Introduction
Even though chemotherapy, radiotherapy and
hematopoietic stem cell transplantation (HSCT) have been used to
treat hematological malignancies, several of these are still
considered incurable. Harnessing the host immune system towards the
targeting of cancer cells for their destruction has been shown to
be effective (1), and therapeutic
vaccines against leukemia-associated antigens (LAAs) are among the
approaches to trigger tumor-specific immune responses.
Identification of LAA and its cytotoxic T lymphocytes (CTL)
epitopes are of major importance for the development of tumor
antigen-specific therapeutic and prophylactic vaccines. A number of
T-cell epitopes from LAAs have been identified, and peptide
vaccines targeting LAAs based on their T-cell epitopes have been
shown to be effective in clinical trials (2).
Epidermal growth factor receptor pathway substrate 8
(Eps8) contains 822 amino acids and is located in chromosome
12p12-p13 in humans. The EPS8 gene encoded for the expression of
proteins with molecular weights of 97 and 68 kDa, referred to as
the two EPS8 isoforms. As a signaling adapter, EPS8 regulates actin
cytoskeleton dynamics and architecture (3) and participates in transduction of
signals from Ras to Rac by activating the Rac-specific guanine
nucleotide exchange factor activity in the form of a trimeric
complex together with SOS1 and ABI1 (4). It involves in the regulation of
processes including dendritic cell migration as well as cancer cell
migration and invasion (5).
Strong evidence demonstrated that EPS8 had oncogenic
potential, and its overexpression was reported in a range of human
malignancies, including breast cancer (6), pituitary tumors (7), pancreatic cancer (8), cervical cancer (9), colon cancer (10), head and neck squamous cell
carcinoma (11), esophageal
cancers (12) and human gliomas
(13), and its amplification is
often associated with tumor progression, acquired drug resistance
and poor prognosis (14). EPS8
upregulation is thus expected to be involved in human
carcinogenesis, implying that this gene may be used as a target in
tumor immunotherapy. A previous study by our group demonstrated an
elevated expression of EPS8 in patients with acute myeloid leukemia
(AML), its overexpression was inversely correlated with overall
survival of patients (15), and
EPS8 protein as a vaccine reagent induced a CTL response in a
murine breast carcinoma model (16). From the aforementioned facts, it
was speculated that this gene may be a prognostic marker and could
be a target for immunotherapy of hematological malignancies.
To the best of our knowledge, no T-cell epitopes of
EPS8 have been reported to date. The aim of the present study was
to identify human leukemia antigen (HLA)-A*0201-restricted epitopes
for EPS8. To achieve this aim, the expression of EPS8 in a range of
tumor cell lines was detected using western blot analysis and
reverse transcription quantitative polymerase chain reaction
(RT-qPCR), and their phenotypes were detected using a direct
immunofluorescence method. Furthermore, screening for the EPS8
protein sequence was performed using various algorithms to predict
the peptide-binding ability to the HLA-A*0201 molecule and its
proteasome cleavage sites at the C-terminus to identify T-cell
epitopes. The findings were validated in vitro by
peptide-binding affinity assay and brefeldin-A (BFA) decay assay.
The functional avidity of candidate peptide-specific CTLs was
evaluated using an enzyme-linked immunosorbent spot (ELISPOT) assay
and a cytotoxicity assay. Four peptides which were CTL epitopes of
EPS8 were identified, and which may be used in vaccine design and
tumor immunotherapy.
Materials and methods
Cell lines
A lymphoblast cell line, designated as T2 (174 ×
CEM. T2), was purchased from the American Type Culture Collection
(cat. no. CRL-1992™; Manassas, VA, USA). This cell line is
transporter associated with antigen processing (TAP)-deficient and
expresses HLA-A2. In the absence of exogenous antigen peptide load,
major histocompatibility complex (MHC) class I expression levels on
its surface are very low due to the poor stability of
non-peptide-loaded MHC class I molecules. The human erythroleukemia
cell line K562 (cat. no. TCHu191), the human acute monocytic
leukemia cell line THP-1 (cat. no. TCHu 57), the colon cancer cell
line SW480 (cat. no. TCHu172) and the human breast tumor cell line
MCF-7 (cat. no. TCHu 74) were purchased from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
The human acute myelogenous leukemia cell line KG1a was provided by
Tianjin Institute of Hematology (Tianjin, China). MCF-7 was
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen
Life Technologies, Grand Island, NY, USA), T2 was cultured in
Iscove’s modlfied Dulbecco’s medium (IMDM; Invitrogen Life
Technologies) containing 20% fetal bovine serum (FBS; GE Healthcare
Life Sciences, Logan, UT, USA), and the other cell lines were all
cultured in RPMI-1640 medium (Invitrogen Life Technologies)
supplemented with 10% FBS, 2 mol/l L-glutamine, 100 IU/ml
penicillin and 100 µg/ml streptomycin (Biological Industries
Israel Beit Haemek Ltd., Beit Haemek, Israel). Cells were
maintained in a humidified 37°C incubator with 5%
CO2.
Reagents
Mouse anti-human HLA-A2 monoclonal antibody (clone
number, BB7.2) conjugated with fluorescein isothiocyanate (FITC)
was purchased from BioLegend (San Diego, CA, USA; cat. no. 343304),
rabbit monoclonal anti-EPS8 antibody (clone number, EPR6112) was
purchased from Abcam Inc. (Cambridge, MA, USA; cat. no. ab12488;
1:1,000 dilution), the secondary goat anti-rabbit immunoglobulin
(Ig)G antibody was purchased from Southern Biotechnology Associates
Inc. (Birmingham, AL, USA; cat. no. 4030-05; 1:2,000 dilution),
mouse anti-GAPDH monoclonal antibody conjugated to horseradish
peroxidase (HRP) was purchased from Kangcheng Bio-Tech (Shanghai,
China; cat. no. KC 5G5; 1:1,000 dilution) and Immobilon western
chemiluminescent HRP substrate was purchased from Merck Millipore
(Billerica, MA, USA). Lymphocyte separation medium was purchased
from Tianjin Haoyang Biological Manufacture Co., Ltd. (Tianjin,
China), recombinant human interleukin (IL)-2 and IL-7 were
purchased from Peprotech (Rocky Hill, NJ, USA), β2-microglubulin
and BFA were purchased from Sigma-Aldrich (St. Louis, MO, USA) and
phytohaemagglutinin-M (PHA) was purchased from Biological
Industries Israel Beit Haemek Ltd (Kibbutz Beit Haemek, Israel).
Cell lysates, proteinase inhibitor phenylmethanesulfonylfluoride
(PMSF) and bicinchoninic acid (BCA) protein assay reagents were all
purchased from Beyotime Institute of Biotechnology (Nanjing,
China), the cytotoxic non-radioactive cytotoxicity assay kit was
purchased from Promega Corp. (Madison, WI, USA), the IFN-γ ELISPOT
kit was purchased from U-CyTech Biosciences (De Uithof, Utrecht,
The Netherlands), TRIzol reagent and SYBR green qPCR super mix were
purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA),
and the primers specific for human EPS8 and GAPDH were synthesized
by Sangon Biotech Co., Ltd (Shanghai, China).
Epitope prediction
To identify the potential T-cell epitopes for EPS8,
the HLA-A*0201 allele was screened, covering a wide range of
populations (17). First, the
protein sequence was screened for the best binding epitopes using
the algorithms SYFPEITHI (http://www.syfpeithi.de/) and Bioinformatics and
Molecular Analysis Section (BIMAS; http://www-bimas.cit.nih.gov/). The cut-off score was
adjusted to >20 for SYFPEITHI and T1/2>100 for
BIMAS, and peptides matching these two criteria and which were
shared by the two algorithms were selected. Second, the NetChop
algorithm (http://www.cbs.dtu.dk/services/NetChop/) was used to
predict whether the selected peptides in the first step would be
cleaved at the C-terminus. Third, protein sequences were analyzed
with five different algorithms for HLA-A*0201, including immune
epitope database (IEDB; http://www.iedb.org/), NetMHC (http://www.cbs.dtu.dk/services/NetMHC/), SVMHC
(http://www.sbc.su.se/~pierre/svmhc/),
EpiJen (http://www.ddgpharmfac.net/epijen/EpiJen/EpiJen.htm)
and Rankpep (http://imed.med.ucm.es/Tools/rankpep.html). Peptides
predicted by at least four algorithms were selected. The known
HLA-A*0201-restricted carcinoembryonic antigen peptide-1 (CAP-1,
YLSGANLNL) was used as a positive control peptide (18), while the HBcAg-derived
HLA-A*2402-restricted peptide HBc117e125 (EYLVSFGVW) was used as a
negative control peptide (19),
and all peptides were synthesized using standard
9-fluorenylmethyl-oxycarbonyl chemistry by the Chinese Peptide
Company (Hangzhou, China). They were of >95% purity as
determined by reverse-phase chromatography and their identity was
confirmed by mass spectrometry. The lyophilized peptide powder was
dissolved according to the manufacturer’s instructions in ultrapure
water or in dimeth-ylsulfoxide (BioLegend), diluted in
phosphate-buffered saline (PBS; Invitrogen Life Technologies) to a
final concentration of 1 mg/ml and stored in aliquots at
−80°C until use.
Peptide binding affinity assay
To evaluate the binding affinity of each candidate
peptide to the HLA-A*0201 molecule, a classic T2 peptide-binding
assay was performed as described by Dervillez et al
(20) with certain modifications.
T2 cells are TAP-deficient and HLA-A*0201-positive, but due to the
poor stability of non-peptide-loaded HLA-A*0201 molecules, its
HLA-A*0201 expression levels on the surface are low. Exogenous
peptides are able to induce the accumulation of HLA-A*0201
molecules, and thus, upregulation of HLA-A*0201 molecules on T2
cells may be detected by florescent intensity exchange which
reflects peptide binding ability to HLA-A*0201 molecules. Briefly,
T2 cells were incubated overnight with peptides (final
concentration, 100 µM) in IMDM serum-free medium containing
human β2-microglobulin (final concentration, 3 µg/ml) in a
humidified 26°C incubator with 5% CO2. Following
18 h, the temperature was raised to 37°C for 2 h. Cells were
harvested by gently transferring into a sterile centrifuge tube and
centrifuged at 200 × g for 5 min. Subsequently, cells were washed
twice, firstly with serum-free IMDM and then with cell staining
buffer (BioLegend; cat. no 420201). The cells were then stained
directly with anti-HLA-A*0201 monoclonal antibody conjugated to
FITC for 30 min at 4°C, and then analyzed using an Elite
flow cytometer (Beckman Coulter, Miami, FL, USA). Three duplicate
wells were set to each group and experiments were repeated three
times; CAP-1 was used as a positive control, HBc117e125 was used as
negative control, and T2 cells without any added peptide was used
as a background control. The fluorescence index (FI) was calculated
using the following formula: FI = [mean fluorescence intensity
(MFI)sampl−MFIbackground] /
MFIbackground, where MFIbackground represents
the value without peptide. FI>1.5 indicated that the peptide had
a high affinity for HLA-A*0201 molecules, 1.0<FI<1.5
indicated that the peptide had moderate affinity for the HLA-A*0201
molecule, and 0.5<FI<1.0 indicated that the peptide had low
affinity for the HLA-A*0201 molecule.
BFA decay assay
A BFA decay assay was performed to evaluate
peptide-HLA-A*0201 complex stability as described by Saini et
al (21) with certain
modifications. Briefly, T2 cells were seeded at 1×106
per well in 24-well plates and cultured with either the candidate
peptides or the control peptide (final concentration, 100
µM) at 26°C overnight in serum-free IMDM medium
containing β2-microglobulin (final concentration, 3 µg/ml)
to accumulate peptide-receptive class I molecules at the cell
surface. Following 18 h of incubation, cells were washed and
incubated with 10 µg/ml BFA dissolved in serum-free IMDM for
1 h at 37°C. Cells were harvested by gently transferring
into a sterile centrifuge tube and centrifuged at 200 × g for 5
min, then washed and resuspended. The cells were then stained with
anti-HLA-A*0201 fluorescent monoclonal antibody and analyzed using
flow cytometry. The calculated MFIs were used as values for the
time-point 0. In another group, cells were treated similarly to
those above; following washing three times, cells were re-suspended
in IMDM medium in the presence of 0.5 µg/ml BFA and
incubated in a humidified 37°C incubator with 5%
CO2. Cells were harvested by gently transferring into a
sterile centrifuge tube at the indicated time points and
centrifuged at 200 × g for 5 min, washed and stained with
anti-HLA-A*0201 fluorescent antibody. The stability of each peptide
bound to HLA-A2 was measured as the DC50-value, which
was defined as an estimate of the time required for a 50%-reduction
of the MFI-value recorded at time 0. The DC50-value was
calculated according to the formula: MFI at indicated time points /
MFI at time 0 × 100%. Three duplicate wells were set for each group
or experiments were repeated three times, with CAP-1 used as
control.
Detection of EPS8 expression and
phenotypic analysis of target cell lines
RT-qPCR
RT-qPCR was performed to detect the relative mRNA
expression levels of EPS8 in T2, MCF-7, K562, SW480, KG1a, TF1a,
Raji and THP-1 cell lines. Briefly, total RNA was extracted using
TRIzol reagent, purity was tested according to the ratio of optical
density (OD)260/OD280, and RNA integrity was
confirmed by 1% agarose gel electrophoresis (Beyotime Institute of
Biotechnology). The RNA was reverse-transcribed using iScript
Select cDNA Synthesis kit (cat. no. 170-8896; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) in which random nonomer primers and oligo
dT primers were included to synthesize cDNA. Purified total RNA was
first heat denatured at 85°C for 5 min, then reaction
systems were established, including RNA (1.0 µg), Oligo(dT)
(0.5 µl), random primers (0.5 µl), dNTP (10 nM),
RNase inhibitor (0.5 µl), buffer (5X, 4 µl) and M-MLV
(0.5 µl) with a total volume of 20 µl. The reaction
conditions were as follows: 30°C, 10 min; 42°C, 60
min; 85°C, 10 min. The quantification of PCR products was
accomplished using Platinum®SYBR® Green qPCR
SuperMix-UDG (cat. no. 11733–038; Invitrogen Life Technologies) The
reaction systems included cDNA, sense and antisense primers of EPS8
and GAPDH, 2X SYBR green qPCR Super mix and dH2O. In
each 20 µl reaction system, 5 µl of cDNA (1:20
dilution) was mixed with sense and antisense primers (0.5 µl
each) of EPS8 and GAPDH, 2X SYBR Green qPCR Super mix (10
µl) and ddH2O (4 µl). The reaction
conditions were as follows: 50°C, 2 min; 95°C, 2 min;
95°C, 15 sec; 60°C, 32 sec, for 40 cycles. The
relative quantitative expressions of EPS8 in each sample were
evaluated by detecting the CT value exchange (ΔCT, ΔΔCT) using the
2−ΔΔCt method. The quality of the cDNA was confirmed by
PCR analysis of GAPDH. All RT-qPCRs using the ABI PRISM 7900
sequence detection system (Applied Biosystems, Foster City, CA,
USA) were performed in triplicate. Gene-specific PCR primers used
to amplify EPS8 and GAPDH were designed as follows: For EPS8,
sense, 5′-GATGGAGGAAGTGCAAGATG-3′ and anti-sense,
5′-GACTGTAACCACGTCTTCACA-3′; for GAPDH, sense,
5′-GGGAAACTGTGGCGTGAT-3′ and antisense,
5′-GAGTGGGTGTCGCTGTTGA-3′.
Western blot analysis
The protein expression levels of EPS8 in the cell
lines were detected using western blot analysis. Cells were
maintained in the indicated culture media and used during their
logarithmic growth phase. Cells were lysed and protein was
extracted from cell lysates using RIPA lysis buffer at 4°C
in the presence of proteinase inhibitor PMSF to protect protein
from degradation. PMSF was dissolved in isopropyl alcohol and
diluted to 100 mmol/l. In the protein extraction procedure, PMSF
was added to the RIPA lysis buffer at a volume ratio of 1:100.
Sample protein concentrations were determined using the BCA protein
assay according to the manufacturer’s instructions prior to
addition of sample loading buffer (Beyotime Institute of
Biotechnology). The supernatant containing protein was mixed with
protein loading buffer (5X) at a ratio of 4:1, and following
boiling in 100°C for 5 min, samples were centrifuged and
stored at −20°C until use. A total of 30 µg of each
sample was electrophoresed on a 10% SDS-polyacrylamide gel and
transferred to a polyvinylidene difluoride (PVDF) membrane (Merck
Millipore). Following blocking in 5% skimmed milk (Beyotime
Institute of Biotechnology) for 1 h at room temperature, the
membrane was incubated for 24 h at 4°C in 5% skimmed milk
with added primary antibody to human EPS8 (dilution rate, 1:1,000).
The membrane was then washed three times with 0.5 % Tris-buffered
saline-Tween 20 (Beyotime Institute of Biotechnology) and incubated
in 5% skimmed milk with added HRP-conjugated secondary antibody
(dilution rate, 1:20,000). Following additional washing, the
membrane was incubated in Immobilon western chemiluminescent HRP
substrate consisting of luminol reagent and peroxide solution and
captured on chemiluminescence-sensitive film. For each sample,
GAPDH was used as a loading control (dilution rate, 1:10,000).
Phenotypic analysis of target cell
lines
Briefly, cells were incubated in recommended medium
containing FBS and antibiotics as described above. During their
logarithmic growth phase, cells were harvested by centrifuging at
200 × g for 5 min and resuspended in Cell Staining buffer.
Following centrifugation at 350 × g for 5 min, the supernatant was
discarded and cells were incubated with 5 µl of Human
TruStain FcX™ (cat. no. 422301; BioLegend) per 100 µl of
cell suspension for 5–10 min at room temperature for Fc receptor
blocking. Subsequently, cells were stained directly with HLA-A2
antibody conjugated with FITC (3–5 µl) and incubated on ice
for 15–20 min in the dark. Following washing twice with Cell
Staining buffer by centrifugation at 350 × g for 5 min, cells were
resuspended in 0.5 ml Cell Staining buffer and 5 µl/million
cells of 7-AAD Viability Staining solution (cat. no. 420403;
BioLegend) were added to exclude dead cells. Cells were incubated
on ice for 3–5 min in the dark and analyzed with a Flow Cytometer.
The HLA-A phenotype of the target cells was detected as described
by Imai et al (22).
Peptide-specific CTL induction
Peptides were used to immunize peripheral blood
mononuclear cells (PBMCs) to induce peptide-specific CTLs as
previously described (23–25). The experiment was approved by the
Institutional Ethics Committee of Southern Medical University
(Guangzhou, China) and informed consent was obtained from all 32
donors. HLA-A*0201 phenotypic analysis of donors was completed
using PCR-sequence-based typing by BGI Tech (Shenzhen, China), and
PBMCs were purified using lymphocyte separation medium.
≥2×106 PBMCs obtained from two HLA-A2.1+ healthy donors
were incubated with the predicted peptides (final concentration, 10
µg/ml) in RPMI-1640 medium. On day three, 20 U/ml IL-2 and
10 ng/ml IL-7 were added. Half the volume of the medium was
replaced with fresh medium containing IL-2 (final concentration, 20
U/ml) and IL-7 (final concentration, 10 ng/ml) every 2–3 days. On
day seven, cells were harvested by gently pipetting and transferred
into sterile centrifuge tubes and centrifuged at 128 × g for 10
min. Following centrifugation, cells were washed and re-suspended
in fresh medium containing IL-2 and IL-7. The PBMCs were repeatedly
stimulated with peptides every 7 days, the stimulated frequency was
at least three in order to harvest a sufficient number of CTLs. At
the same time, the antigen signal could be fully presented to
CD8+ T-cells in PBMCs. After 3 weeks of stimulation,
these cells were used for the cytotoxicity assay. The main cells in
total PBMCs were CD8+ T-cells. Following three times of
peptide stimulation, the CTLs were harvested and used for
functional analysis. Whenever necessary, cells were divided.
IFN-γ ELISPOT assay
To further evaluate peptide immunogenicity, an IFN-γ
ELISPOT assay was performed according to the manufacturer’s
instructions. In brief, ~4×106/ml/well (or more) PBMCs
were stimulated with peptides at a final concentration of 10
µg/ml. On day three, IL-2 (final concentration, 10 U/ml) was
added to each well. On day five, half of the volume of medium was
replaced with fresh medium containing IL-2 with a final
concentration of 10 U/ml. On day seven, PBMCs were stimulated with
peptides again as above. On day nine, the microliter plates were
coated overnight at 4°C with anti-IFN-γ monoclonal antibody.
On day 10, cells were harvested for experiments. Peptide at a final
concentration of 10 µg/ml was added to ~2×105
PBMCs per well in 100 µl X-VIVO 15 medium (Lonza Group Ltd.,
Auckland, New Zealand) and cultured at 37°C in a humidified
incubator with 5% CO2. Following 20–26 h of incubation,
contents of all wells were discarded, and following washing,
streptavidin-HRP solution was added into each well, and plates were
incubated for 1 hour at 37°C. The PVDF membrane was washed
thoroughly, 3-amino-9 ethylcarbazole substrate solution was added
into each well and plates were incubated for 25 min at room
temperature in the dark. Plates were then washed with demineralized
water and air-dried at room temperature. Following the completion
of the experiments, spot-forming cells (SFCs) were counted by
Dakota Biotechnology company (Shenzhen, China). For all
experiments, PHA stimulation was used for the positive control,
PBMCs without added peptide were used as the negative control, and
serum-free X-VIVO 15 medium from (Hebei Lonzer Chemicals Co., Ltd.,
Handan, China) was used as the background control. Experiments for
each group were performed in triplicate.
Cytotoxicity assay
The functional avidity of peptide-specific CTLs was
evaluated using a lactate dehydrogenase (LDH) release assay. For
this, the non-radioactive cytotoxicity assay kit was used to detect
the lysis effects of CTLs on target cells. Briefly, the induced
CTLs were used as effective cells, and MCF-7 cells (HLA-A*0201+,
EPS8+), T2 cells pulsed with corresponding peptides, T2 cells
pulsed with irrelevant peptide, T2 cells only, THP-1 cells
(HLA-A*0201−, EPS8−) and K562 cells (HLA-A*0201−, EPS8+) were used
as target cells. The ratio of effective cells to target cells was
set to 100:1, a constant number of target cells (5,000 cells/well)
was added to the effectors, and at the same time, controls were set
according to the manufacturer’s instructions. Following
centrifugation at 250 × g for 4 min, cells were incubated in a
humidified 37°C incubator with 5% CO2 in 100
µl phenol red-free culture medium containing 5% FBS for 4–6
h. Fourty-five minutes prior to supernatant harvest, lysis solution
(10X) was added to target cell maximum LDH release control and
cells were cultivated continually in the same condition.
Subsequently, cells were centrifuged at 250 × g for 4 min, 50
µl supernatant was transferred to the corresponding well of
a flat-bottomed 96-well enzymatic assay plate for LDH measurement.
Experiments were performed in three duplicate wells and repeated
three times. The percent cytotoxicity was determined using the
following formula: %Cytotoxicity = [Experimental−Effector
spontaneous−Target spontaneous] / [Target maximum−Target
spontaneous] ×100%.
Statistical analysis
SPSS 17.0 software (SPSS, Inc, Chicago, IL, USA) was
used for statistical analysis. One-way analysis of variance was
used for comparing differences between groups. P<0.05 was
considered to indicate statistically significant differences.
Results
In silico analysis of EPS8 protein for
potential candidate peptides
To identify potential CTL epitopes of EPS8, the two
algorithms SYFPEITHI and BIMAS were used to predict the potential
peptides with high binding ability to the HLA-A*0201 molecule,
which was a limiting step, resulting in a small number of peptides,
and four peptides shared by those two algorithms were selected. The
algorithm NetChop was then used to predict whether the peptides
would be cleaved at the C-terminus, and only peptides with cleavage
sites at the C-terminus were selected. Finally, five different
algorithms were used to further confirm the selected peptides. Four
9-mer peptides with starting positions in the EPS8 protein sequence
at 455, 276, 360 and 92, were finally selected (Table I). The positive and negative
control peptides are also listed in the table.
 | Table ICharacteristics of in silico
predicted EPS8 CTL epitopes restricted to the HLA-A*0201 allele and
control peptides. |
Table I
Characteristics of in silico
predicted EPS8 CTL epitopes restricted to the HLA-A*0201 allele and
control peptides.
| Peptidea | Sequence | Score
| FIk |
DC50l |
|---|
| SYFPEITHIb | BIMASc | NetChopd | IEDBe | NetMHCf | Rankpepg/proteasomeh | EpiJeni | SVMHCj |
|---|
| 455 | QLAESVANV | 30 | 655.88 | + | 0.7 | SB | 108.0/+ | 0.78 | 1.04 | 2.22 | >8 h |
| 92 | KLLDAKGKV | 26 | 480.07 | + | 2.1 | WB | 80.0/+ | 1.03 | 0.53 | 1.02 | 2–4 h |
| 276 | ILDDIEFFI | 21 | 927.86 | + | 0.3 | SB | 68.0/+ | 0.45 | 0.34 | 1.07 | 6–8 h |
| 360 | FLFTPLNMV | 28 | 2722.68 | + | 0.3 | SB | − | 0.32 | 1.06 | 2.05 | >8 h |
| CAP 1m | YLSGANLNL | – | – | − | – | – | − | – | – | 2.19 | 6–8 h |
| HBc117e125n | EYLVSFGVW | – | – | − | – | – | − | – | – | 0.36 | – |
In vitro analysis of peptide affinity and
binding stability to the HLA-A*0201 molecule
Evidence suggested that peptide affinity to MHC
molecules is often correlated with its immunogenicity (26). Therefore, the TAP-deficient and
HLA-A*0201-positive cell line T2 was used to detect peptide
affinity for the HLA-A*0201 molecule. When exogenous peptides were
added, the MFI of T2 cells increased significantly (Fig. 1A). Of the four candidate peptides,
peptide 455 and 360 had the highest affinity to HLA-A*0201, with
MFIs of 312.73±3.14 and 296.02±5.31, and FIs of 2.22 and 2.05,
respectively. The FI values of the two peptides were almost as high
as those of the positive control (FI=2.19). For peptides 276 and
92, the MFI was 200.77±2.60 and 195.96±1.35, and their FI was 1.07
and 1.02, respectively, indicating moderate affinity for the
HLA-A*0201 molecule (P<0.001). The FI of the negative control
was only 0.36 (Table I), and no
weak-affinity peptides were found in the four candidate peptides
according to the definition of peptide affinity for the HLA-A*0201
molecule.
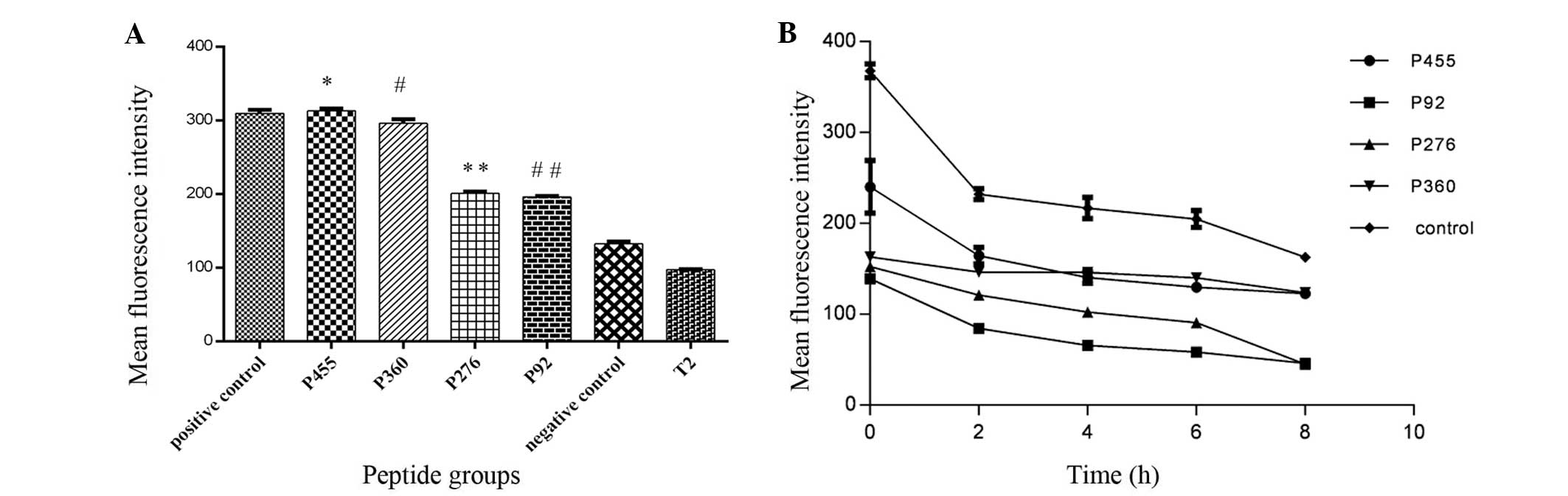 | Figure 1Affinity and stability of peptide
binding to HLA-A*0201. (A) Affinity of candidate epitopes to the
HLA-A*0201 molecule. *P<0.01, P455 vs. P276, P360,
P92, negative control and T2. #P<0.01, P360 vs. P276,
P92, negative control, positive control and T2.
**P<0.01, P276 vs. negative control, positive control
and T2. ##P<0.01, P92 vs. negative control, positive
control and T2. Values are expressed as the mean ± standard
deviation (n=3). (B) Binding stability of candidate epitopes to
HLA-A*0201 molecule. HLA, human leukemia antigen. |
In the BFA decay assay, with increasing time, the
MFI of T2 cells in each group decreased (Fig. 1B), and the DC50-value
was calculated to reflect the peptide/MHC complex stability; the
longer the time, the more stable the complex. Complex stability of
peptide 455/HLA-A*0201 and peptide 360/HLA-A*0201 was the highest,
as their DC50-values were all >8 h, followed by
peptide 276 with DC50<8 h, while the stability of
peptide 92/MHC complex was lowest with a DC50 of 2–4 h
(Table I).
EPS8 expression detection and phenotypic
analysis of target cell lines
To identify candidate target cells, RT-qPCR was used
to detect the mRNA expression of EPS8; furthermore, the protein
expression of EPS8 was detected by western blot analysis. Among the
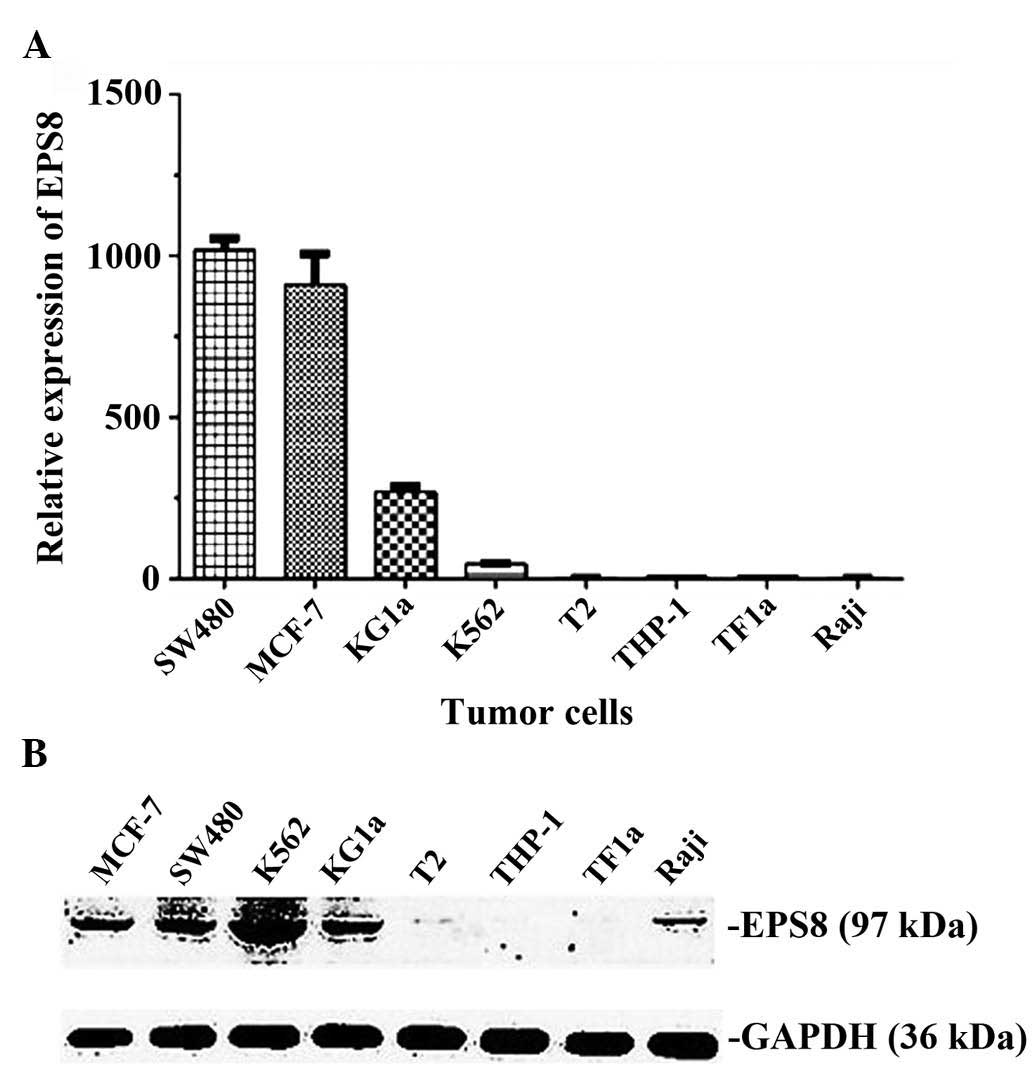
eight cell lines, MCF-7, SW480, KG1a and K562 were EPS8-positive at
the mRNA expression level (Fig.
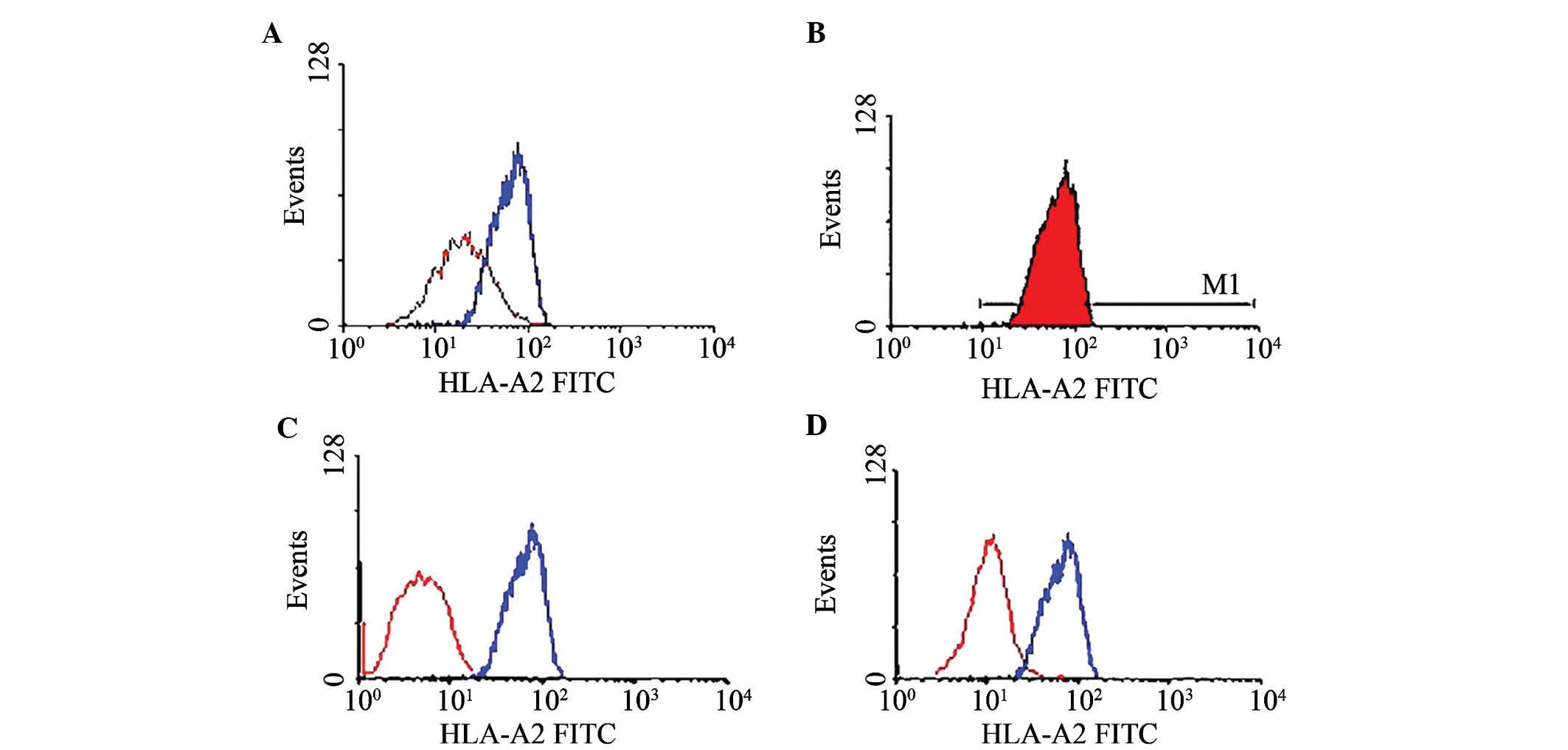
2A) and the protein expression level (Fig. 2B). The HLA-A phenotype of the
target cells was detected by direct immunofluorescence antibody
staining and flow cytometric analysis. Of the eight cell lines,
only MCF-7 and T2 were HLA-A*0201-positive (Fig. 3A and B).
CTL epitopes of EPS8 stimulate PBMCs to
express IFN-γ
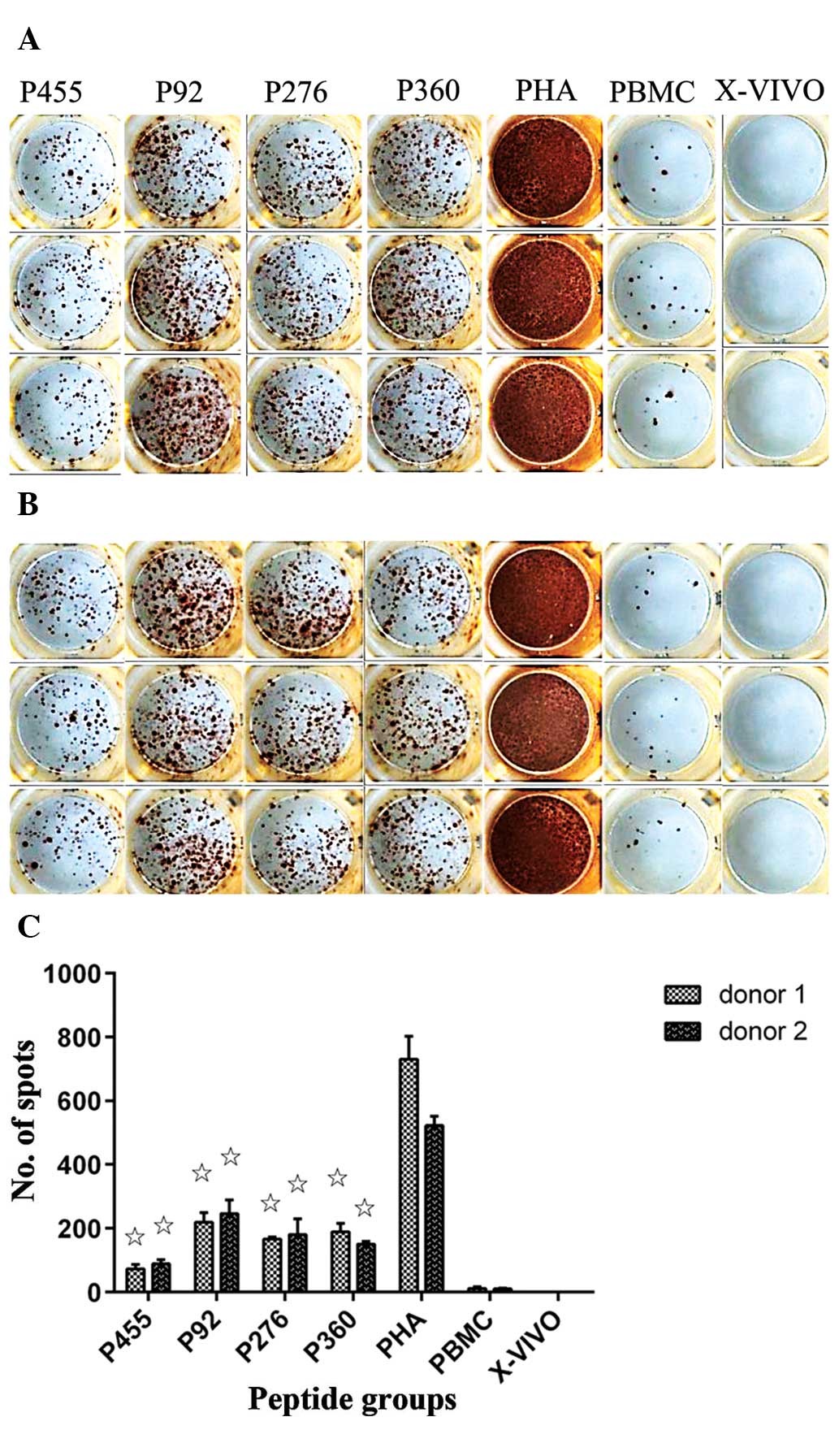
To determine the immunogenicity of peptides, PBMCs
were stimulated in a 10-day procedure as described in the Materials
and methods section, followed by ELISPOT analysis. PBMCs separated
from two HLA-A*0201-positive healthy volunteers were repeatedly
stimulated with peptides, and the IFN-γ secretion of the stimulated
PBMCs in response to the peptides was assessed for the two donors
(Fig. 4). The PBMCs of the two
donors responded positively to stimulation with all of the four
predicted peptides, as indicated by an increased number of SFCs
following stimulation. Of note, PBMCs responded strongly to
stimulation with the relatively weak binder peptide 92 (218±30.4
peptide-specific spots/2×105 cells), but elicited a
response of lower intensity to strong binder peptide 455 (73.0±14.2
spots/2×105 cells) (Fig.
4C) (P<0.001).
The four peptide-specific CTLs
specifically lyse EPS8 expression target cells in an
HLA-A*0201-restricted manner
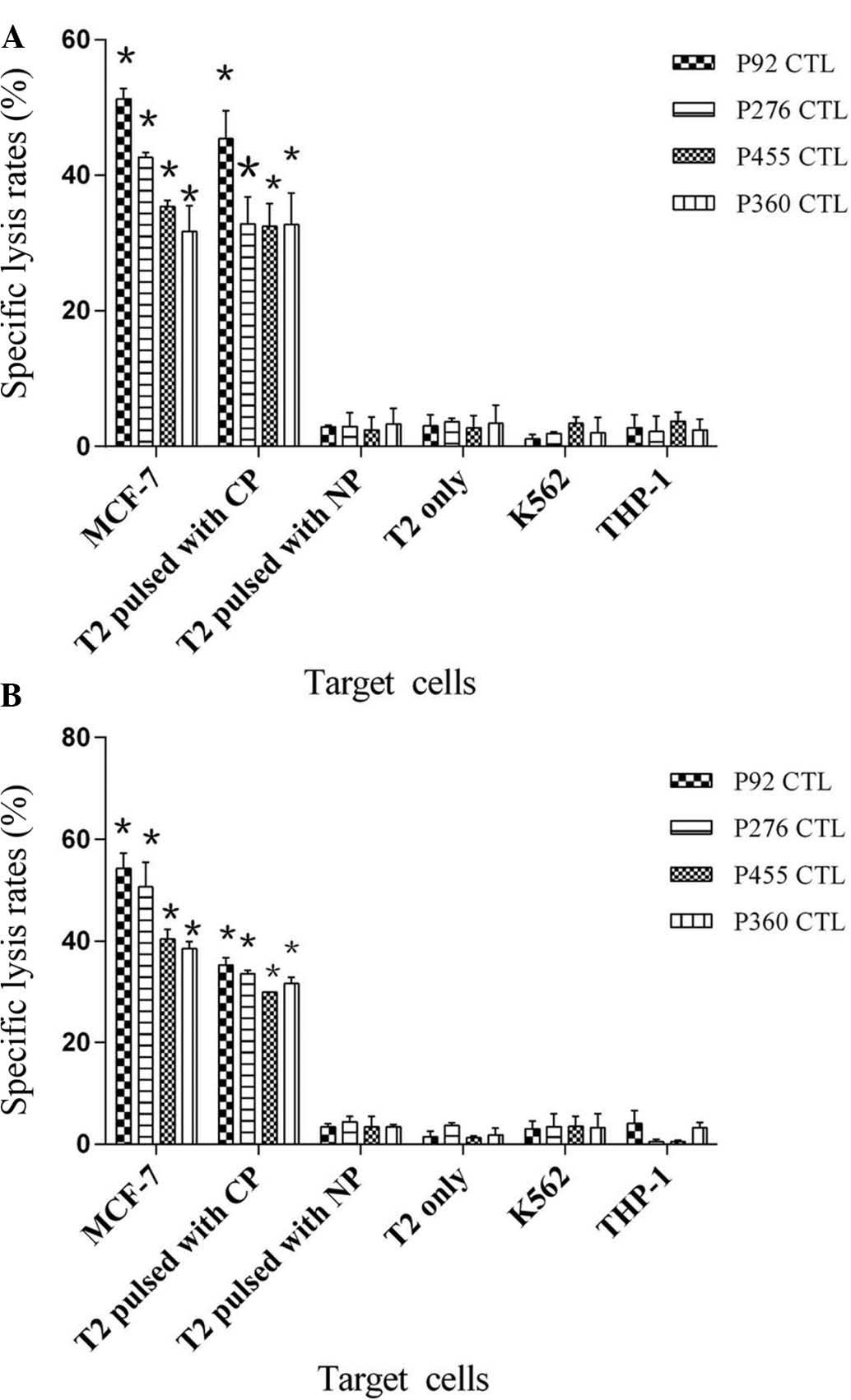
To characterize the functional avidity of
peptide-specific CTLs, an LDH release assay was performed to
evaluate their cytotoxic effects on MCF-7 and T2 cells pulsed with
cognate peptides. As shown in Fig.
5, the four peptide-specific CTLs efficiently lysed MCF-7 as
well as T2 cells pulsed with corresponding peptides but could
almost not lyse T2 cells pulsed with irrelevant peptide, T2 only,
K562 or THP-1 cells at an effector to target cell ratio of 100:1
(P<0.05). Of them, peptide 92-specific CTLs had the highest
lysis rates of MCF-7 and T2 cells pulsed with the corresponding
peptide, and the average lysis rate of MCF-7 cells was 51.28±1.54%
(P=0.001; donor 1) (Fig. 5A) and
54.22±3.01% (P=0.001; donor 2) (Fig.
5B), respectively, while the lysis rate of T2 cells loaded with
the corresponding peptide was 45.41±4.16% (P=0.023; donor 1) and
35.23±1.50%, (P=0.001; donor 2), respectively. The cytotoxic
effects of peptide- induced CTLs on target cells were EPS8
expression-specific and restricted by the HLA-A*0201 molecule as
the effectors only target HLA-A*0201 positive cells.
Discussion
Concepts for treating hematologic malignancies by
harnessing the immune system have been established due to defects
of current treatment methods (27). HSCT is the only potential method to
cure hematological malignancies, but relapse rates remain high,
particularly in patients transplanted during relapse or in second
or subsequent remission (28).
Therefore, efforts are required to seek for novel avenues which are
tumor-specific and have synergy with current treatment methods;
from this viewpoint, therapeutic vaccines against LAAs are an
optimal choice.
Mounting evidence demonstrated the existence of LAAs
(29); as most LAAs are not muted,
their immunogenicity is often poor; however, at the same time, the
immunity evoked by current therapeutic vaccines targeting LAAs is
far from optimal. Accordingly, isolating specific components that
have immunogenicity and can induce a strong immune response are
rational choices for vaccines. Therefore, the most important task
in the discovery of cancer vaccines is to identify novel TAAs and
their CTL epitopes in order to make additional therapeutic targets
available which are closely associated with tumor incidence and
progression. Reverse immunology has been commonly used to identify
epitopes of LAAs (30).
The present study was the first, to the best of our
knowledge, to report EPS8 as a potential TAA and identify its
HLA-A*0201-restricted epitopes. EPS8 was reported to be expressed
at elevated levels in numerous human malignancies and its
overexpression has been reported to be sufficient to transform
non-tumorigenic human cells into a tumorigenic phenotype (31). Its expression in normal tissue
types is relatively low, and it has a pivotal role in
tumorigenesis, tumor proliferation, invasion and metastasis;
furthermore, the expression levels of EPS8 correlated with disease
severity and overall survival of patients (32). Thus, EPS8 is a TAA and may be
utilized as a target in immunotherapy of cancer as well as in
hematological malignancies.
In the present study, a reverse immunology method
was used to identify HLA-A*0201-restricted epitopes from EPS8. In
the prediction phase, to appropriately select the T-cell epitopes
contained in this protein, the focus was on three main events of
the intracellular generation of peptides presented in HLA class I
molecules; these HLA class I ligands are called CTL epitopes only
when immunogenic (33). The first
event is enzymatic digestion of protein by cytosolic peptidases,
leading to the release of the epitope or epitope precursor in the
protein. The second important event is required to prevent peptides
from further cytosolic degradation via the TAP, only peptides that
escape from cytosolic degradation and finally enter into
endoplasmic reticulum can bind with HLA class I molecules. The
third event is the assembly of peptides with HLA class I heavy and
light chains in the ER for cell surface presentation (34–39).
Peptides processed and presented through these three steps have the
highest potential to be CTL epitopes. In the final step of the
in silico analysis, five different popular algorithms were
used to further confirm the selected peptides, leading to the final
selection of four 9-mer peptides. It is worth mentioning that the
aim of the present study was to identify dominant T-cell epitopes.
For this, a combined prediction using SYFPEITHI and BIMAS was
utilized, resulting in only four peptides, which significantly
saved time by narrowing the validation scope. However, this does
not indicate that there are only four CTL epitopes within the EPS8
protein sequence; due to stringent restrictions arising from the
selection criteria, several potential subdominant epitopes may have
been ignored. BIMAS and SYFPEITHY are based on the principle of
binding motifs/quantitative matrices (39), and in spite of numerous novel
algorithms that have emerged, they remain to be commonly used in
current studies. The main reason for this may be the large dataset
they comprise, which is important, as most predictions are
dataset-driven-the bigger the dataset, the more accurate the
prediction. One weakness of the motifs or quantitative matrices is
the ignorance of the contribution of the overall peptide structure
to binding. To overcome the limitations of motif-based predictions,
more powerful methods based on tools including ANN, PSSM, SMM and
SVMHC were used. It was reported that a sensitivity of 80% and a
specificity of 80% can be achieved with ANN (40). Together with proteasomal cleavages
and TAP translocation prediction, it was speculated that the anchor
residues of selected peptides could chemically complement the main
pockets of the peptide-binding groove of the HLA molecule.
The predicted peptides are not always true binders,
and several false positive peptides may be selected (41); therefore, experiments are required
to validate their immunogenicity. Peptide affinity to HLA molecules
is a key event, and a correlation between HLA binding and
immunogenicity is often observed (42). The eradication of tumor cells by
T-cells requires high-affinity targeted peptide-MHC interactions,
which lead to efficient cross-presentation of antigens. Their
binding affinity to the HLA-A*0201 molecule was evaluated using the
TAP-deficient cell line T2. When exogenous peptides were added, the
HLA-A*0201 molecule on the surface of T2 accumulated, resulting in
a change in fluorescent intensity, which changes in the MFI
reflecting their binding affinity to the HLA-A*0201 molecule. Of
the four peptides, peptide 455 and 360 had the highest affinity to
the HLA-A*0201 molecule, while peptide 276 and 92 had a relatively
low binding affinity to the HLA-A*0201 molecule. This meant that
peptide 455 and 360 were most likely to be CTL epitopes.
However, certain peptides with high binding affinity
to HLA class I molecules may be non-immunogenic (43), as they fail to form stable
complexes with HLA class I molecules (44). Although peptide immunogenicity was
found to correlate significantly with high affinity, the stability
of peptide interaction with MHC-I correlated better with
immunogenicity than affinity (45). Following the identification of the
CTL epitopes, the present study assessed the peptide/MHC complex
stability by using the BFA decay assay. BFA inhibits protein
synthesis in cultured cells and reversibly inhibits the
intracellular translocation of proteins to the cell surface for
secretion or expression (46,47);
therefore, the accumulated complex remained stable on the cells.
When peptide-pulsed T2 cells were continuously cultured in the
presence of BFA at 37°C, their MFI on the cells decreased,
the peptide/HLA-A*0201 molecule complex dissociated naturally, and
DC50-values indicated that the 455 and 360/HLA-A*0201
molecule complexes were more stable than those of peptide 276 and
peptide 92. This meant that peptide 455 and 360 may be more
immunogenic than peptide 276 and peptide 92.
High binding affinity and stable binding of peptides
to MHC in vitro does not necessarily imply that these
peptides can be naturally presented. To test the natural
presentation of the candidate epitopes, the target cells were first
screened by RT-qPCR and western blot to detect the EPS8 expression,
and their HLA-A phenotype was detected by direct immune
fluorescence antibody staining and flow cytometric analysis. Three
tumor cell lines and T2 were selected as target cells. Most of
epitope identification methods which have been used to date require
the induction of peptide-specific CTLs, followed by their
functional avidity evaluation; PBMCs separated from healthy donors
are commonly used to evaluate the immunogenicity of a peptide
(48). Two HLA-A*0201-positive
donors were identified among healthy volunteers using an external
high-resolution method. Peptide-specific CTLs were induced by
immunizing the PBMCs of two healthy HLA-A*0201-positive donors
through at least two rounds of weekly stimulations with peptides
and in the presence of IL-2 and IL-7, the number of
peptide-specific CTLs was sufficiently increased.
Following successful induction of specific CTLs, an
IFN-γ ELISPOT assay was used to quantify the occurrence of
T-lymphocyte cells secreting IFN-γ after stimulation with cognate
peptide. The results demonstrated that the four peptide-specific
CTLs all responded to the corresponding peptides and secreted
IFN-γ; their effective SFCs met the criteria of the positive
standard, with the number of SFCs of peptide 92 specific CTL being
the highest. The results for the two donors were consistent, which
meant that peptide 92 was more immunogenic than the other three
peptides.
Finally, the recognition efficiency of
peptide-specific CTLs to target cells was evaluated using a
non-radioactive cytotoxicity assay, and in the two volunteers, all
peptide-specific CTLs could lyse MCF-7 (HLA-A*0201+, EPS8+) and T2
cells pulsed with the same peptides, but could almost not lyse
either HLA-A*0201-negative or EPS8-negative cells at the effectors
to targets ratio of 100:1 (P<0.05). This indicated that their
cytotoxicity was EPS8 expression-specific and in an
HLA-A*0201-restricted fashion, the immunogenicity of peptide 92 was
the strongest. Together with the ELISPOT results, it was
demonstrated that the four predicted epitopes were naturally
presented, and that they were CTL epitopes of EPS8.
In the present study it was observed that peptide
binding affinity and peptide/HLA complex stability did not
correlate well with their intensity of immunogenicity; for example,
irrespective of its low affinity or complex stability with
HLA-A*0201 molecule, the relatively weak binder peptide 92
stimulated PBMCs more strongly to secret the highest amount of
IFN-γ than the strong binder peptide 455, which stimulated the
least amount of IFN-γ secretion by the PBMCs. At the same time,
compared to the mean lysis rates of CTLs induced by other peptides,
the mean lysis rate of peptide 92-induced CTLs was the highest to
MCF-7 and T2 pulsed with corresponding peptide. The reason of this
may be based on the interaction dynamics of the T-cell receptor
(TCR)-peptide/MHC complex, which is closely associated with T-cell
activation and subsequently its differential fate (49). There is a physiological affinity
range, which is optimal to activate T-cells, and a plateau of
T-cell functional activity above a defined affinity threshold has
been reported, suggesting that all clustered TCRs will be occupied
above the defined threshold, and further increases in affinity do
not contribute to T-cell functions; therefore, above a defined
threshold, TCRs with higher avidity may not be at an advantage over
lower-avidity TCRs in the exertion of cytotoxic function (50). Furthermore, negative feedback
mechanisms, including programmed death 1 and lymphocyte activation
gene-3 (CD223), would further curtail the potency of high avidity
TCRs (51–53). The discrepancy observed in the
present study may be ascribed to the peptide affinity of
high-affinity binding peptides above the defined affinity
threshold. Another reason may be the supra-optimal antigen dose.
Evidence suggested that culturing of CTLs in vitro in the
presence of high- or low- dose antigen leads to polarization of low
and high-avidity responses, respectively (54,55).
In regard to low- or intermediate-affinity peptides, the peptide
dose used for CTL induction in the present study may have been
supra-optimal for high-affinity peptides, with the optimal TCR
stimulation peptide dose for maintenance of high-avidity T-cells
ex vivo being 1 ng/ml and resulting in retained avidity,
proliferation and ability to kill specific targets. By contrast,
the supra-optimal TCR stimulation peptide dose (10 µg/ml
peptide) for high-avidity T-cell maintenance ex vivo
resulted in reduced avidity and failure to kill tumor cells
(56). The present study
highlights the importance of optimal stimulation for the induction
and maintenance of high-avidity CTL. Due to certain limitations,
the present study did not further explore the best affinity to HLA
molecules, which is in favor of peptide-specific CTL induction and
maintenance.
Furthermore, the experiments of the present study
had additional drawbacks: First, due to limitations to the amount
of blood samples, the optimal cytokine concentration and their
dosing intervals for best induction and maintenance of
peptide-specific CTL were not explored. Second, background signals
of target cells that lacked tumor antigens or restriction were
observed, implying that non-specific killing effects still existed.
The reason for this may be ascribed to the use of bulk CTL instead
of CD8+ T-cell from the colon as effectors. Furthermore, CD8+ T
cells were not sorted from PBMCs for CTL induction at the beginning
and designated antigen-presenting cells such as dendritic cells
(DCs) were not used; this may have resulted in inadequate antigen
processing and presentation. Despite these drawbacks, from an
economic and practical point of view, the methods used in the
present study are an economical and rapid method for epitope
validation. Furthermore, total PBMCs include a variety of cell
types, including monocytes and DCs, which can be used as
antigen-presenting cells (APC) (57).
It is worth mentioning that certain CTL epitopes
identified in vitro may be less immunogenic in vivo,
which may be due to inadequate antigen processing and presentation
by APC (58). Therefore, a
subsequent study by our group will test the immunogenicity of the
epitopes in vivo using transgene mice.
In conclusion, the present study identified four
HLA-A*0201-restricted, EPS8-derived epitopes, which are located at
the starting positions of 92, 276, 360 and 455 in the EPS8 protein
sequence. They had good binding affinity for the HLA-A*0201
molecule and their peptide/HLA-A*0201 complex stability was high.
At the same time, these peptides promoted lymphocyte proliferation,
and their specific CTLs responded to the corresponding peptides and
secreted IFN-γ. They were also able to recognize and lyse
ESP8-expressing target tumor cells in an HLA-A*0201-restricted
manner, and they were naturally processed and presented on tumor
cells. Among them, peptide 92 was the most promising as an
antigenic epitope to target tumor cells due to its higher
immunogenicity. Future studies will be required to investigate the
clinical utility of the identified epitopes by using blood samples
from patients with hematological malignancies.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81372249 and
81300431), Foundation of the Ministry of Education of China for
Returned Scholars, Research Fund for the Doctoral Program of Higher
Education of The Ministry of National Education China (grant no.
20114433110012), The Project of Department of Education of
Guangdong Province (grant no. 2012KJCX0025), Key Project of Science
and Technology of Guangzhou city (no. 12C22121595) and Natural
Science Foundation of Guangdong Province, China (grant no.
S2013040014449).
References
|
1
|
Zigler M, Shir A and Levitzki A: Targeted
cancer immunotherapy. Curr Opin Pharmacol. 13:504–510. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Casalegno-Garduño R, Schmitt A and Schmitt
M: Clinical peptide vaccination trials for leukemia patients.
Expert Rev Vaccines. 10:785–799. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaggi F, Disanza A, Milanesi F, Di Fiore
PP, Menna E, Matteoli M, Gov NS, Scita G and Ciliberto A: The
Eps8/IRSp53/VASP network differentially controls actin capping and
bundling in filopodia formation. PLOS Comput Biol. 7:e10020882011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Disanza A, Carlier MF, Stradal TE, Didry
D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP and
Scita G: Eps8 controls actin-based motility by capping the barbed
ends of actin filaments. Nat cell Biol. 6:1180–1188. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Werner A, Disanza A, Reifenberger N,
Habeck G, Becker J, Calabrese M, Urlaub H, Lorenz H, Schulman B,
Scita G, et al: SCFFbxw5 mediates transient degradation of actin
remodeller Eps8 to allow proper mitotic progression. Nat cell Biol.
15:179–188. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao J, Weremowicz S, Feng B, Gentleman RC,
Marks JR, Gelman R, Brennan C and Polyak K: Combined cDNA array
comparative genomic hybridization and serial analysis of gene
expression analysis of breast tumor progression. Cancer Res.
66:4065–4078. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu M, Shorts-Cary L, Knox AJ,
Kleinsmidt-DeMasters B, Lillehei K and Wierman ME: Epidermal growth
factor receptor pathway substrate 8 is overexpressed in human
pituitary tumors: Role in proliferation and survival.
Endocrinology. 150:2064–2071. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Welsch T, Endlich K, Giese T, Büchler MW
and Schmidt J: Eps8 is increased in pancreatic cancer and required
for dynamic actin-based cell protrusions and intercellular
cytoskeletal organization. Cancer Lett. 255:205–218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen YJ, Shen MR, Chen YJ, Maa MC and Leu
TH: Eps8 decreases chemosensitivity and affects survival of
cervical cancer patients. Mol Cancer Ther. 7:1376–1385. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC,
Wang ST, Huang CC, Chow NH and Leu TH: Eps8 facilitates cellular
growth and motility of colon cancer cells by increasing the
expression and activity of focal adhesion kinase. J Biol Chem.
282:19399–19409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Patel V, Miyazaki H, Gutkind JS
and Yeudall WA: Role for EPS8 in squamous carcinogenesis.
Carcinogenesis. 30:165–174. 2009. View Article : Google Scholar
|
|
12
|
Bashir M, Kirmani D, Bhat HF, Baba RA,
Hamza R, Naqash S, Wani NA, Andrabi KI, Zargar MA and Khanday FA:
P66shc and its downstream Eps8 and Rac1 proteins are upregulated in
esophageal cancers. Cell Commun Signal. 8:132010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ding X, Zhou F, Wang F, Yang Z, Zhou C,
Zhou J, Zhang B, Yang J, Wang G, Wei Z, et al: Eps8 promotes
cellular growth of human malignant gliomas. Oncol Rep. 29:697–703.
2013.
|
|
14
|
Gorsic LK, Stark AL, Wheeler HE, Wong SS,
Im HK and Dolan ME: EPS8 inhibition increases cisplatin sensitivity
in lung cancer cells. PLoS One. 8:e822202013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Cai SH, Xiong WY, He YJ, Deng L
and Li YH: Real-time quantitative polymerase chain reaction assay
for detecting the eps8 gene in acute myeloid leukemia. Clin Lab.
59:1261–1269. 2013.
|
|
16
|
He YJ, Zhou J, Zhao TF, Hu LS, Gan JY,
Deng L and Li Y: Eps8 vaccine exerts prophylactic antitumor effects
in a murine model: A novel vaccine for breast carcinoma. Mol Med
Rep. 8:662–668. 2013.PubMed/NCBI
|
|
17
|
Li S, Li H, Chen B, Lu D, Deng W, Jiang Y,
Zhou Z and Yang Z: Identification of novel HLA-A 0201-restricted
cytotoxic T lymphocyte epitopes from Zinc Transporter 8. Vaccine.
31:1610–1615. 2013. View Article : Google Scholar
|
|
18
|
Wang B, Chen H, Jiang X, Zhang M, Wan T,
Li N, Zhou X, Wu Y, Yang F, Yu Y, et al: Identification of an
HLA-A*0201-restricted CD8+ T-cell epitope SSp-1 of SARS-CoV spike
protein. Blood. 104:200–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo YJ, Wang KY and Sun SH: Identification
of an HLA-A*0201-restricted CD8(+) T-cell epitope encoded within
Leptospiral immunoglobulin like protein. A Microbes Infect.
12:364–373. 2010. View Article : Google Scholar
|
|
20
|
Dervillez X, Qureshi H, Chentoufi AA, Khan
AA, Kritzer E, Yu DC, Diaz OR, Gottimukkala C, Kalantari M,
Villacres MC, et al: Asymptomatic HLA-A*02:01-restricted epitopes
from herpes simplex virus glycoprotein B preferentially recall
polyfunctional CD8+ T-cells from seropositive asymptomatic
individuals and protect HLA transgenic mice against ocular herpes.
J Immunol. 191:5124–5138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saini SK, Ostermeir K, Ramnarayan VR,
Schuster H, Zacharias M and Springer S: Dipeptides promote folding
and peptide-binding of MHC class I molecules. Proc Natl Acad Sci
USA. 110:15383–15388. 2013. View Article : Google Scholar
|
|
22
|
Imai K, Hirata S, Irie A, Senju S, Ikuta
Y, Yokomine K, Harao M, Inoue M, Tomita Y, Tsunoda T, et al:
Identification of HLA-A2-restricted CTL epitopes of a novel
tumour-associated antigen, KIF20A, overexpressed in pancreatic
cancer. Br J Cancer. 104:300–307. 2011. View Article : Google Scholar :
|
|
23
|
Wu YH, Gao YF, He YJ, Shi RR, Zhai MX, Wu
ZY, Sun M, Zhai WJ, Chen X and Qi YM: A novel cytotoxic T
lymphocyte epitope analogue with enhanced activity derived from
cyclooxygenase-2. Scand J Immunol. 76:278–285. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu B, Chen Z, Cheng X, Lin Z, Guo J, Jia
Z, Zou L, Wang Z, Hu Y, Wang D, et al: Identification of
HLA-A*0201-restricted cytotoxic T lymphocyte epitope from TRAG-3
antigen. Clin Cancer Res. 9:1850–1857. 2003.PubMed/NCBI
|
|
25
|
Imahashi N, Nishida T, Ito Y, Kawada J,
Nakazawa Y, Toji S, Suzuki S, Terakura S, Kato T, Murata M, et al:
Identification of a novel HLA-A*24:02-restricted adenovirus
serotype 11-specific CD8+ T-cell epitope for adoptive
immunotherapy. Mol Immunol. 56:399–405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paul S, Weiskopf D, Angelo MA, Sidney J,
Peters B and Sette A: HLA class I alleles are associated with
peptide-binding repertoires of different size, affinity, and
immunogenicity. J Immunol. 191:5831–5839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gajewski TF: Cancer immunotherapy. Mol
Oncol. 6:242–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weber G, Gerdemann U, Caruana I, Savoldo
B, Hensel NF, Rabin KR, Shpall EJ, Melenhorst JJ, Leen AM, Barrett
AJ, et al: Generation of multi-leukemia antigen-specific T cells to
enhance the graft-versus-leukemia effect after allogeneic stem cell
transplant. Leukemia. 27:1538–1547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vesely MD and Schreiber RD: Cancer
immunoediting: Antigens, mechanisms, and implications to cancer
immunotherapy. Ann NY Acad Sci. 1284:1–5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mishra S and Sinha S: Immunoinformatics
and modeling perspective of T cell epitope-based cancer
immunotherapy: A holistic picture. J Biomol Struct Dyn. 27:293–306.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matoskova B, Wong WT, Salcini AE, Pelicci
PG and Di Fiore PP: Constitutive phosphorylation of eps8 in tumor
cell lines: Relevance to malignant transformation. Mol Cell Biol.
15:3805–3812. 1995.PubMed/NCBI
|
|
32
|
Li YH, Xue TY, He YZ and Du JW: Novel
oncoprotein EPS8: A new target for anticancer therapy. Future
Oncol. 9:1587–1594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kessler JH and Melief CJ: Identification
of T-cell epitopes for cancer immunotherapy. Leukemia.
21:1859–1874. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vigneron N and Van den Eynde BJ: Insights
into the processing of MHC class I ligands gained from the study of
human tumor epitopes. Cell Mol Life Sci. 68:1503–1520. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dönnes P and Kohlbacher O: Integrated
modeling of the major events in the MHC class I antigen processing
pathway. Protein Sci. 14:2132–2140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Castelli M, Cappelletti F, Diotti RA,
Sautto G, Criscuolo E, Dal Peraro M and Clementi N: Peptide-based
vaccinology: Experimental and computational approaches to target
hyper-variable viruses through the fine characterization of
protective epitopes recognized by monoclonal antibodies and the
identification of T-cell-activating peptides. Clin Dev Immunol.
2013:5212312013. View Article : Google Scholar
|
|
37
|
Lundegaard C, Nielsen M and Lund O: The
validity of predicted T-cell epitopes. Trends Biotechnol.
24:537–538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sieker F, May A and Zacharias M:
Predicting affinity and specificity of antigenic peptide binding to
major histocompatibility class I molecules. Curr Protein Pept Sci.
10:286–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lundegaard C, Lund O, Buus S and Nielsen
M: Major histocompatibility complex class I binding predictions as
a tool in epitope discovery. Immunology. 130:309–318. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Viatte S, Alves PM and Romero P: Reverse
immunology approach for the identification of CD8 T-cell-defined
antigens: Advantages and hurdles. Immunol Cell Biol. 84:318–330.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kessler JH, Mommaas B, Mutis T, Huijbers
I, Vissers D, Benckhuijsen WE, Schreuder GM, Offringa R, Goulmy E,
Melief CJ, et al: Competition-based cellular peptide binding assays
for 13 prevalent HLA class I alleles using fluorescein-labeled
synthetic peptides. Hum Immunol. 64:245–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cole DK, Edwards ES, Wynn KK, et al:
Modification of MHC anchor residues generates heteroclitic peptides
that alter TCR binding and T cell recognition. J Immunol.
185:2600–2610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Assarsson E, Sidney J, Oseroff C, et al: A
quantitative analysis of the variables affecting the repertoire of
T cell specificities recognized after vaccinia virus infection. J
Immunol. 178:7890–7901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jørgensen KW, Rasmussen M, Buus S and
Nielsen M: NetMHCstab-predicting stability of peptide-MHC-I
complexes; impacts for cytotoxic T lymphocyte epitope discovery.
Immunology. 141:18–26. 2014. View Article : Google Scholar
|
|
45
|
Harndahl M, Rasmussen M, Roder G, Dalgaard
Pedersen I, Sørensen M, Nielsen M and Buus S: Peptide-MHC class I
stability is a better predictor than peptide affinity of CTL
immunogenicity. Eur J Immunol. 42:1405–1416. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Strous GJ, van Kerkhof P, van Meer G,
Rijnboutt S and Stoorvogel W: Differential effects of brefeldin A
on transport of secretory and lysosomal proteins. J Biol Chem.
268:2341–2347. 1993.PubMed/NCBI
|
|
47
|
Misumi Y, Misumi Y, Miki K, Takatsuki A,
Tamura G and Ikehara Y: Novel blockade by brefeldin A of
intracellular transport of secretory proteins in cultured rat
hepatocytes. J Biol Chem. 261:11398–11403. 1986.PubMed/NCBI
|
|
48
|
Okuyama R, Aruga A, Hatori T, Takeda K and
Yamamoto M: Immunological responses to a multi-peptide vaccine
targeting cancer-testis antigens and VEGFRs in advanced pancreatic
cancer patients. OncoImmunology. 2:e270102013. View Article : Google Scholar
|
|
49
|
Laugel B, van den Berg HA, Gostick E, Cole
DK, Wooldridge L, Boulter J, Milicic A, Price DA and Sewell AK:
Different T cell receptor affinity thresholds and CD8 coreceptor
dependence govern cytotoxic T lymphocyte activation and tetramer
binding properties. J Biol Chem. 282:23799–23810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhong S, Malecek K, Johnson LA, Yu Z,
Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J,
Corse E, et al: T-cell receptor affinity and avidity defines
antitumor response and autoimmunity in T-cell immunotherapy. Proc
Natl Acad Sci USA. 110:6973–6978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Corse E, Gottschalk RA, Krogsgaard M and
Allison JP: Attenuated T cell responses to a high-potency ligand in
vivo. PLoS Biol. 8:e10004812010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Slansky JE and Jordan KR: The Goldilocks
model for TCR-too much attraction might not be best for vaccine
design. PLoS Biol. 8:e10004822010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hebeisen M, Baitsch L, Presotto D,
Baumgaertner P, Romero P, Michielin O, Speiser DE and Rufer N:
SHP-1 phosphatase activity counteracts increased T cell receptor
affinity. J Clin Invest. 123:1044–1056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alexander Miller MA, Leggatt GR and
Berzofsky JA: Selective expansion of high- or low-avidity cytotoxic
T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl
Acad Sci USA. 93:4102–4107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zeh HJ 3rd, Perry-Lalley D, Dudley ME,
Rosenberg SA and Yang JC: High avidity CTLs for two self antigens
demonstrate superior in vitro and in vivo antitumor efficacy. J
Immunol. 162:989–994. 1999.PubMed/NCBI
|
|
56
|
Brentville VA, Metheringham RL, Gunn B and
Durrant LG: High avidity cytotoxic T lymphocytes can be selected
into the memory pool but they are exquisitely sensitive to
functional impairment. PLoS One. 7:e411122012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ho WY, Nguyen HN, Wolfl M, Kuball J and
Greenberg PD: In vitro methods for generating CD8+ T-cell clones
for immunotherapy from the naïve repertoire. J Immunol Methods.
310:40–52. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kang YJ, Zeng W, Song W, Reinhold B, Choi
J, Brusic V, Yamashita T, Munshi A, Li C, Minvielle S, et al:
Identification of human leucocyte antigen (HLA)-A*0201-restricted
cytotoxic T lymphocyte epitopes derived from HLA-DOβ as a novel
target for multiple myeloma. Br J Haematol. 163:343–351. 2013.
View Article : Google Scholar : PubMed/NCBI
|