Introduction
Liver cancer, the most common form of which is
hepatocellular carcinoma (HCC), is the fifth most common malignant
tumor and the third leading cause of cancer-associated mortality
worldwide. Each year ~500,000 new cases of HCC are diagnosed
worldwide (1,2). Surgical and nonsurgical therapeutic
treatments include tumor resection, liver transplantation,
radiofrequency (thermal) ablation, percutaneous ethanol injection
and transarterial chemoembolization (3). Despite these methods, the prognosis
for HCC patients remains poor and tumor recurrence rates remain
high (4). Therefore, novel
therapeutic approaches are required to improve the treatment
outcome for patients with HCC (5).
Tumor adoptive immunotherapy has been demonstrated
to have potential as an adjunct treatment to control the disease.
This approach can be efficiently employed for the eradication of
residual cancer cells and prevention or delay of tumor relapse (NEW
1 – 6). With the development of tumor adoptive immunology, progress
has been achieved in preclinical studies and clinical practice
(7). Dendritic cells (DCs) and
cytokine-induced killer cells (CIKs) have been demonstrated to
possess high in vitro and in vivo antitumor and
cytotoxic activities against HCC cells (8–10).
DCs are the most potent antigen-capturing and antigen-presenting
cells, with the ability to capture, process and present tumor
antigens to naïve cells, and stimulate a marked immune response
against these antigens. The antigen-presenting ability of DCs makes
them attractive vehicles for the delivery of therapeutic tumor
vaccines and provides a suitable platform for vaccine development
(11). In 2010, the first
DC-associated cancer vaccine for prostate cancer therapy received
approval from the U.S. Food and Drug Administration (12). CIKs are obtained from human
peripheral blood mononuclear cells stimulated by interferon
(IFN)-γ, interleukin (IL)-2 and cluster of differentiation (CD)3
monoclonal antibodies. CIKs can express the surface markers of T
cells and natural killer (NK) cells (13). The characteristic
CD3+CD56+ CIKs phenotype has been
demonstrated to exhibit a major histocompatibility complex
(MHC)-unrestricted tumor killing ability, in vitro and in
medical practice (14). The CIKs
that possess the ability to attack tumor cells are expressed on the
cell surface of CD3/CD56. In addition, CIKs have superior antitumor
activity against a variety of cancer types, evident by their
co-culturing with antigen-loaded DCs. Therefore, as a nontoxic,
efficient and adoptive immunotherapeutic strategy, the use of a
vaccine of DCs co-cultured with CIKs may increase the potential of
specific immune response against HCC.
Studies performed by the authors of the present
study and by other researchers have investigated the expression,
function and regulation of carcinoembryonic antigen glypican 3
(GPC3) which has been found to be overexpressed in HCC tissues and
may serve as a potential diagnostic biomarker and therapeutic
target for this disease (15–17).
GPC3, a 70 kDa protein of 580 amino acids, is a heparan sulfate
proteoglycan that is positioned on the cell surface using a
mechanism involving a glycosylphosphatidylinositol anchor. In
addition, GPC3 promotes the growth of HCC cells through the
stimulation of the canonical Wnt signaling pathway (18). In HCC tumors, GPC3 is overexpressed
and correlates with poor prognosis, as well as functioning as a
secretory protein released from the cell membrane surface to the
extracellular environment (19).
Therefore, GPC3 may serve as a tumor-associated antigen (TAA)
target for immunotherapy against HCC. Considering the
aforementioned properties, the present study analyzed the
effectiveness of CIKs co-cultured with autologous GPC3-transduced
DCs against GPC3-expressing HCC cells, in vitro and in
vivo. The present study aims to provide new insight into the
design of DC-based tumor vaccine strategies for personalizing
adoptive immunotherapy.
Materials and methods
Animals and cell line
Nude mice (age, 6–8 weeks) were purchased from the
Academy of Military Medical Science (Beijing, China). The mice were
housed under specific pathogen-free conditions. All the experiments
were performed according to the National Institutes of Health Guide
for Care and Use of Laboratory Animals (National Institutes of
Health, Bethesda, MD, USA) and were approved by the Bioethics
Committee of Tianjin First Central Hospital (Tianjin, China). The
human HCC cell line, HepG2 (GPC3-expressing cell line), was
purchased from the American Type Culture Collection (Rockville, MD,
USA) and maintained in the Key Laboratory for Critical Care
Medicine of the Ministry of Health (Tianjin, China). The cells were
cultured in complete RPMI 1640 medium [RPMI 1640 (Invitrogen Life
Technologies, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Gibco Life Technologies,
Carlsbad, CA, USA), 100 U/ml penicillin and 100 mg/ml streptomycin
(Sigma-Aldrich, St. Louis, MO, USA)] at 37°C in a 5% CO2
atmosphere.
Generation of DCs and CIKs
DCs and CIKs were generated from peripheral blood
mononuclear cells (PBMCs) of consenting healthy volunteers
according to our protocol approved by the ethics committee of
Tianjin First Central Hospital. DCs and CIKs were generated as
described previously (20).
Briefly, PBMCs were isolated from whole blood by Ficoll density
gradient centrifugation using a commercially lymphocyte separation
medium (Sigma-Aldrich) and centrifuged at 400 × g for 25 min (NEW 2
– 21). Next, the cells were allowed to adhere in six-well plates
(Corning Life Sciences Tewksbury, MA, USA) at a density of
5×106 cells/ml for 2 h at 37°C in complete RPMI 1640
medium. The adherent and non-adherent cells were collected for
generating DCs and CIKs, respectively. To generate DCs, the
adherent cells were cultured in complete RPMI 1640 medium with
1,000 U/ml recombinant human (rh) granulocyte-macrophage
colony-stimulating factor and 500 U/ml rhIL-4 (R&D Systems,
Minneapolis, MN, USA) at 37°C in a humidified atmosphere of 5%
CO2 and immature DCs were obtained. The medium along
with the necessary cytokines were replaced every three days. On day
6, a further 1,000 U/ml tumor necrosis factor (TNF)-α was added to
the DC sample to induce maturation. To generate CIKs, the
non-adherent PBMCs were prepared and grown in complete RPMI 1640
medium with 1,000 U/ml rhIFN-γ. After 24-h incubation, 50 ng/ml
mouse anti-human CD3 monoclonal antibody and 1,000 U/ml IL-2 were
added. The CIKs were incubated at 37°C in a humidified atmosphere
of 5% CO2 and subcultured every three days with cytokine
replenishment.
Transduction of DCs with the GPC3
gene
The recombinant plasmid green fluorescent protein
(pGFP)-GPC3 eukaryotic expression vector was constructed and
maintained at the Key Laboratory for Critical Care Medicine of the
Ministry of Health (Tianjin, China). Briefly, the pDONR223-GPC3
plasmid (full length GPC3 cDNA) was ligated into a pcDNA-DEST53
vector containing GFP (Invitrogen Life Technologies) with
recombinase. The recombinant pGFP-GPC3 was amplified in E.
coli DH5α competent cells and isolated with Takara MiniBEST
plasmid purification kit (Takara Bio, Inc., Otsu, Japan). The
correct pGFP-GPC3 plasmid sequence was verified using DNA analysis.
The DCs were transduced using the Amaxa®
Nucleofector® apparatus (Lonza Cologne GmbH, Cologne,
Germany), according to the manufacturer’s instructions. Briefly, on
day 6, 5×106 immature DCs were cultured in serum-free
growth medium (Gibco Life Technologies) without antibiotics prior
to nucleofection. The cells were gently resuspended in 100 μl human
electroporation buffer (Lonza Cologne GmbH) at a concentration of
2×106 cells/100 μl and then transferred to a sterile
Amaxa® nucleofection cuvette (Lonza Cologne GmbH).
Subsequently, the immature DCs were incubated with 2 μg pEGFP-GPC3
or empty vector containing GFP. The cells were electroporated using
of the appropriate nucleofection program (as recommended in the
manufacturer’s instructions) and immediately transferred into
six-well plates containing fresh pre-warmed culture medium at 37°C
with the necessary cytokine (TNF-α) and serum. DCs were incubated
at 37°C for 24 h to induce maturation and were termed as the
DCs-GPC3 group. DCs transduced with pcDNA3 (DC-pcDNA3) were used as
the control group. After 24 h of incubation, DCs-GPC3 viability was
assessed using trypan blue exclusion (Sigma-Aldrich) and the
transfection efficiency of the cells was assessed by the extent of
GFP expression using Ni-U fluorescence microscopy (Nikon
Corporation, Tokyo, Japan) and fluorescence-activated cell sorting
(FACS) flow cytometric analysis was performed using a FACSCalibur
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The DCs
were then collected for subsequent experiments.
GPC3 expression in DCs-GPC3
The expression of GPC3 in DCs-GPC3 was detected at
the transcriptional and translational levels. Following
transfection for 48 h, DCs-GPC3 were collected and the total RNA or
total protein was prepared for detection by TaqMan reverse
transcription-polymerase chain reaction (RT-PCR) or western
blotting, respectively. Non-transduced mature DCs and DCs-pcDNA3
were evaluated in parallel as controls. Primer Premier V5.0
software was used to design the primers according to human gene
sequences (GenBank database, www.ncbi.nlm.nih.gov/genbank). Primers were
synthesized by Integrated DNA Technologies (Coralville, IA, USA).
The PCR primers used for GPC3 were as follows: forward,
5′-AGAGGCCTTTGAAATTGT-3′, and reverse 5′-AAATACTTTCAGGTCACGTC-3′;
and the probe 5′-FAM-ATGCCAAGAACTACACCAATGCTAMRA-3′ (22). The conditions for each PCR reaction
were as follows: 15 min at 95°C, followed by 40 cycles of
denaturation for 20 sec at 95°C and annealing/extension for 60 sec
at 60°C. The level of expression was represented as
2−ΔCt, where ΔCt was calculated as: (copy number of
target molecule)/(copy number of β-actin). For western blot
analysis, the proteins were resolved on an SDS denaturing
polyacrylamide gel and then transferred onto nitrocellulose
membranes (EMD Millipore, Billerica, MA, USA). A primary rabbit
anti-human polyclonal antibody to GPC3 (sc-11395; 1:500) or an
endogenous control β-actin (sc-7210; 1:500) were incubated with the
membranes first and then with horseradish peroxidase-conjugated
goat anti-rabbit IgGFc secondary antibodies (sc-2004; 1:2,000) (All
antibodies from Santa Cruz Biotechnology Inc., Santa Cruz, CA,
USA). Protein expression was assessed by enhanced chemiluminescence
(EMD Millipore), and the bands were captured using a FluorChem FC2
Imaging System (ProteinSimple, San Jose, CA, USA).
Phenotypic characterization
To ensure that the DCs were mature, DCs-GPC3 and DCs
were collected on day 8 and resuspended in cold FACS buffer. The
cells were immunostained with fluorescein isothiocyanate
(FITC)-labeled or phycoerythrin (PE)-labeled mouse monoclonal
antibodies against human CD80, CD83, CD86, human leukocyte antigen
(HLA)-DR or an isotype control. All the monoclonal antibodies used
in this study were obtained from BD Pharmingen, San Diego, CA, USA.
The cells were incubated with antibodies on ice for 30 min, washed
twice with phosphate-buffered saline (PBS) and resuspended. Next,
phenotypic characterization was performed by flow cytometric
analysis using the FACSCalibur flow cytometer (BD Biosciences).
CIKs were harvested on day 8 and co-cultured for a further seven
days with autologous DCs, DCs-pcDNA3 or DCs-GPC3 at a ratio of 1:5
to produce DCs-CIKs, DCs-pcDNA3-CIKs or DCs-GPC3-CIKs,
respectively. Subsequently, CIKs were harvested on day 14 and their
cytotoxicity was assessed. Phenotypic characterization of CIKs was
conducted with antibodies against CD3, CD8 and CD56 (BD
Pharmingen). Next, the cells were incubated with the corresponding
antibodies on ice for 15 min and then washed with PBS, and flow
cytometric analysis was performed.
Analysis of IFN-γ-secreting CIKs
IFN-γ-secreting cells were detected on day 7 of the
co-culture using intracellular staining and flow cytometry
(23). Briefly, the aforementioned
proliferative CIKs, DCs-CIKs, DCs-pcDNA3-CIKs or DCs-GPC3-CIKs were
suspended in complete RPMI 1640 and stimulated for 4 h with 25
ng/ml phorbol 12-myristate 13-acetate, 1 μM ionomycin and 2 μM
monensin (Sigma-Aldrich). Following washing with PBS, the cells
were stained with FITC-conjugated mouse anti-human CD3 monoclonal
antibody (BD Pharmingen) for 30 min at 4°C, washed with PBS and
then permeabilized with FACS permeabilizing solution (BD
Pharmingen) for a further 10 min at room temperature. The samples
were incubated with PE-labelled mouse anti-human INF-γ monoclonal
antibody (BD Pharmingen) for 30 min at room temperature in the
dark, washed with PBS and analyzed by flow cytometry.
Cytotoxicity assay
A nonradioactive cytotoxicity assay kit (Promega
Corp., Madison, WI, USA), lactate dehydrogenase (LDH) release, was
used to measure the cytotoxic activity on target cells, according
to the manufacturer’s instructions. Briefly, the target cells,
GPC3-expressing HepG2, were plated in triplicate in 96-well culture
plates and incubated with the various effector cells (CIKs,
DCs-CIKs, DCs-pcDNA3-CIKs and DCs-GPC3-CIKs) with an effector to
target (E/T) ratio of 20:1 or 50:1. Maximal release of LDH was
performed by completely lysing target cells. Target cells without
effector cells were used as negative controls (spontaneous
release). Cytotoxicity was calculated as follows: percentage
cytotoxicity (%) = [(experimental release - spontaneous release of
effector cells - spontaneous release of target cells) / (maximal
release of target cells - spontaneous release of target cells)]
×100.
Animal testing
Tumors were generated by subcutaneous inoculation
with 1×107 HepG2 cells in 0.2 ml of PBS into the right
flank of each nude mouse with a 100% incidence rate on day 7. The
mice were randomly divided into four groups (n=4 each) and
subjected to treatment. Subsequently, the four groups were injected
locally with DCs-GPC3-CIKs (1×107 cells), DCs-CIKs
(1×107 cells), CIKs (1×107 cells) or PBS
(control), at the area where the tumor cells had been inoculated,
for five consecutive times. The subcutaneous tumor volume was
measured using a caliper and estimated as follows: tumor volume
(mm3) = 0.5 × length × width2.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Statistical analysis was performed using Student’s t-test
or one-way analysis of variance. Statistical analyses were
performed using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA).
P≤0.05 was considered to indicate a statistically significant
difference.
Results
Transduction of DCs with a eukaryotic
expression vector
DCs were transduced with pGFP-GPC3 to analyze the
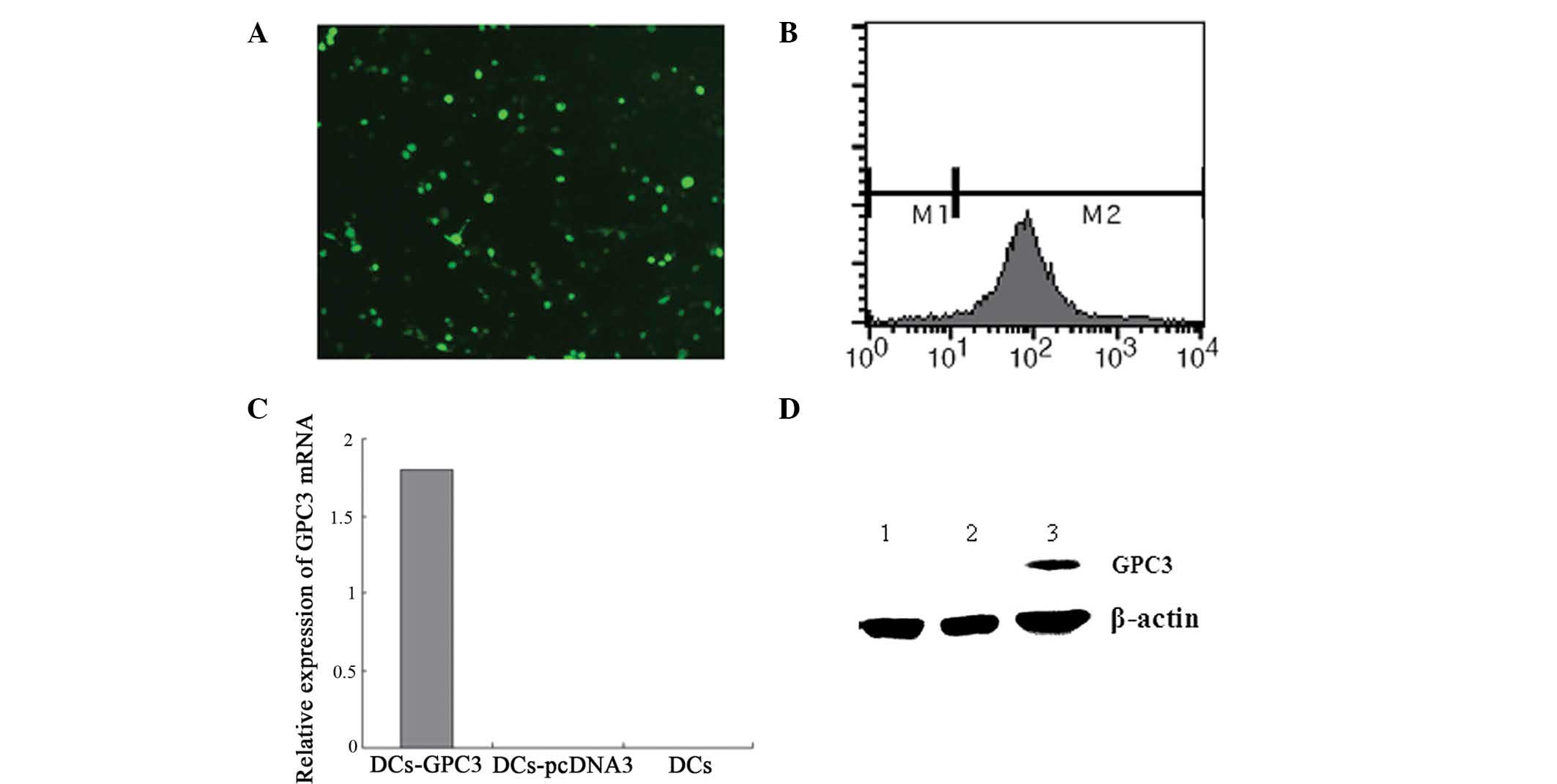
transduction efficiency. Positive GFP expression was detected in
~51% DCs, as determined using fluorescence microscopy (Fig. 1A) and flow cytometry (Fig. 1B). At 48 h after transduction,
RT-PCR and western blot assays were performed to detect the
expression of GPC3 in DCs. GPC3 was specifically detected in the
DCs-GPC3, but not in the DCs-pcDNA3 or DCs (Fig. 1C and D).
Phenotypic characteristics of DCs
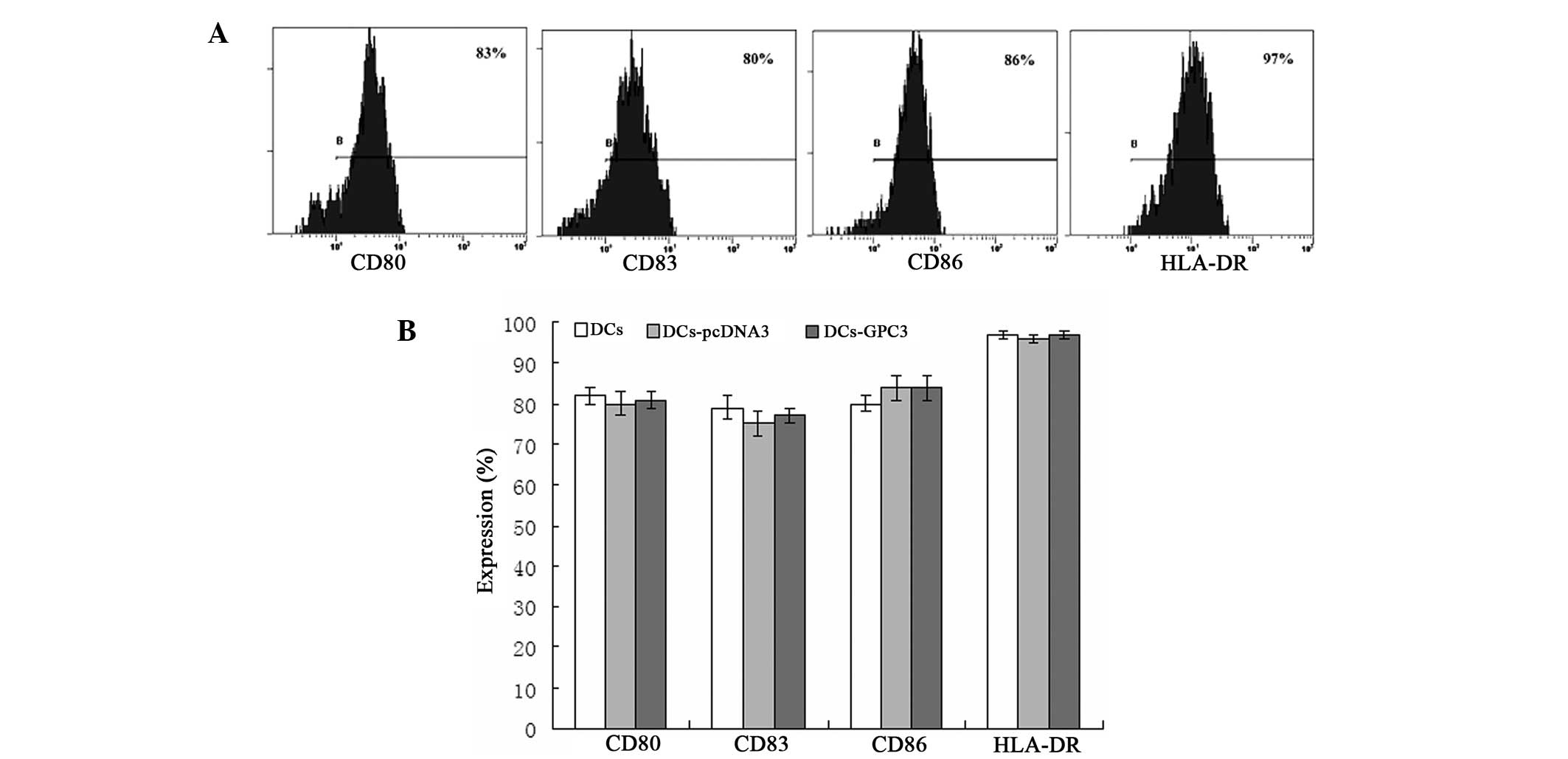
The phenotypes of transduced and non-transduced
mature DCs were analyzed using flow cytometry and used to detect
whether transduction may affect DC differentiation and maturation
in vitro. The results revealed that all transduced DCs
exhibited a complete mature DC phenotype following stimulation with
TNF-α. No statistically significant differences were observed in
the expression levels of CD80, CD83, CD86 and HLA-DR between the
mature DCs and transduced DCs (P>0.05; Fig. 2), indicating that gene transduction
did not alter the DC surface phenotypes.
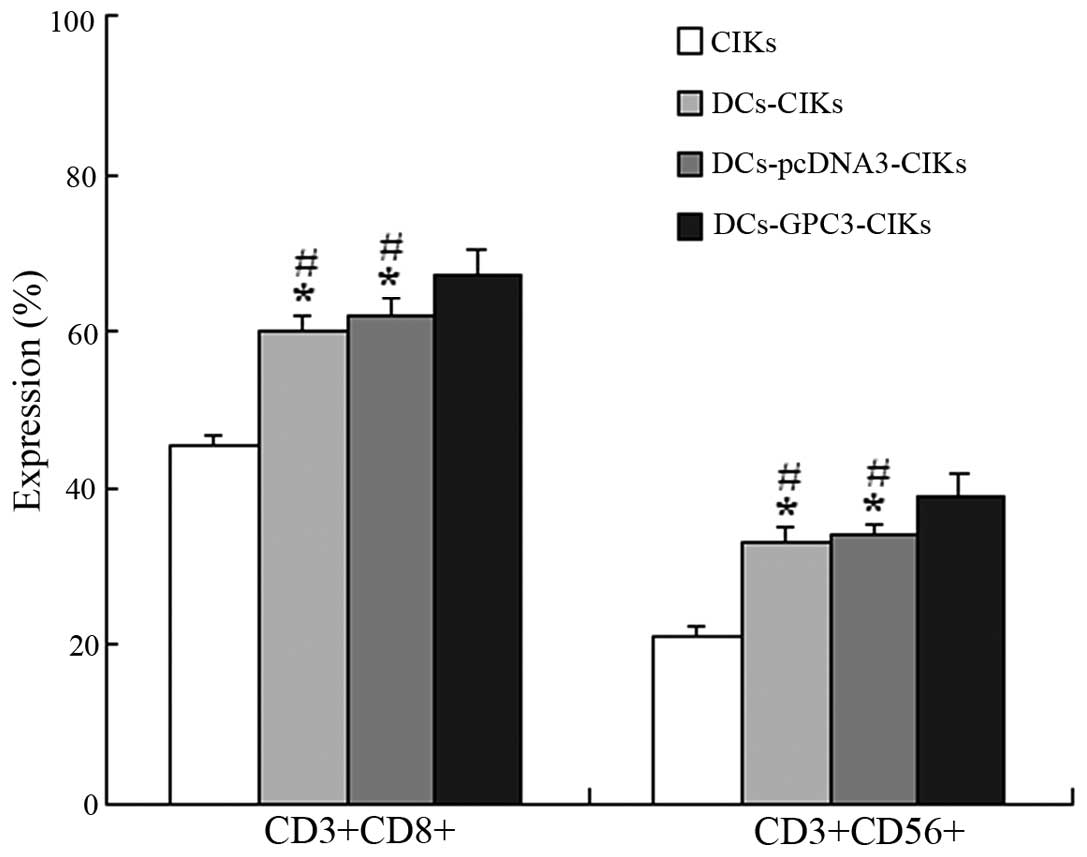
Phenotypic characteristics of CIKs
The proportion of CD3+CD8+ and
CD3+CD56+ cells was found to be significantly
higher in DCs-CIKs and DCs-pcDNA3-CIKs compared with the autologous
CIKs alone (P<0.01; Fig. 3). In
addition, the proportion of CD3+CD8+ and
CD3+CD56+ cells was significantly higher in
the DCs-GPC3-CIKs compared with the DCs-CIKs and DCs-pcDNA3-CIKs
(P<0.05; Fig. 3).
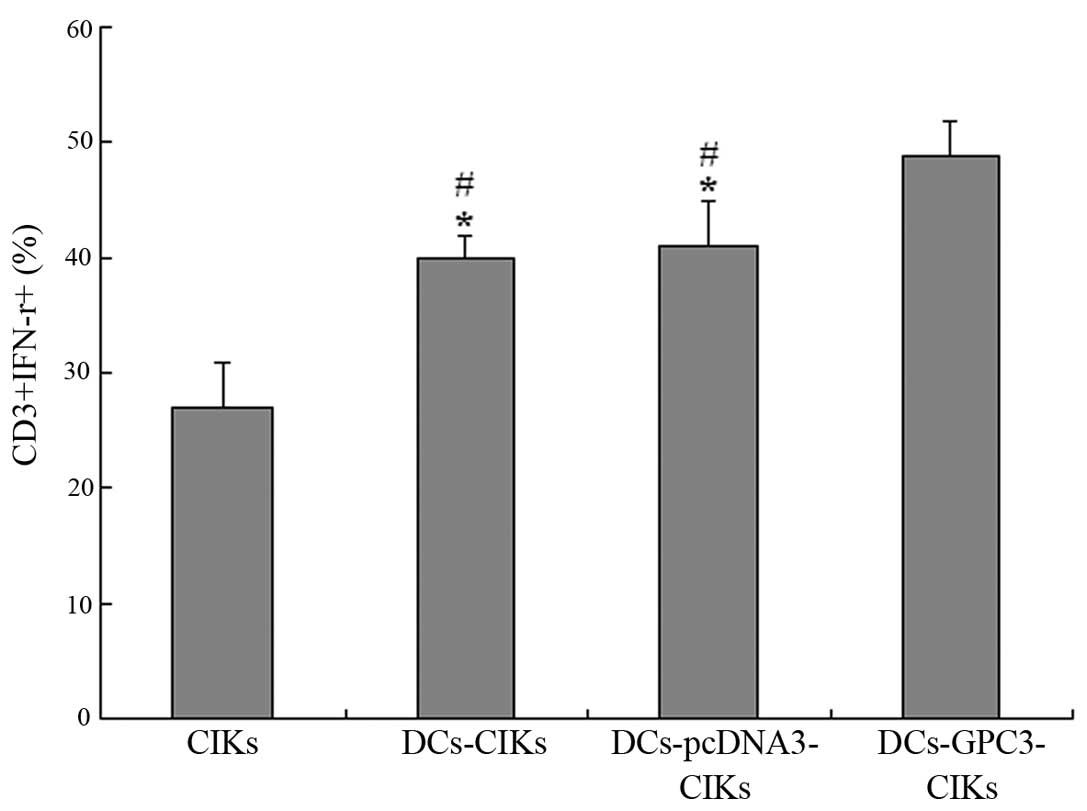
Intracellular IFN-γ secretion
Flow cytometric analysis was used to detect the
intracellular IFN-γ secretion, representing specific activation, by
autologous CIKs, CIKs co-cultured with DCs that were transduced
with an empty vector or GPC3. When CIKs were co-cultured with
DCs-GPC3, 49% CIKs were found to secret IFN-γ. By contrast, 41 and
40% CIKs secreted IFN-γ when co-cultured with DCs-pcDNA3 or DCs,
respectively (P<0.01). In addition, 27% non-transduced CIKs were
found to be CD3+ and IFN-γ+ (P<0.01;
Fig. 4). These results indicated
that DCs transduced with GPC3 and matured with TNF-α were able to
process and present GPC3 protein, resulting in the effective
induction of functional CIKs, indicated by IFN-γ production.
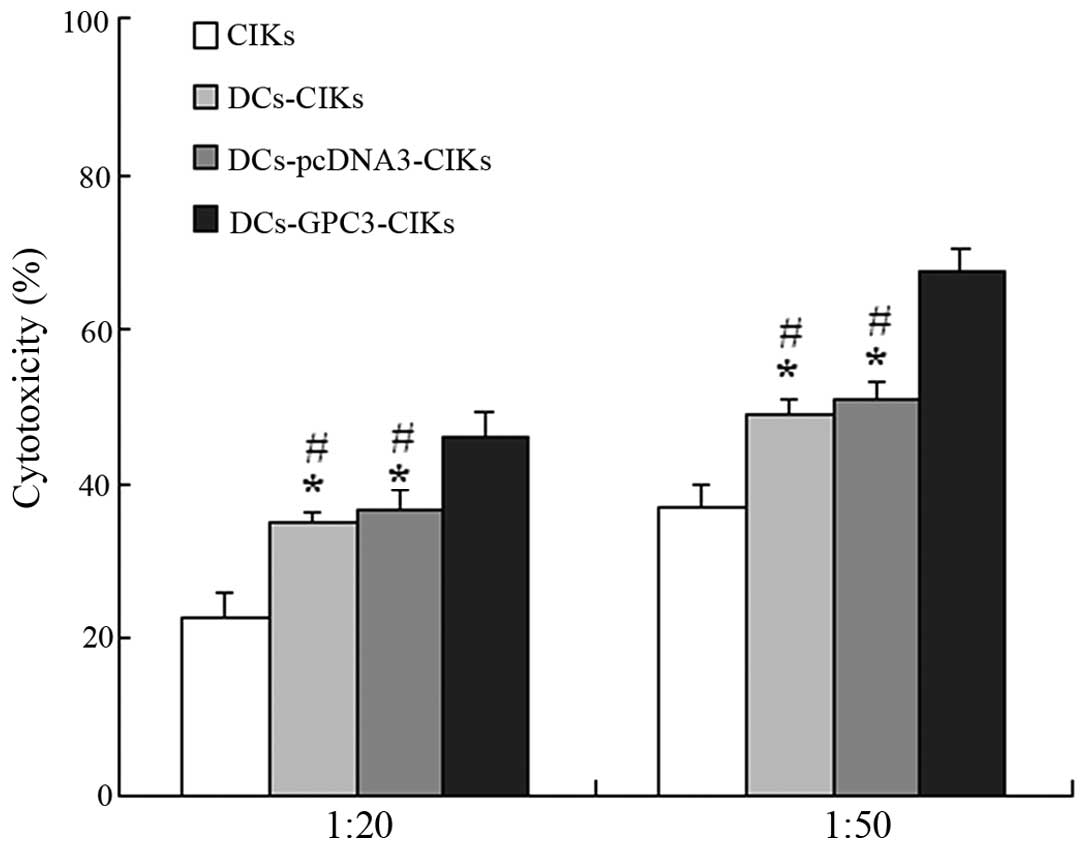
Induction of marked specific cytotoxic
activity against HCC cells
In the LDH cytotoxic analysis, HepG2 cells were used
as the target cells at various E/T ratios (20:1 and 50:1) to
evaluate the specific cytotoxic activity. The results demonstrated
that the cytotoxic activity against GPC3-expressing HepG2 cells was
considerably increased in DCs-GPC3-CIKs compared with the other
effector cells at the two E/T ratios (P<0.01; Fig. 5). Furthermore, the cytotoxic
activity in DCs-pcDNA3-CIKs and DCs-CIKs was higher when compared
with the CIKs alone (P<0.01; Fig.
5).
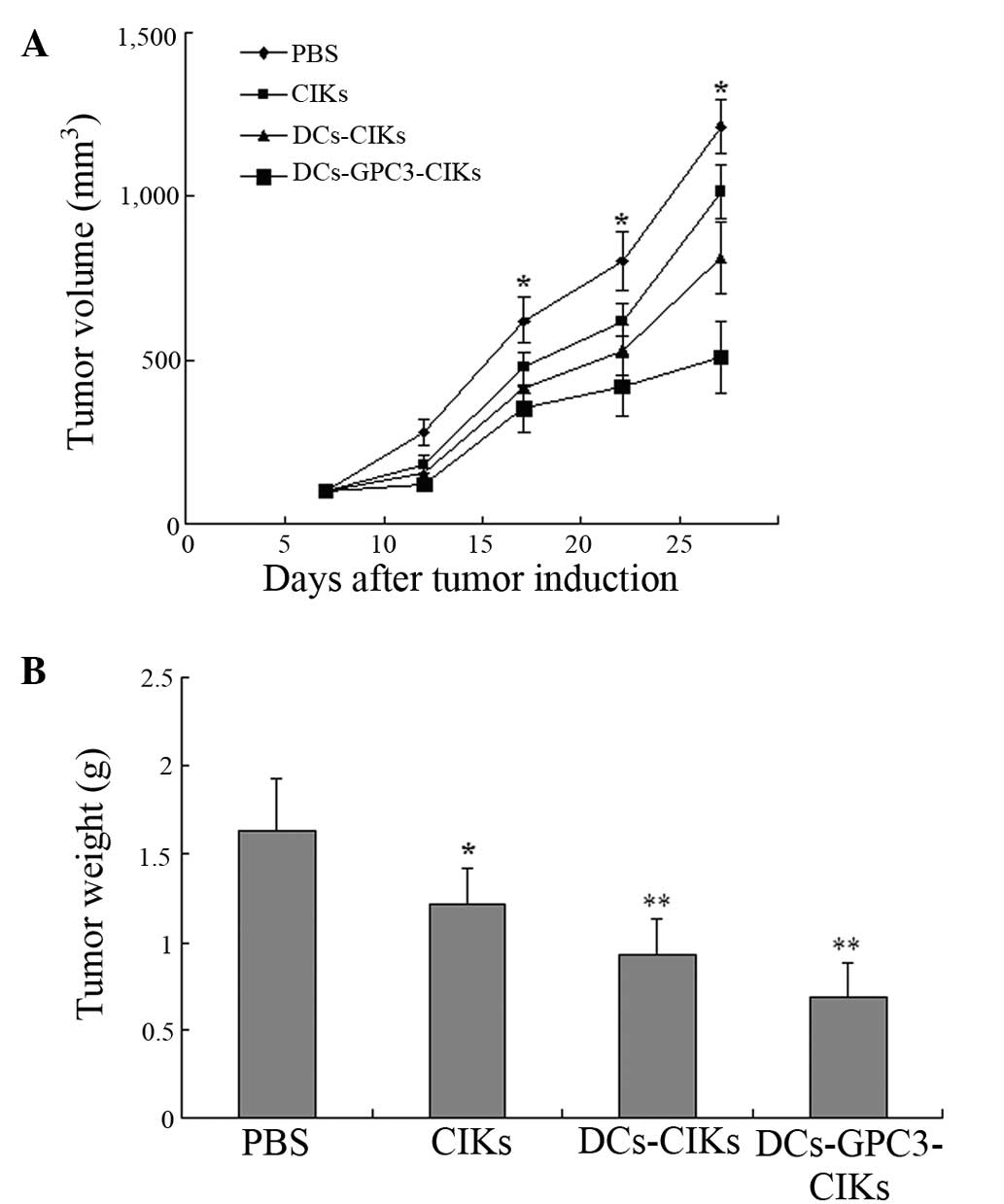
Inhibitory effects on HepG2 cell-induced
tumor growth in vivo
The inhibitory effects of each effector cell on
HepG2 cell-induced tumor growth in tumor-bearing nude mice are
shown in Fig. 6. The tumor volume
and weight were found to be the highest in the DCs-GPC3-CIKs group,
followed by the DCs-CIKs, CIKs and PBS control groups. These
results indicated that DCs-GPC3-CIKs were the most effective in
inhibiting the growth of HepG2 cells in vivo.
Discussion
Personalized adoptive immunotherapy may allow for
more precise and optimal treatment, in order to lower the
recurrence and metastasis rates of malignant tumors (24). In the present study, an
immunotherapy was developed, aiming to target GPC3-expressing HCC
cells for the treatment of HCC. The present study identified
several key points in the development of personalized adoptive
immunotherapy for HCC. An interaction was detected between DCs
transduced with the GPC3 gene and CIKs, resulting in augmentation
of special cytotoxicity of CIK subsets against GPC3-expressing HCC
cells in vitro. In addition, the results revealed that gene
nucleofection may be a promising approach for TAA loading.
Furthermore, HepG2-induced tumor growth in vivo was found to
be effectively inhibited by DCs-GPC3-CIKs. Finally, the marked
inhibitory potential of DCs-GPC3-CIKs on HepG2-induced tumor growth
may be associated with antitumor cytokines, such as IFN-γ.
Identifying reliable biomarkers is essential in
order to personalize the cancer treatment of each patient through a
baseline assessment of tumor gene expression and/or immune profile
to optimize the therapy for the highest therapeutic success
possibility (25). Several
features of the expression of GPC3 on the surface of HCC cells
indicate that novel immunotherapeutic approaches for HCC may be
generated by targeting the GPC3 protein. Firstly, GPC3 is a
membrane protein overexpressed in 70–90% of HCC cases and five HCC
previously investigated cells lines (HepG2, Huh7, Hep3B, MHCC97-H
and SMMC-7721) (25,26). In addition, GPC3 is not expressed
in normal and cirrhotic liver tissues or in benign hepatic lesions.
A previous functional analysis revealed that GPC3 promotes HCC cell
migration and invasion, which may lead to tumor progression
(27). Finally,
clinicopathological studies have indicated that GPC3 expression
correlates with poorly-differentiated HCC tumors with intrahepatic
metastasis, which is a leading cause of post-surgical recurrence
and reduced patient survival rates (16,29).
The feasibility of GPC3 targeting for antibody or DC-based
immunotherapy has been investigated in a number of studies
(16,30–32).
The induction of a specific GPC3 immune response is
a crucial factor for the design of immunotherapeutic strategies
against cancer. Application of DC-based immunotherapy strategies is
promising; however, enhancing the immunogenicity of DCs is
essential. In addition, CIKs that exhibit nonspecific cytotoxicity
against tumor targets but lack antitumor specificity are required.
The co-cultivation of TAA-loaded DCs with autologous CIKs generates
effective antitumorigenic cells (DCs-TAA-CIKs) and appears to
compensate for their individual deficiencies and enhance their
marked and specific antitumor immune effects (33). In the present study, nucleofector
technology was selected, as previous studies have identified that
nucleofection is an efficient and safe nonviral method of gene
transfer into DCs without affecting the pivotal properties of DCs,
which then may be used as cellular vehicles for the delivery of TAA
(34,35). Despite the efficient transduction
and high GPC3 expression achieved in DCs, the results of the
present study revealed that all transduced DCs exhibited a complete
mature DC phenotype following stimulation with TNF-α, which
demonstrated that the nucleofector and gene expression did not
affect the DC’s maturation and functional change toward effective
presentation of specific antigens.
In order to develop an effective cancer therapy, at
least three important aspects must be achieved. These aspects
include obtaining sufficient effector cells, producing effector
cells with a high cytotoxic activity against tumor cells and
generating effector cells with specific cytotoxicity for the target
cell. In the present study, an in vitro experiment was used
to compare the autologous CIKs, DCs-CIKs, DCs-pcDNA3-CIKs and
DCs-GPC3-CIKs. Co-culturing with autologous DCs-CIKs was found to
enhance the cytotoxic activity against HepG2 cells compared with
CIKs alone. In addition, DCs-GPC3-CIKs induced the highest cell
death activity on HepG2 cells compared with other effector cells.
This increment in the CIK specific cytotoxic activity when
co-cultured with DCs transduced with the GPC3 gene is possibly due
to an increment in the proportion of CD3+CD8+
and CD3+CD56+ cells plus a larger number of
cells actively differentiating and proliferating. Another feature
of CIKs is the production of effector cytokines, including IFN-γ,
which enable the effector cells to potentially tilt the immune
response toward the type I T helper cell and type I CD8+
T cell direction (NEW 3 - 36). In addition, the increased secretion
of IFN-γ may further irritate autocrine CIK subsets (37). The present results demonstrated
that CIKs were strongly stimulated by DCs-GPC3 with high levels of
IFN-γ production, suggesting the presence of a possible mechanism
of cytotoxicity. These results are in accordance with previously
reported data (15,38), which demonstrated that GPC3 mRNA
transfected DCs generated functional GPC3-reactive T cells, as
revealed by IFN-γ production and effective lysis of GPC3-expressing
HCC cells. With a substantial increase in cytotoxicity on a per
cell basis and a higher proliferative response, CIKs presented a
>70-fold increase in total cytolytic activity per culture when
compared with other T-lymphocytes generated from peripheral blood,
including lymphokine-activated killer cells and NK cells (13). The results of the current study
indicated that co-cultivation of GPC3-loaded DCs with autologous
CIKs may provide specific anti-HCC effective cells.
A previous study demonstrated that inhibition of the
GPC3 expression of HCC cells through RNA interference reduced the
tumorigenicity in nude mice, which indicated that GPC3 may be a
potential molecular target in HCC therapy (28). In further in vivo
experiments, the present study identified that tumor nodule
formation in nude mice, induced by HepG2 cells, was suppressed
significantly following treatment with DCs-GPC3-CIKs, indicating
that specific CIKs induced by DCs-GPC3 targeting of GPC3-expressing
HCC cells exhibited strong antitumor activity against HepG2
xenografts in mice.
In conclusion, the personalization of immunotherapy
using DCs-GPC3-CIKs may provide an adjuvant treatment method to
conventional therapeutic modalities, decreasing the recurrence
rates and improving the overall survival rates of HCC patients. The
precise mechanism of growth inhibition requires further examination
in future studies.
Acknowledgements
This study was supported by State-funded
Construction Projects - Key Specialized Subject of Clinical
Laboratory Medicine (grant no. 2013-544), National Natural Science
Foundation of China (grant nos. 81470982 and 81402322), the
Technology Foundation of Tianjin Municipal Health Bureau (grant no.
2014K028) and the National High-Tech R&D Program (863 Program)
of China (grant no. 2012AA021001).
References
|
1
|
Schütte K, Bornschein J and Malfertheiner
P: Hepatocellular carcinoma - epidemiological trends and risk
factors. Dig Dis. 27:80–92. 2009. View Article : Google Scholar
|
|
2
|
Mittal S and El-Serag HB: Epidemiology of
hepatocellular carcinoma: consider the population. J Clin
Gastroenterol. 47(Suppl): S2–S6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vivarelli M, Montalti R and Risaliti A:
Multimodal treatment of hepatocellular carcinoma on cirrhosis: An
update. World J Gastroenterol. 19:7316–7326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishikawa H, Arimoto A, Wakasa T, et al:
Effect of transcatheter arterial chemoembolization prior to
surgical resection for hepatocellular carcinoma. Int J Oncol.
42:151–160. 2013.
|
|
5
|
Greten TF, Duffy AG and Korangy F:
Hepatocellular carcinoma from an immunologic perspective. Clin
Cancer Res. 19:6678–6685. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie F, Zhang X, Li H, et al: Adoptive
immunotherapy in postoperative hepatocellular carcinoma: a systemic
review. PLoS One. 7:e428792012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Raval RR, Sharabi AB, Walker AJ, et al:
Tumor immunology and cancer immunotherapy: summary of the 2013 SITC
primer. J Immunother Cancer. 2:142014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weng DS, Zhou J, Zhou QM, et al: Minimally
invasive treatment combined with cytokine-induced killer cells
therapy lower the short-term recurrence rates of hepatocellular
carcinomas. J Immunother. 31:63–71. 2008. View Article : Google Scholar
|
|
9
|
Yu X, Xia W, Zhang T, et al: Enhanced
cytotoxicity of IL-24 gene-modified dendritic cells co-cultured
with cytokine-induced killer cells to hepatocellular carcinoma
cells. Int J Hematol. 92:276–282. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su S, Zhou H, Xue M, et al: Anti-tumor
efficacy of a hepatocellular carcinoma vaccine based on dendritic
cells combined with tumor-derived autophagosomes in murine models.
Asian Pac J Cancer Prev. 14:3109–3116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Palucka K and Banchereau J: Cancer
immunotherapy via dendritic cells. Nat Rev Cancer. 12:265–277.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kantoff PW, Higano CS, Shore ND, et al:
IMPACT Study Investigators: Sipuleucel-T immunotherapy for
castration-resistant prostate cancer. N Engl J Med. 363:411–422.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang J, Wu C and Lu B: Cytokine-induced
killer cells promote antitumor immunity. J Transl Med. 11:832013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sangiolo D: Cytokine induced killer cells
as promising immunotherapy for solid tumors. J Cancer. 2:363–368.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O’Beirne J, Farzaneh F and Harrison PM:
Generation of functional CD8+ T cells by human dendritic
cells expressing glypican-3 epitopes. J Exp Clin Cancer Res.
29:482010. View Article : Google Scholar
|
|
16
|
Wang YL, Zhu ZJ, Teng DH, et al:
Glypican-3 expression and its relationship with recurrence of HCC
after liver transplantation. World J Gastroenterol. 18:2408–2414.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Filmus J and Capurro M: Glypican-3: a
marker and a therapeutic target in hepatocellular carcinoma. FEBS
J. 280:2471–2476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao W and Ho M: The role of glypican-3 in
regulating Wnt in hepatocellular carcinomas. Cancer Rep. 1:14–19.
2011.PubMed/NCBI
|
|
19
|
Qi XH, Wu D, Cui HX, et al: Silencing of
the glypican-3 gene affects the biological behavior of human
hepatocellular carcinoma cells. Mol Med Rep. 10:3177–3184.
2014.PubMed/NCBI
|
|
20
|
Wang YF, Kunda PE, Lin JW, et al:
Cytokine-induced killer cells co-cultured with complete tumor
antigen-loaded dendritic cells, have enhanced selective
cytotoxicity on carboplatin-resistant retinoblastoma cells. Oncol
Rep. 29:1841–1850. 2013.PubMed/NCBI
|
|
21
|
Huls MH, Figliola MJ, Dawson MJ, et al:
Clinical application of Sleeping Beauty and artificial antigen
presenting cells to genetically modify T cells fromperipheral and
umbilical cord blood. J Vis Exp. 72:e500702013.
|
|
22
|
Wang Y, Shen Z, Zhu Z, et al: Clinical
values of AFP, GPC3 mRNA in peripheral blood for prediction of
hepatocellular carcinoma recurrence following OLT: AFP, GPC3 mRNA
for prediction of HCC. Hepat Mon. 11:195–199. 2011.PubMed/NCBI
|
|
23
|
Wang YL, Zhang YY, Zhou YL, et al:
T-helper and T-cytotoxic cell subsets monitoring during active
cytomegalovirus infection in liver transplantation. Transplant
Proc. 36:1498–1499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wayteck L, Breckpot K, Demeester J, et al:
A personalized view on cancer immunotherapy. Cancer Lett.
352:113–125. 2014. View Article : Google Scholar
|
|
25
|
Ascierto PA, Kalos M, Schaer DA, et al:
Biomarkers for immunostimulatory monoclonal antibodies in
combination strategies for melanoma and other tumor types. Clin
Cancer Res. 19:1009–1020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu SJ, Qi CY, Xiao WK, et al: Glypican-3
is a potential prognostic biomarker for hepatocellular carcinoma
after curative resection. Surgery. 154:536–544. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu C, Yan Z, Zhou L and Wang Y: A
comparison of glypican-3 with alpha-fetoprotein as a serum marker
for hepatocellular carcinoma: a meta-analysis. J Cancer Res Clin
Oncol. 139:1417–1424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ruan J, Liu F, Chen X, et al: Inhibition
of glypican-3 expression via RNA interference influences the growth
and invasive ability of the MHCC97-H human hepatocellular carcinoma
cell line. Int J Mol Med. 28:497–503. 2011.PubMed/NCBI
|
|
29
|
Chen IP, Ariizumi SI, Nakano M, et al:
Positive glypican-3 expression in early hepatocellular carcinoma
predicts recurrence after hepatectomy. J Gastroenterol. 49:117–125.
2014. View Article : Google Scholar :
|
|
30
|
Zhu AX, Gold PJ, El-Khoueiry AB, et al:
First-in-man phase I study of GC33, a novel recombinant humanized
antibody against glypican-3, in patients with advanced
hepatocellular carcinoma. Clin Cancer Res. 19:920–928. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sawada Y, Yoshikawa T, Fujii S, et al:
Remarkable tumor lysis in a hepatocellular carcinoma patient
immediately following glypican-3-derived peptide vaccination: an
autopsy case. Hum Vaccin Immunother. 9:1228–1233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tada Y, Yoshikawa T, Shimomura M, et al:
Analysis of cytotoxic T lymphocytes from a patient with
hepatocellular carcinoma who showed a clinical response to
vaccination with a glypican-3-derived peptide. Int J Onco.
43:1019–1026. 2013.
|
|
33
|
Thanendrarajan S, Nowak M, Abken H, et al:
Combining cytokine-induced killer cells with vaccination in cancer
immunotherapy: more than one plus one? Leuk Res. 35:1136–1142.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Artusio E, Hathaway B, Stanson, et al:
Transfection of human monocyte-derived dendritic cells with native
tumor DNA induces antigen-specific T-cell responses in vitro.
Cancer Biol Ther. 5:1624–1631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Landi A, Babiuk LA and van Drunen
Littel-van den Hurk S: High transfection efficiency, gene
expression, and viability of monocyte-derived human dendritic cells
after nonviral gene transfer. J Leukoc Biol. 82:849–860. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Q, Wang L, Luo C, et al: Phenotypic
and functional characterization of cytokine-induced killer cells
derived from preterm and term infant cord blood. Oncol Rep.
32:2244–2252. 2014.PubMed/NCBI
|
|
37
|
Sun Z, Shi L, Zhang H, et al: Immune
modulation and safety profile of adoptive immunotherapy using
expanded autologous activated lymphocytes against advanced cancer.
Clin Immunol. 138:23–32. 2011. View Article : Google Scholar
|
|
38
|
Guo DW, Zhang SY, Hou XZ, et al: Glypican3
in genetically modified human monocyte-derived dendritic cells
induced specific cytotoxity against glypican3 overexpressing human
hepatocellular carcinoma cells in vitro. Saudi Med J. 29:1235–1240.
2008.PubMed/NCBI
|