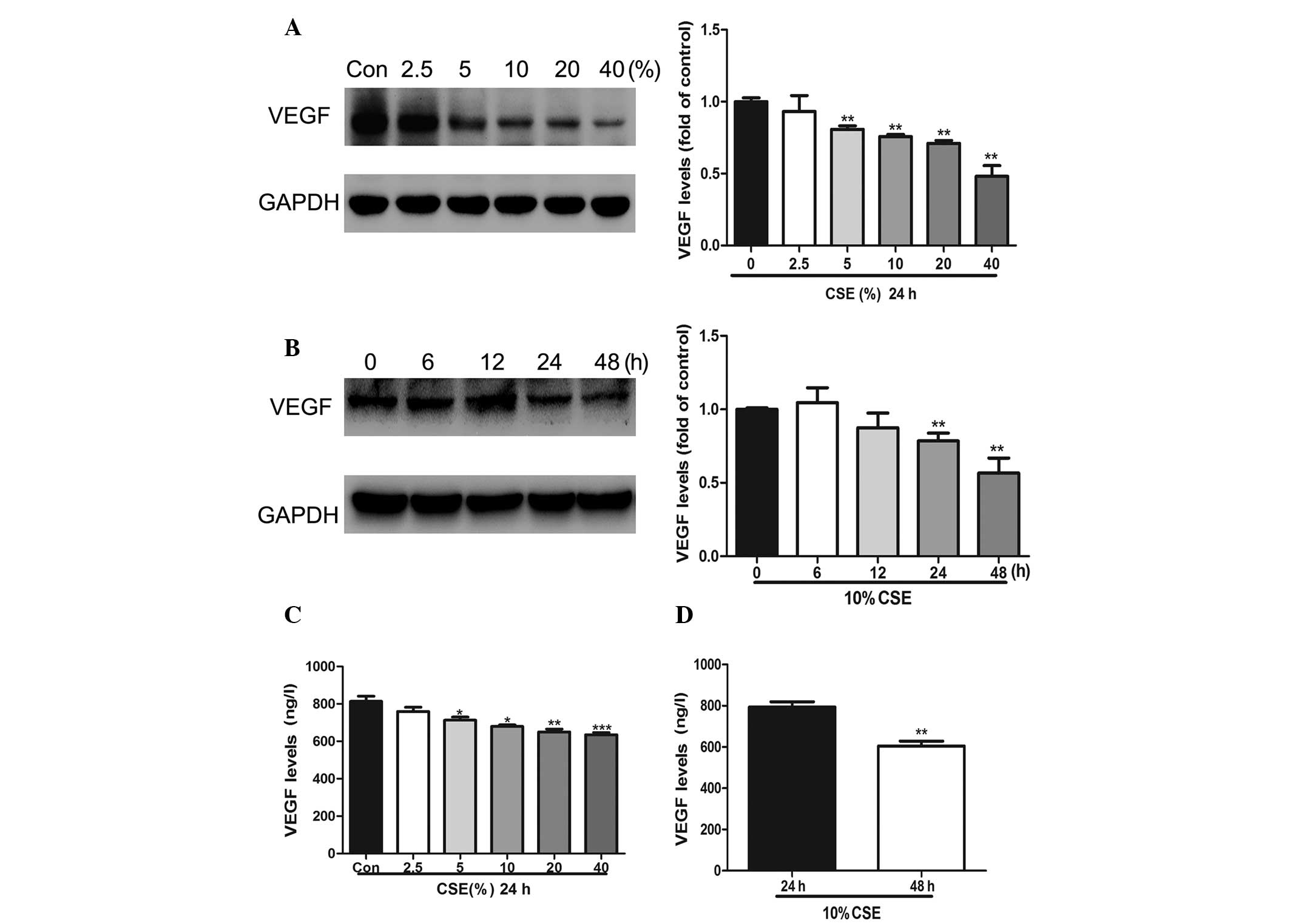
CSE attenuates the expression of VEGF in
a dose- and time-dependent manner
In order to determine the effects of CSE on VEGF
protein expression, A549 cells were treated with 2.5, 5, 10, 20 and
40% CSE for 24 h (Fig. 2A and C)
or with 10% CSE for 6, 12, 24 and 48 h (Fig. 2B) or 10% CSE for 24 and 48 h
(Fig. 2D). Western blot analysis
and ELISA demonstrated that the CSE treatment significantly reduced
the levels of VEGF expression in a dose- and time-dependent
manner.
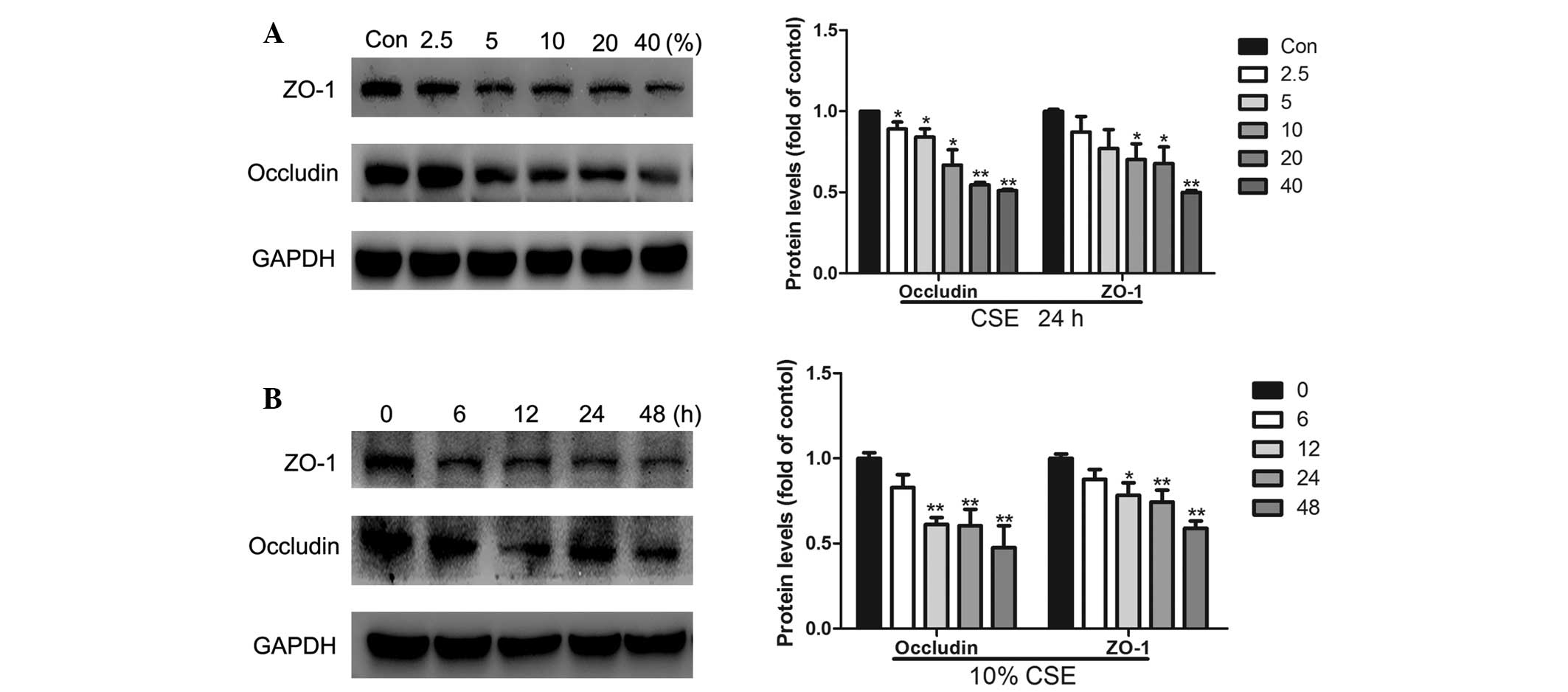
CSE attenuates the expression of ZO-1 and
occludin in a dose- and time-dependent manner
The expression of ZO-1 and occludin was investigated
using western blotting. A549 cells were treated with 2.5, 5, 10, 20
and 40% CSE for 24 h or with 10% CSE for 6, 12, 24 and 48 h.
Western blot analysis revealed that CSE treatment significantly
reduced the levels of ZO-1 and occludin expression in a dose- and
time-dependent manner (Fig. 3). At
doses of 2.5, 5, 10, 20 and 40%, the decreases in occludin protein
expression were 11, 16, 33, 45 and 49% and at doses of 10, 20 and
40%, the decreases in ZO-1 protein expression were 30, 32 and 50%,
respectively (Fig. 3A). When the
A549 cells were treated with 10% CSE for 12, 24 and 48 h, occludin
protein expression decreased by 39, 40 and 52% and ZO-1 protein
expression decreased by 22, 26 and 42% (Fig. 3B).
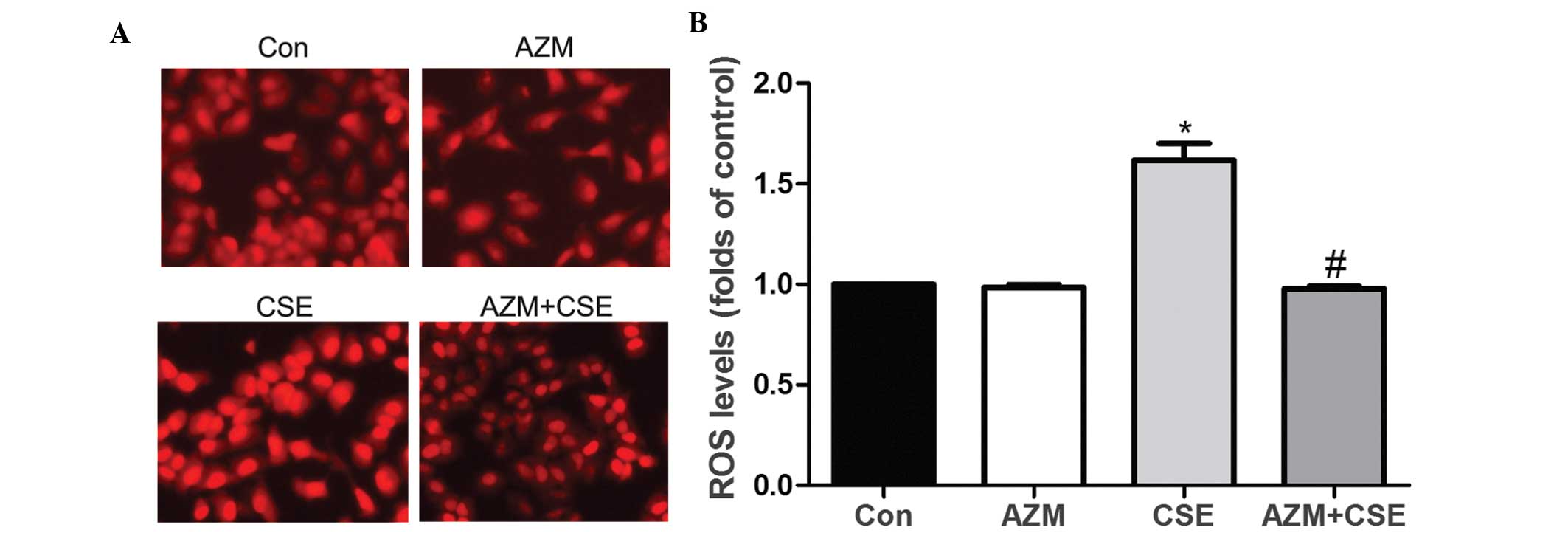
AZM suppresses CSE-induced ROS increase
in A549 cells
To determine whether AZM affected CSE-induced ROS
increases in A549 cells, the cells were divided into four groups,
including a control group, an AZM group, a CSE group and a
CSE-supplemented AZM group (CSE+AZM). The A549 cells were
pretreated with AZM (10 μg/ml) for 2 h and subsequently treated
with 10% CSE for 24 h. Fluorescence staining (Fig. 4A) and flow cytometry (Fig. 4B) illustrated that A549 cells
incubated with CSE had significantly increased ROS generation. ROS
generation induced by CSE was decreased by AZM in the A549
cells.
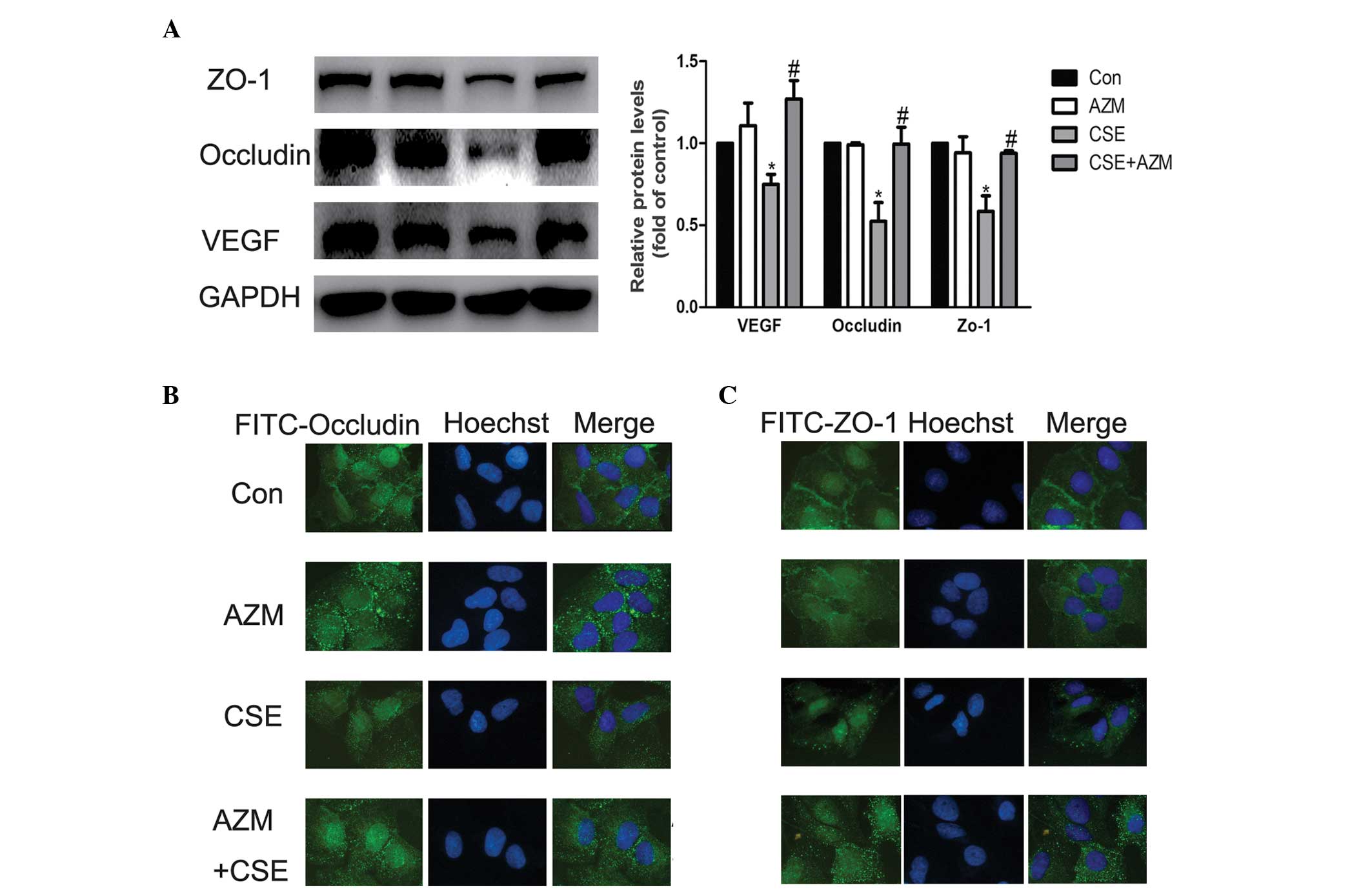
AZM reverses the alterations in VEGF,
ZO-1 and occludin induced by CSE
Our preliminary experiments revealed that AZM was
important at 10 μg/ml in the alterations observed in VEGF, ZO-1 and
occludin induced by CSE, which is also the physiological
concentration, at 2 h prior to being incubated with CSE (data not
shown) (30). Following incubation
with SF-DMEM, A549 cells were pretreated with AZM (10 μg/ml) 2 h
prior to being incubated with CSE. Following a 24 h period, VEGF
was examined using western blotting. Downregulation of VEGF induced
by CSE was attenuated by AZM at 10 μg/ml (Fig. 5A). In addition, western blotting
and immunofluorescence analyses revealed that A549 cells exposed to
CSE significantly decreased the expression of Occludin and ZO-1,
while AZM inhibited these changes induced by CSE (Fig. 5A–C). These results indicated that
AZM pretreatment for 2 h at a dose of 10 μg/ml was able to
significantly reverse the changes in the expression of VEGF,
Occludin and ZO-1 induced by CSE.
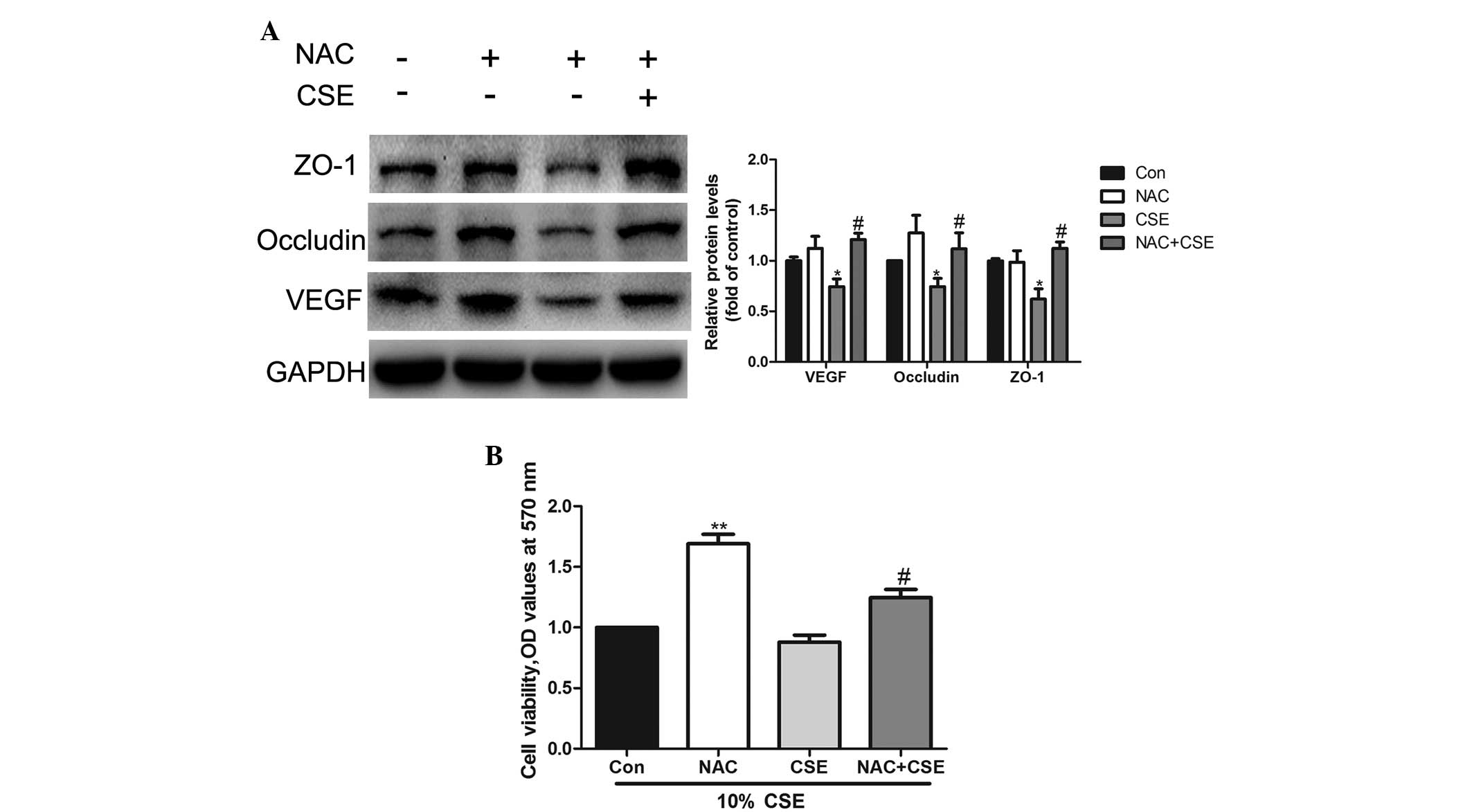
NAC reverses the effect of CSE on
occludin and ZO-1 expression and cell viability
The A549 cells were pretreated with 5 mM NAC (an
antioxidant) for 2 h prior to CSE treatment. Western blot analysis
demonstrated that NAC restores occludin and ZO-1 expression during
CSE treatment (Fig. 6A). In
addition, the data indicated that cell viability was increased by
NAC (Fig. 6B).
Discussion
CS is the main etiological factor contributing to
respiratory disorders in the lung, including COPD and idiopathic
pulmonary fibrosis (IPF) (7–9),
which is characterized by the irreversible damage of lung
epithelial cells. CS is a rich source of ROS with oxidative damage
being the main pathogenic factor of CS (31). The chemical composition of CS is
complex; therefore, it is not easy to predict which compounds or
combinations of compounds may be involved in its effects. In the
present study, an extract of CS, CSE, was used to imitate CS. Among
the various cell types in the lung, AECs appear to be a major
target for oxidant injury (2,3). It
is well established that alveolar type II epithelial (AEC II) cells
are stem cells of the alveolar epithelium (16,32).
Following oxidant injury, the rapidity of initiation of AEC II
proliferation is crucial for sufficient healing. A549 cells possess
numerous features of AEC II cells (35) and a number of studies investigating
CS-associated oxidant injury have used A549 cells as a model of AEC
II (3,36–39).
Therefore, in the present study, A549 cells were used as a model of
AEC II, to examine the protective effects of AZM on CSE injury.
Excessive apoptosis of epithelial cells is an
important factor in the pathogenesis of IPF and COPD. The results
reported in the present study demonstrate that CSE affects A549
cell viability in a dose- and time-dependent manner, indicating
that CSE has a deleterious effect on human AEC viability. A
concentration of 10% CSE was selected for subsequent experiments as
10% CSE produced a maximal decrease in VEGF, ZO-1 and occludin
proteins without apparent cell damage.
Lung epithelial barrier injury is one of the early
pathological alterations induced by CS (5–7).
Cell-cell junctions are important in maintaining cell and tissue
polarity and integrity (40). Once
the integrity of the epithelial barrier is damaged, opportunities
for atmospheric components and pathogens to enter into the
circulation and the interstitial and alveolar cavities increase,
leading to the destruction of the alveolar walls and pulmonary
edema (8,16). Epithelial integrity depends on the
regulation of junctional complexes, including TJs. ZO-1 and
occludin are markers of epithelial cells. When epithelial cells are
damaged, typical features of the cell are disrupted and the
expression of ZO-1 and occludin is decreased. A previous study
demonstrated that the expression of ZO-1 decreased in smokers and
patients with COPD (41). In
animal models of acute lung and intestinal epithelial cell injury,
TJ changes were associated with the downregulation of occludin and
ZO-1 protein expression (42–44).
In addition, ZO-1 and occludin are integral to TJs and CS may
increase the permeability of AECs, which may be associated with the
destruction of the TJ proteins (16). Numerous studies have revealed that
ZO-1 and occludin expression alterations are associated with
changes in the cytoskeleton (32–34).
Additionally, the decrease of ZO-1 and occludin affects the
connections between cells. Previous studies have demonstrated that
AZM may increase transepithelial electrical resistance (45) and prevent disintegration of the TJ
proteins in the airway epithelium exposed to Pseudomonas
aeruginosa (46). AZM may
inhibit epithelial-mesenchymal transition (EMT) in primary human
small and large airway epithelial cells induced by transforming
growth factor-β1 (47), causing
cells to maintain their epithelial characteristics. However,
whether AZM may reverse the alterations in ZO-1 and occludin
proteins induced by CSE has not, to the best of our knowledge, been
investigated previously. In the present study, it was observed that
treatment with CSE significantly reduced ZO-1 and occludin
expression in A549 cells in a dose- and time-dependent manner. It
remains uncertain whether the reduction in ZO-1 and occludin and
the protective effect of AZM is the result of changes in cell
viability. As cell viability decreased, the expression of ZO-1 and
occludin was reduced. However, it is unlikely that the decreased
ZO-1 and occludin expression resulted from a direct cytotoxic
effect of CSE as no significant reduction in viability of A549
cells was observed following incubation with 10% CSE for 24 h. The
decreased ZO-1 and occludin expression was most likely secondary to
CSE-induced alterations in cell structure. This conclusion is
similar to that made when analyzing the effect of phenol on the
barrier function of a human intestinal epithelial cell line
(48) and glucose degradation
products on human peritoneal mesothelial cells (49).
Smoking does not only affect the epithelial cell
structure, but also the function of a series of epithelial cells.
CS exposure is associated with reduced expression of VEGF and VEGF
receptor (VEGFR)-2 in the lungs of patients with severe emphysema
and in rodent lungs (21,50). In addition, the decrease in VEGF
has been observed in the destruction of alveolar wall components,
including microvasculature (20).
Previous studies have suggested a beneficial role for VEGF in
tissue repair and proliferation (51). VEGF may be an anti-apoptotic agent
in the lung (52). The inhibition
of VEGF results in increased markers of oxidative stress, alveolar
enlargement and alveolar cell apoptosis in animals (53–55).
VEGF is known to be a secreted protein (56). In our preliminary experiments,
ELISA was used to measure the expression of VEGF. The results
demonstrated that CSE attenuated the expression of VEGF in
supernatant in a dose- and time-dependent manner. However, at 6 and
12 h, the level of VEGF was not able to be detected as VEGF did not
reach the minimum concentration of the ELISA kits. The ELISA
results were consistent with the western blotting results,
suggesting that the content inside the cell is associated with
exocrine function, therefore, in the preliminary experiments
western blotting was used. The present study demonstrated that
treatment with CSE significantly reduced VEGF expression in A549
cells in a dose- and time-dependent manner. It was also revealed
that AZM pretreatment may reverse CSE-induced VEGF decreases for
the first time, to the best of our knowledge. This indicated that
AZM may be able to protect against epithelial injury induced by
CSE. A possible explanation for this protective effect may be that
AZM reduces the acute onset of COPD. AZM not only reversed the
decrease in VEGF expression induced by CSE but also reduced the
effects of CSE on epithelial cell integrity in the A549 cells,
suggesting a protective response.
It has previously been reported that numerous
chemical components in CS may induce ROS production (55). Oxidative stress is one of the
classical signals of cell injury and may alter ZO-1 and occludin
protein expression and localization (57,58).
Taken together, the damaging effects of CSE on AECs may be
initiated by oxidative stress. NAC is a well-known antioxidant
(59) and pretreating A549 cells
with 5 mM NAC for 2 h prior to exposure to CSE resulted in a
significant increase in cell viability. In addition, western
blotting indicated that the decreased expression of ZO-1 and
occludin was reversed following NAC pretreatment. ROS are crucial
factors that result in oxidative stress. Thus, augmentation of
intracellular ROS may be one of the major factors contributing to
CSE-induced injury in A549 cells. The present study demonstrated,
for the first time to the best of our knowledge, that AZM decreases
ROS generation induced by CSE. AZM, a macrolide antibiotic widely
used in clinical practice, was reported to suppress neutrophil ROS
release (27). In a cystic
fibrosis airway epithelial cell line, AZM significantly reduced the
activity of glutathione transferase (28). These findings indicate that AZM has
antioxidant potential. However, whether AZM is able to inhibit the
ROS generation induced by CSE in AECs has not been reported. CSE
exposure caused oxidative stress, as revealed by the increased
levels of ROS production. In the present study, CSE induced a
significant increase in ROS levels. AZM pretreatment was used prior
to CSE stimulation in A549 cells and it was observed that AZM may
alleviate the changes in ROS production.
A number of controversial areas of this topic
require further investigation. The establishment of functional TJs
and the formation of impermeable monolayers in A549 cells has been
questioned (60). However, a
previous study demonstrated that the A549 cell line expresses ZO-1
with 16HBE14o-, a human bronchial epithelial cell line and Calu-3
cells in in vitro models and forms functional TJs (61). CS exposure has been associated with
the increased permeability of A549 cells (62). In acute lung injury cell models
exposed to thrombin, the permeability of A549 cells was
hypothesized to be associated with ZO-1 and occludin (63). At the same time, as an epithelial
marker, ZO-1 expression is decreased during EMT following injury
(41) as the cells lose their
epithelial cell character. These findings implicate the structural
integrity of A549 cells associated with ZO-1 and occludin. In the
present study, the cells formed confluent layers at the time of the
experiments. It was demonstrated that the expression of ZO-1 and
occludin is associated with the structural integrity of A549 cells.
A structural change affects normal cell function. Thus, AZM has a
similar protective effect on A549 cells. VEGF is a well-known
permeabilizing factor and a downregulator of occludin and ZO-1 in
vascular endothelial cells. The present study demonstrated that the
change in VEGF is consistent with the expression of ZO-1 and
occludin, which may involve more complex regulatory mechanisms
requiring further investigation. Based on the present results, it
is hypothesized that VEGF is expressed as several splice variants
(64) and VEGFR also has different
subtypes. The complex interactions may lead to the various
downstream signaling pathways. Finally, although AZM protected A549
cells from the effects of CSE, whether this effect is true of human
adult type I/II cells remains to be elucidated. Further
investigation into the in vivo effects of AZM in animals is
required to verify these findings.
In conclusion, the present study demonstrated that
AZM is able to reverse smoke-induced aberrant expression of VEGF
and structural proteins in A549 cells. Furthermore, AZM is able to
inhibit the ROS production induced by CSE. Due to the widespread
use of AZM in a clinical setting, its protective effect against
oxidative stress caused by CS and as it is easily obtained, the
present results may contribute towards new methods of treatment of
smoking-associated disease, supporting that the present study has
implications for clinical care.
References
|
1
|
Hartman TE, Tazelaar HD, Swensen SJ and
Müller NL: Cigarette smoking: CT and pathologic findings of
associated pulmonary diseases. Radiographics. 17:377–390. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tuder RM, Wood K, Taraseviciene L, Flores
SC and Voekel NF: Cigarette smoke extract decreases the expression
of vascular endothelial growth factor by cultured cells and
triggers apoptosis of pulmonary endothelial cells. Chest.
117:241S–242S. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoshino Y, Mio T, Nagai S, Miki H, Ito I
and Izumi T: Cytotoxic effects of cigarette smoke extract on an
alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol
Physiol. 281:L509–L516. 2001.PubMed/NCBI
|
|
4
|
Kasahara Y, Tuder RM,
Taraseviciene-Stewart L, et al: Inhibition of VEGF receptors causes
lung cell apoptosis and emphysema. J Clin Invest. 106:1311–1319.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung KF and Adcock IM: Multifaceted
mechanisms in COPD: inflammation, immunity and tissue repair and
destruction. Eur Respir J. 31:1334–1356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park JW, Ryter SW and Choi AM: Functional
significance of apoptosis in chronic obstructive pulmonary disease.
COPD. 4:347–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van Driessche W, Kreindler JL, Malik AB,
Margulies S, Lewis SA and Kim KJ: Interrelations/cross talk between
transcellular transport function and paracellular tight junctional
properties in lung epithelial and endothelial barriers. Am J
Physiol Lung Cell Mol Physiol. 293:L520–L524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XY, Rahman I, Donaldson K and MacNee W:
Mechanisms of cigarette smoke induced increased airspace
permeability. Thorax. 51:465–471. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Serikov VB, Leutenegger C, Krutilina R, et
al: Cigarette smoke extract inhibits expression of peroxiredoxin V
and increases airway epithelial permeability. Inhal Toxicol.
18:79–92. 2006. View Article : Google Scholar
|
|
10
|
Oshitani N, Watanabe K, Nakamura S,
Fujiwara Y, Higuchi K and Arakawa T: Dislocation of tight junction
proteins without F-actin disruption in inactive Crohn’s disease.
Int J Mol Med. 15:407–410. 2005.PubMed/NCBI
|
|
11
|
Mankertz J, Tavalali S, Schmitz H, et al:
Expression from the human occludin promoter is affected by tumor
necrosis factor alpha and interferon gamma. J Cell Sci. 113(Pt 11):
2085–2090. 2000.PubMed/NCBI
|
|
12
|
van Baarlen P, Troost FJ, van Hemert S, et
al: Differential NF-kappaB pathways induction by Lactobacillus
plantarum in the duodenum of healthy humans correlating with immune
tolerance. Proc Natl Acad Sci USA. 106:2371–2376. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fanning AS, Jameson BJ, Jesaitis LA and
Anderson JM: The tight junction protein ZO-1 establishes a link
between the transmembrane protein occludin and the actin
cytoskeleton. J Biol Chem. 273:29745–29753. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yadav UC, Naura AS, Aguilera-Aguirre L, et
al: Aldose reductase inhibition prevents allergic airway remodeling
through PI3K/AKT/GSK3beta pathway in mice. PLoS one. 8:e574422013.
View Article : Google Scholar
|
|
15
|
Azghani AO: Pseudomonas aeruginosa and
epithelial permeability: role of virulence factors elastase and
exotoxin A. Am J Respir Cell Mol Biol. 15:132–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Olivera D, Knall C, Boggs S and Seagrave
J: Cytoskeletal modulation and tyrosine phosphorylation of tight
junction proteins are associated with mainstream cigarette
smoke-induced permeability of airway epithelium. Exp Toxicol
Pathol. 62:133–143. 2010. View Article : Google Scholar
|
|
17
|
Voelkel NF, Vandivier RW and Tuder RM:
Vascular endothelial growth factor in the lung. Am J Physiol Lung
Cell Mol Physiol. 290:L209–L221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mura M, Han B, Andrade CF, et al: The
early responses of VEGF and its receptors during acute lung injury:
implication of VEGF in alveolar epithelial cell survival. Crit
Care. 10:R1302006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thaikoottathil JV, Martin RJ, Zdunek J,
Weinberger A, Rino JG and Chu HW: Cigarette smoke extract reduces
VEGF in primary human airway epithelial cells. Eur Respir J.
33:835–843. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kasahara Y, Tuder RM, Cool CD, Lynch DA,
Flores SC and Voelkel NF: Endothelial cell death and decreased
expression of vascular endothelial growth factor and vascular
endothelial growth factor receptor 2 in emphysema. Am J Respir Crit
Care Med. 163:737–744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marwick JA, Stevenson CS, Giddings J, et
al: Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling
complex in rat lungs and patients with COPD: morphological impact
of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol.
290:L897–L908. 2006. View Article : Google Scholar
|
|
22
|
Culić O, Eraković V and Parnham MJ:
Anti-inflammatory effects of macrolide antibiotics. Eur J
Pharmacol. 429:209–229. 2001. View Article : Google Scholar
|
|
23
|
Shinkai M, Henke MO and Rubin BK:
Macrolide antibiotics as immunomodulatory medications: proposed
mechanisms of action. Pharmacol Ther. 117:393–405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vanaudenaerde BM, Vos R, Meyts I, et al:
Macrolide therapy targets a specific phenotype in respiratory
medicine: from clinical experience to basic science and back.
Inflamm Allergy Drug Targets. 7:279–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rubin BK: Immunomodulatory properties of
macrolides: overview and historical perspective. Am J Med.
117(Suppl 9A): 2S–4S. 2004.PubMed/NCBI
|
|
26
|
Zuckerman JM: The newer macrolides:
azithromycin and clarithromycin. Infect Dis Clin North Am.
14:449–462. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levert H, Gressier B, Moutard I, et al:
Azithromycin impact on neutrophil oxidative metabolism depends on
exposure time. Inflammation. 22:191–201. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bergamini G, Cigana C, Sorio C, et al:
Effects of azithromycin on glutathione S-transferases in cystic
fibrosis airway cells. Am J Respir Cell Mol Biol. 41:199–206. 2009.
View Article : Google Scholar
|
|
29
|
Popovic M, Janicijevic-Hudomal S,
Kaurinovic B, Rasic J and Trivic S: Antioxidant effects of some
drugs on ethanol-induced ulcers. Molecules. 14:816–826. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saint-Criq V, Rebeyrol C, Ruffin M, et al:
Restoration of chloride efflux by azithromycin in airway epithelial
cells of cystic fibrosis patients. Antimicrob Agents Chemother.
55:1792–1793. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Faux SP, Tai T, Thorne D, Xu Y, Breheny D
and Gaca M: The role of oxidative stress in the biological
responses of lung epithelial cells to cigarette smoke. Biomarkers.
14(Suppl 1): 90–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rao R: Oxidative stress-induced disruption
of epithelial and endothelial tight junctions. Front Biosci.
13:7210–7226. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
You K, Xu X, Fu J, et al: Hyperoxia
disrupts pulmonary epithelial barrier in newborn rats via the
deterioration of occludin and ZO-1. Respir Res. 13:362012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schweitzer KS, Hatoum H, Brown MB, et al:
Mechanisms of lung endothelial barrier disruption induced by
cigarette smoke: role of oxidative stress and ceramides. Am J
Physiol Lung Cell Mol Physiol. 301:L836–L846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Willis BC and Borok Z: TGF-beta-induced
EMT: mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pierson T, Learmonth-Pierson S, Pinto D
and van Hoek ML: Cigarette smoke extract induces differential
expression levels of beta-defensin peptides in human alveolar
epithelial cells. Tob Induc Dis. 11:102013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Noriyasu A, Konishi T, Mochizuki S, et al:
Menthol-enhanced cytotoxicity of cigarette smoke demonstrated in
two bioassay models. Tob Induc Dis. 11:182013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fukano Y, Yoshimura H and Yoshida T: Heme
oxygenase-1 gene expression in human alveolar epithelial cells
(A549) following exposure to whole cigarette smoke on a direct in
vitro exposure system. Exp Toxicol Pathol. 57:411–418. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang T, Chen M and Sun T: Simvastatin
attenuates TGF-beta1-induced epithelial-mesenchymal transition in
human alveolar epithelial cells. Cell Physiol Biochem. 31:863–874.
2013. View Article : Google Scholar
|
|
40
|
Heijink IH, Brandenburg SM, Postma DS and
van Oosterhout AJ: Cigarette smoke impairs airway epithelial
barrier function and cell-cell contact recovery. Eur Respir J.
39:419–428. 2012. View Article : Google Scholar
|
|
41
|
Milara J, Peiró T, Serrano A and Cortijo
J: Epithelial to mesenchymal transition is increased in patients
with COPD and induced by cigarette smoke. Thorax. 68:410–420. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Han X, Fink MP, Yang R and Delude RL:
Increased iNOS activity is essential for intestinal epithelial
tight junction dysfunction in endotoxemic mice. Shock. 21:261–270.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mazzon E and Cuzzocrea S: Role of
TNF-alpha in lung tight junction alteration in mouse model of acute
lung inflammation. Respir Res. 8:752007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Noth R, Lange-Grumfeld J, Stuber E, et al:
Increased intestinal permeability and tight junction disruption by
altered expression and localization of occludin in a murine graft
versus host disease model. BMC Gastroenterol. 11:1092011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Asgrimsson V, Gudjonsson T, Gudmundsson GH
and Baldursson O: Novel effects of azithromycin on tight junction
proteins in human airway epithelia. Antimicrob Agents Chemother.
50:1805–1812. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Halldorsson S, Gudjonsson T, Gottfredsson
M, Singh PK, Gudmundsson GH and Baldursson O: Azithromycin
maintains airway epithelial integrity during Pseudomonas aeruginosa
infection. Am J Respir Cell Mol Biol. 42:62–68. 2010. View Article : Google Scholar
|
|
47
|
Banerjee B, Musk M, Sutanto EN, et al:
Regional differences in susceptibiity of bronchial epithelium to
mesenchymal transition and inhibition by the macrolide antibiotic
azithromycin. PLoS one. 7:e523092012. View Article : Google Scholar
|
|
48
|
McCall IC, Betanzos A, Weber DA, Nava P,
Miller GW and Parkos CA: Effects of phenol on barrier function of a
human intestinal epithelial cell line correlate with altered tight
junction protein localization. Toxicol Appl Pharmacol. 241:61–70.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Leung JC, Chan LY, Li FF, et al: Glucose
degradation products downregulate ZO-1 expression in human
peritoneal mesothelial cells: the role of VEGF. Nephrol Dial
Transplant. 20:1336–1349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Suzuki M, Betsuyaku T, Nagai K, et al:
Decreased airway expression of vascular endothelial growth factor
in cigarette smoke-induced emphysema in mice and COPD patients.
Inhal Toxicol. 20:349–359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mura M, dos Santos CC, Stewart D and Liu
M: Vascular endothelial growth factor and related molecules in
acute lung injury. J Appl Physiol 1985. 97:1605–1617. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kasahara Y, Iwai K, Yachie A, et al:
Involvement of reactive oxygen intermediates in spontaneous and
CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood.
89:1748–1753. 1997.PubMed/NCBI
|
|
53
|
Tuder RM, Zhen L, Cho CY, et al: Oxidative
stress and apoptosis interact and cause emphysema due to vascular
endothelial growth factor receptor blockade. Am J Respir Cell Mol
Biol. 29:88–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang K, Rossiter HB, Wagner PD and Breen
EC: Lung-targeted VEGF inactivation leads to an emphysema phenotype
in mice. J Appl Physiol 1985. 97:1559–1566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Onoue S, Ohmori Y, Endo K, Yamada S,
Kimura R and Yajima T: Vasoactive intestinal peptide and pituitary
adenylate cyclase-activating polypeptide attenuate the cigarette
smoke extract-induced apoptotic death of rat alveolar L2 cells. Eur
J Biochem. 271:1757–1767. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chatterjee S, Heukamp LC, Sioba M, et al:
Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis
in lung cancer. J Clin Invest. 123:1732–1740. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
González-Mariscal L, Quirós M and
Diaz-Coránguez M: ZO proteins and redox-dependent processes.
Antioxid Redox Signal. 15:1235–1253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Blasig IE, Bellmann C, Cording J, et al:
Occludin protein family: oxidative stress and reducing conditions.
Antioxid Redox Signal. 15:1195–1219. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen RM, Chou MW and Ueng TH: Induction of
cytochrome P450 1A1 in human hepatoma HepG2 cells by
6-nitrochrysene. Toxicol Lett. 117:69–77. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Winton HL, Wan H, Cannell MB, et al: Cell
lines of pulmonary and non-pulmonary origin as tools to study the
effects of house dust mite proteinases on the regulation of
epithelial permeability. Clin Exp Allergy. 28:1273–1285. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fu LS, Ko YH, Lin KW, Hsu JY, Chu JJ and
Chi CS: Dioscorin protects tight junction protein expression in
A549 human airway epithelium cells from dust mite damage. J
Microbiol Immunol Infect. 42:457–463. 2009.
|
|
62
|
Li XY, Donaldson K, Rahman I and MacNee W:
An investigation of the role of glutathione in increased epithelial
permeability induced by cigarette smoke in vivo and in vitro. Am J
Respir Crit Care Med. 149:1518–1525. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Puig F, Fuster G, Adda M, et al:
Barrier-protective effects of activated protein C in human alveolar
epithelial cells. PLoS one. 8:e569652013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tischer E, Mitchell R, Hartman T, et al:
The human gene for vascular endothelial growth factor. Multiple
protein forms are encoded through alternative exon splicing. J Biol
Chem. 266:11947–11954. 1991.PubMed/NCBI
|