Introduction
It is now believed that autophagy has broader
importance in regulating growth and maintaining homeostasis in
multicellular organisms (1). In
the course of autophagy, an isolation membrane forms, which is
thought to arise from a vesicular compartment, invaginates and
sequesters cytoplasmic constituents to form a double or
multimembranous structure, known as the autophagosome. The outer
membrane of the autophagosome fuses with the lysosome to deliver
the inner membranous vesicle to the lumen of the degradative
compartment. Degradation of the sequestered material generates
nucleotides, amino acids and free fatty acids that are recycled for
macromolecular synthesis and adenosine triphosphate generation
(2). At the level of the organism
and the cell, autophagy can, paradoxically, have pro-death or
pro-survival functions depending on the context. Autophagy mainly
has an adaptive role to protect organisms during periods of
enhanced cellular distress (3).
However, the pro-survival functions of autophagy may be deleterious
and lead to cell death, which is termed type II cell death
(4).
One of the most-studied mammalian autophagy proteins
that falls into the category of vesicle nucleation is beclin-1
(5). Beclin-1 interacts with class
III phosphoinositide 3 kinase (PI3K), Vps34, and is involved in
autophagic vesicle nucleation. However, under favorable
physiological conditions, anti-apoptotic members of the B-cell
lymphoma 2 (Bcl-2) family, including Bcl-2 and Bcl-extra large
(Bcl-XL), interact with beclin-1 (through the BH3 domain), thus
inhibiting its proautophagic function (4,6,7). It
has been reported that beclin-1 is involved in several other
biological functions and in pathological conditions, including
heart disease, pathogen infection, development and
neurodegeneration (8).
Cd is a significant toxic and carcinogenic element
that is widely present in the environment (9). This heavy metal can cause
neurological and behavioral impairment, leading to memory deficits
(10), alteration of social
contact (11), olfactory
dysfunction (12), synaptic
function and neurotransmission (13,14).
Most studies conducted using cell models have focused on the
induction of severe apoptosis and necrosis by Cd (15,16)
and, as a result, the role of autophagy has remained elusive. The
present study sought to determine whether autophagy is responsible
for the cytotoxicity of Cd and investigated the mechanisms
underlying autophagic pathways in rat cerebral cortical
neurons.
Materials and methods
Materials
Cd (CH3COO)2×3H2O
(Cd), poly-L-lysine, chloroquine (CQ), rapamycin (RAP) and MTT were
purchased from Sigma-Aldrich (St. Louis, MO, USA). NEUROBASAL™
medium, B27 supplement and Dulbecco’s modified Eagle’s medium
(DMEM)-F12 (1:1) were purchased from Invitrogen Life Technologies
(Carlsbad, CA, USA), while PI3K inhibitor LY294002 (LY). Monoclonal
rabbit anti-class III PI3K (cat. no. 3358), anti-Bcl-2 (cat. no.
2870), anti-β-actin (cat. no. 4970), horseradish peroxidase
(HRP)-conjugated goat anti-rabbit (cat. no. 7074) and goat
anti-mouse (cat. no. 7056) immunoglobulin (Ig)G antibodies, and
fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG (cat.
no. 4412) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Monoclonal mouse anti-beclin-1 antibody (cat.
no. sc48341) was purchased from Santa Cruz Biotechnology (Dallas,
TX, USA). Polyclonal rabbit anti-LC3B antibody (cat. no. L7543) was
purchased from Sigma-Aldrich.
Cell isolation and culture
The present study was performed in strict accordance
with the recommendations of the Guide for the Care and Use of
Laboratory Animals of the National Research Council. All procedures
described in the present study were reviewed and approved by the
Animal Care and Use Committee of Yangzhou University [Yangzhou,
China; approval ID: SYXK (Su) 2007-0005].
Primary cerebral cortical neurons were isolated from
13 embryos removed from pregnant Sprague-Dawley rats (Laboratory
Animal Center in Yangzhou University, Yangzhou, China) at 17–18
days of gestation as described previously (17). The cells were used for experiments
after five days of culture.
Immunofluorescence
Cortical neuronal cells were seeded at a density of
2.5×105 cells/ml in six-well plates containing a glass
coverslip in each well and treated with 0 or 20 μmol/l Cd
for 4 or 6 h. After being washed with phosphate-buffered saline
(PBS; pH 7.2–7.4), cells were fixed with 4% paraformaldehyde
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China) in PBS
for 30 min at 4°C. Cells were then permeabilized for 10 min with
0.5% Triton X-100 (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) in PBS and blocked with bovine serum albumin
(Beijing Solarbio Science & Technology Co., Ltd.) in PBS for 20
min at room temperature. Cells were incubated with anti-LC3B rabbit
polyclonal antibody (diluted 1:150) in blocking solution for 2 h at
room temperature, washed in blocking solution and then stained with
FITC-conjugated anti-rabbit IgG (diluted 1:200) in blocking
solution for 1 h. Cell nuclei were then stained by DAPI
(Sigma-Aldrich). After being washed, samples were examined under a
fluorescence microscope (Leica DMI 3000B; Leica Microsystems,
Wetzlar, Germany).
Electron microscopy
For transmission electron microscopy (TEM), after
being treated with 0 and 20 μmol/l Cd for 4 h, the cells
were trypsinized, collected and centrifuged. The pellets were
immediately fixed in ice-cold glutaraldehyde (2.5% in 0.1 mol/l
cacodylate buffer, pH 7.4; Nanjing Jiancheng Bioengineering
Institute) for 24 h, and the cells were post-fixed in osmium
tetroxide (Tianjing Dengke Chemical Reagent Co., Ltd., Tianjing,
China). After dehydration with a graded series of ethanol (Nanjing
Jiancheng Bioengineering Institute), the samples were rinsed in
propylene oxide (Tianjing Dengke Chemical Reagent Co., Ltd.) and
impregnated with epoxy resins (Tianjing Dengke Chemical Reagent
Co., Ltd.). The ultrathin sections were contrasted with uranyl
acetate and lead citrate (Tianjing Dengke Chemical Reagent Co.,
Ltd.) for electron microscopy. Electron micrographs were captured
using a PHILIPS CM-120 (Philips, Eindhoven, The Netherlands)
transmission electron microscope.
Western blot analysis
After treatment, cells were washed twice with cold
PBS and extracted using radioimmunoprecipitation lysis buffer
(Beijing Solarbio Science & Technology Co., Ltd.) on ice for 30
min, sonicated for 10 sec and centrifuged at 12,000 × g for 10 min
at 4°C. The protein content was determined using a bicinchoninic
acid protein assay kit (Beyotime Institute of Biotechnology,
Shanghai, China). Lysates were diluted with 6X SDS sample buffer
(Beijing Solarbio Science & Technology Co., Ltd.) and boiled
for 10 min. Equal amounts of protein were separated using 12–15%
SDS-PAGE and transferred onto nitrocellulose membranes (Beijing
Solarbio Science & Technology Co., Ltd.). After being blocked
at room temperature with 5% non-fat milk in Tris-buffered saline
with 0.1% Tween-20 (Beijing Solarbio Science & Technology Co.,
Ltd.), the membranes were incubated overnight at 4°C with the
primary antibodies to LC3B (1:1,000), beclin-1 (1:2,000), class III
PI3K (1:1,000), Bcl-2 (1:1,000) and β-actin (1:2,000), followed by
incubation with the appropriate secondary antibody conjugated to
HRP (1:7,500) at room temperature for 2 h. The membranes were
visualized using an enhanced chemiluminescence detection kit
(Thermo Fisher Scientific, Waltham, MA, USA) and then exposed to
X-ray film (Carestream Health, Xiamen, China). The relative band
size was determined by standard scanning densitometry and values
were normalized to β-actin.
MTT assay for cell viability
Cells were seeded at a density of 2×105
cells/ml in a flat-bottom- 96-well plate. After treatment, MTT
solution (5.0 g/l in PBS) was added to the culture media at a final
concentration of 500 μg/ml, and the cells were incubated for
4 h at 37°C. The supernatant was then discarded, and 150 μl
dimethyl sulfoxide (Tianjing Dengke Chemical Reagent Co., Ltd.) was
added to dissolve the formazan. The absorbance was measured at
570/630 nm using a microplate reader (Sunrise; Tecan Group Ltd,
Männedorf, Switzerland).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Significance was assessed by one-way analysis of
variance following appropriate transformation to normalized data
and equalized variance where necessary. Statistical analysis was
performed using SPSS 19.0 (International Business Machines, Armonk,
USA). P<0.05 was considered to indicate a statistically
significant difference between values.
Results
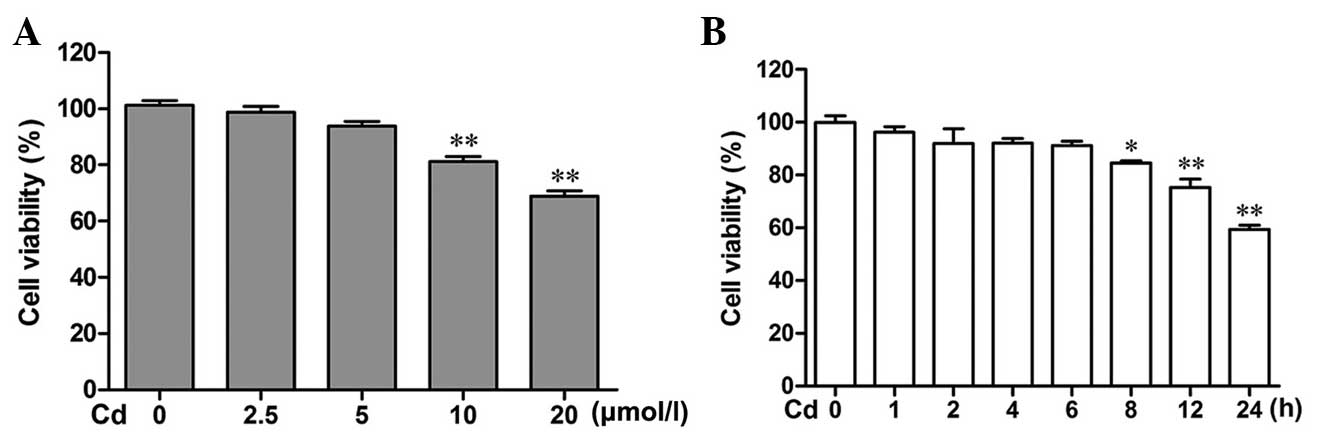
Cd decreases the viability of cerebral
cortical neurons
The effect of Cd on the viability of cerebral
cortical neuron cells was investigated by MTT assays. As shown in
Fig. 1A and B, Cd treatment caused
a decrease in cell viability in a dose- and time-dependent manner.
Cd caused a significant decrease in cell viability at 10 and 20
μmol/l when applied for 24 h, compared with that in the
control group (P<0.01). Treatment with 20 μmol/l Cd for
0, 1, 2, 4 and 6 h had no inhibitory effect. However, 8, 12 and 24
h after treatment with 20 μmol/l Cd, an inhibitory effect
was obvious (P<0.05, P<0.01 and P<0.01, respectively).
Activation of autophagy in cerebral
cortical neurons after Cd treatment
Autophagy induction in Cd-treated cerebral cortical
neuron cells was determined via detection of LC3 using western blot
analysis, immunofluorescence staining and TEM. As shown in Fig. 2A, the levels of LC3-II protein
increased after Cd treatment at 10 and 20 μmol/l
(P<0.01). In the case of treatment with 20 μmol/l Cd,
there was a significant increase in LC3-II protein levels after
treatment for 4 h, 6 h (P<0.01) and 8 h (P<0.05), but not
after shorter (2 h) or longer (12 and 24 h) treatments (Fig. 2B). The distribution of endogenous
LC3-II staining in cells prior to and after 4 h of Cd treatment was
monitored by indirect immunofluorescence staining (Fig. 2C). Specific punctate distribution
of endogenous LC3-II was observed in cortical neurons treated with
Cd; more dots appeared in Cd-treated neurons than in control
neurons (P<0.01; Fig. 2D).
Single-membrane vacuoles, whorls of membranous material and other
ultrastructures were observed in Cd-treated neurons (4 h), which
were not present in control neurons (Fig. 2E; a–c). However, after Cd treatment
for 24 h, signs of apoptosis, including chromatin condensation
along the nuclear envelope and vacuole formation in the cytoplasm,
were also observed (Fig. 2E; d–f).
These results indicated that Cd treatment induced autophagy and
apoptosis in cortical neurons.
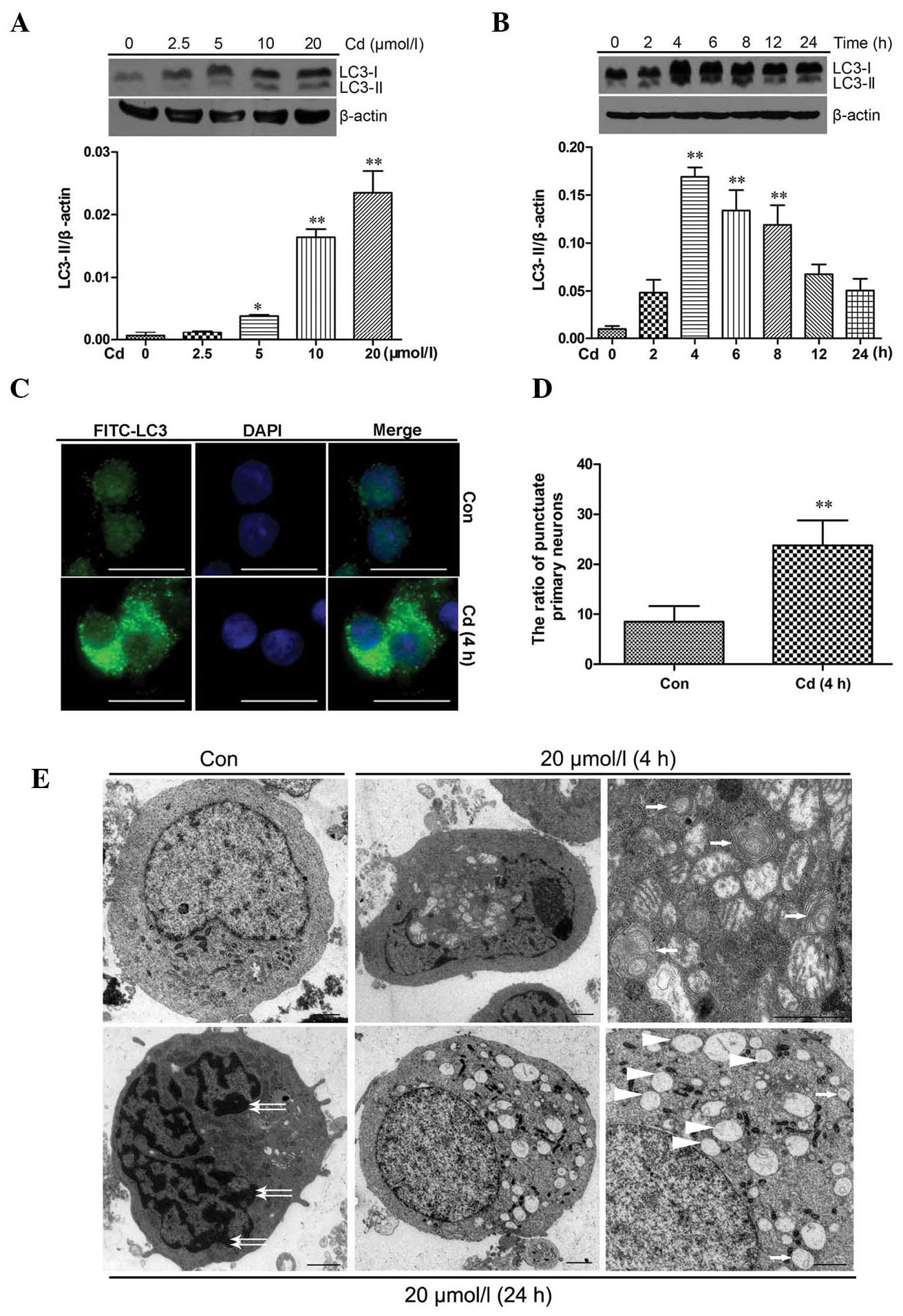 | Figure 2Activation of autophagy in cerebral
cortical neurons after Cd treatment. Cerebral cortical neurons were
treated with (A) 0–20 μmol/l Cd for 4 h or (B) with 20
μmol/l Cd for different durations, and cell lysates were
analyzed by western blot analysis to detect LC3 expression (upper
panels of A and B). Relative band widths were analyzed, quantified
and normalized to β-actin levels (lower panels of A and B). Values
are expressed as the mean ± standard deviation from at least three
independent experiments. *P<0.05,
**P<0.01 vs. control group. (C) Cerebral cortical
neurons were treated with 20 μmol/l Cd for 4 h and subjected
to indirect immunofluorescence staining (scale bar, 10 μm).
(D) Histogram showing the number of cerebral cortical neurons with
punctate FITC-LC3 staining totaling ~300 cells. Values are
expressed as the mean ± standard deviation
(**P<0.01). (E) Transmission electron microscopy
images of cerebral cortical neurons. (a) Control cell; (b and c)
cells treated with 20 μmol/l Cd for 4 h at low and high
power, respectively. The lower panels show cells treated with 20
μmol/l Cd for 24 h at (d and e) low power and (f) high
power. Autophagosomes are indicated by white arrows in (c) and (f),
whereas apoptotic changes such as chromatin condensation (white
double-headed arrows) and cytoplasmic vacuolization (white
arrowheads) are highlighted in (d) and (f), respectively (scale
bar, 1 μm). FITC, fluorescein isothiocyanate; LC-3,
microtubule-associated protein 1A/1B-light chain 3; Con,
control. |
Induction of autophagy reduces Cd-induced
cytotoxicity
Numerous studies have demonstrated that autophagy
can serve as a protective response to prevent heavy metal-induced
cell death (18). CQ, an inhibitor
of autophagy, can inhibit the autophagic process by preventing
lysosome-autophagosome fusion, which leads to a remarkable
accumulation of autophagic vacuoles (19). As LC3-II is associated with
autophagic vacuoles, CQ treatment also induces intense LC3-II
accumulation (20). RAP, an
inducer of autophagy, has been shown to induce autophagy by
inhibiting the mammalian target of rapamycin pathway (21). Thus, CQ and RAP were employed in
the present study to detect the effect of autophagy on Cd-induced
cytotoxicity. The results indicated that CQ and RAP markedly
increased LC3-II production (Fig.
3A–D). An MTT assay was used to monitor the effects of CQ and
RAP on cell viability. Treatment with CQ or RAP alone did not
significantly inhibit cell viability, but in combination with Cd,
CQ significantly augmented the decrease in cell viability observed
with Cd alone (Fig. 3E). However,
RAP markedly reduced the Cd-induced decrease in cell viability
(Fig. 3F), confirming that
Cd-mediated autophagy has a cytoprotective effect.
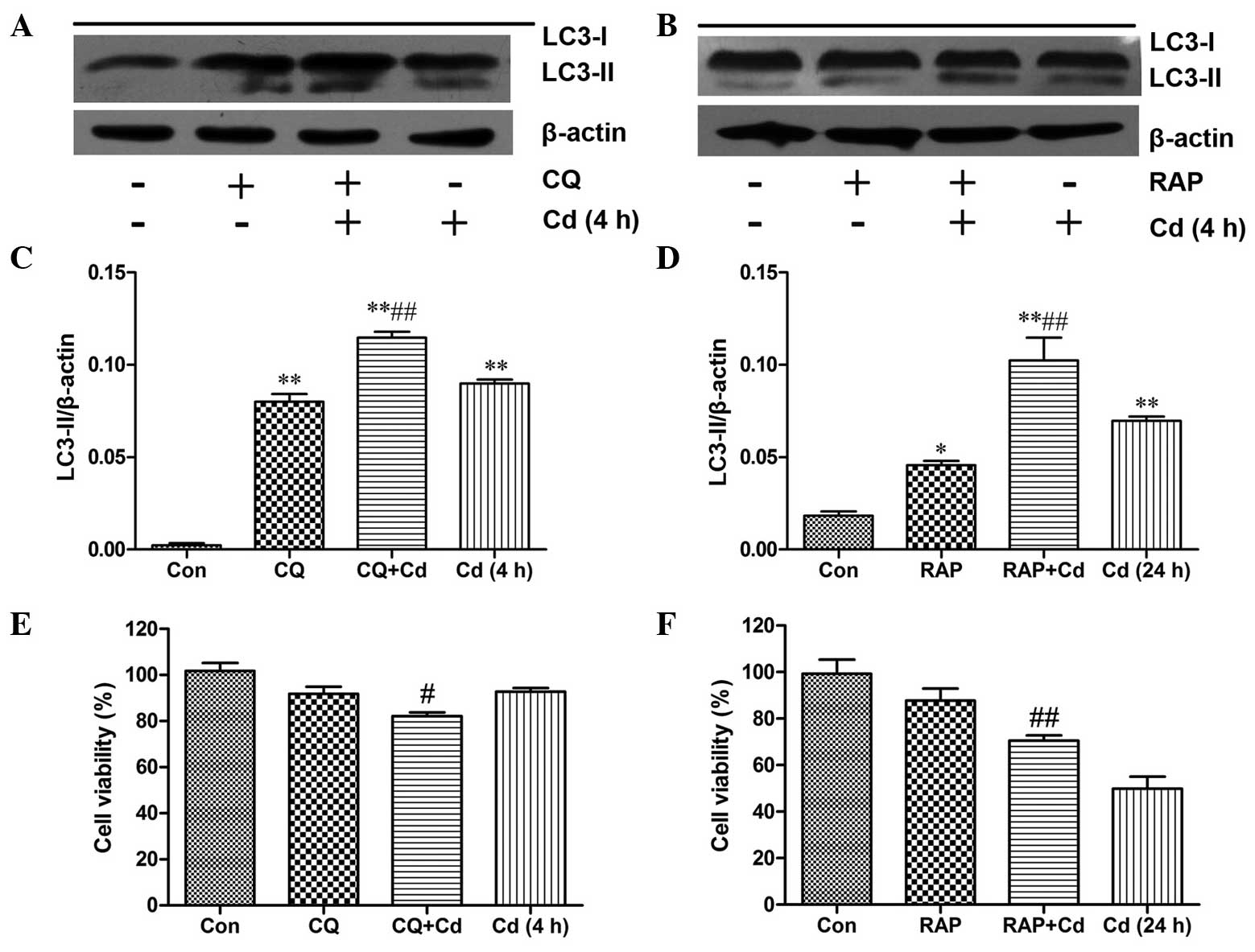 | Figure 3Induction of autophagy reduces the
Cd-mediated cytotoxicity. (A) Cerebral cortical neurons were
pre-treated with 5 μmol/l CQ for 0.5 h, followed by
treatment with 20 μmol/l Cd for 4 h, or (B) pre-treated with
100 nmol/l RAP for 24 h, followed by treatment with 20
μmol/l Cd for another 24 h. Western blot analysis was then
performed to measure LC3 protein expression. The blots were also
probed for β-actin as a loading control. Relative band widths were
analyzed, quantified and normalized to β-actin levels (n=3). (C and
D) MTT assays were performed to determine cell viability following
treatment with Cd alone, (E) in combination with CQ or (F) in
combination with RAP. Values are expressed as the mean ± standard
deviation (n=4–6). *P<0.05, **P<0.01,
compared with control group; #P<0.05,
##P<0.01 compared to the respective Cd group. LC-3,
microtubule-associated protein 1A/1B-light chain 3; Rap, rapamycin;
CQ, chloroquine; Con, control. |
The class III PI3K/beclin-1/Bcl-2
signaling pathway contributes to autophagy activation
To investigate the signaling pathways involved in
the induction of autophagy in Cd-treated cerebral cortical neuron
cells, class III PI3K, beclin-1 and Bcl-2 were examined by
immunoblot analysis. In cells treated with 20 μmol/l Cd for
0–24 h (Fig. 4A and B), the
protein levels of beclin-1 and class III PI3K were significantly
upregulated and peaked at 12 h after Cd treatment. Bcl-2 levels
were initially decreased and then slightly increased; overall,
however, Bcl-2 levels exhibited a downward trend within 24 h. The
results suggested that the class III PI3K/beclin-1/Bcl-2 pathway
may be involved in the autophagy of cerebral cortical neuronal
cells.
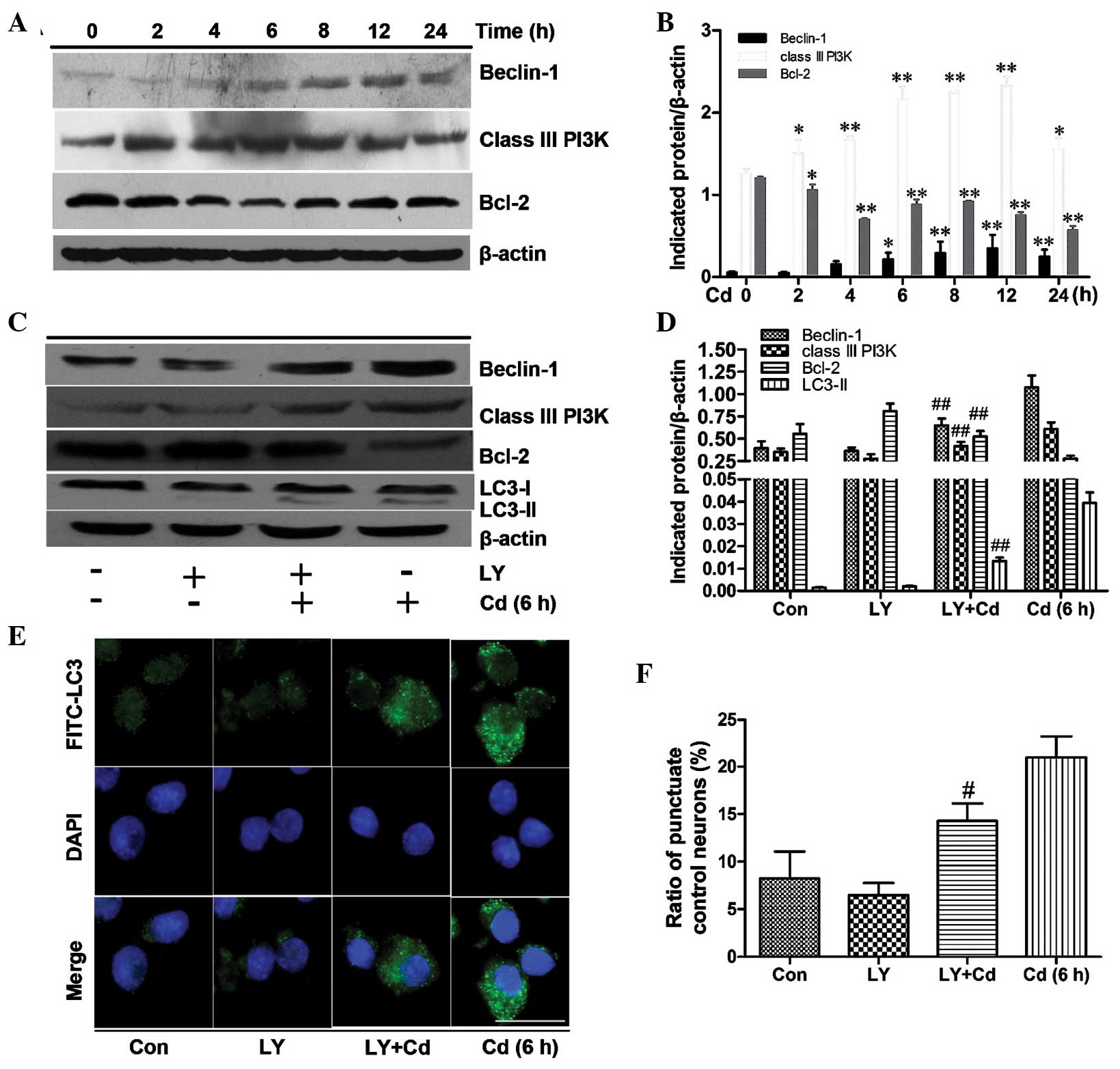 | Figure 4The class III PI3K/beclin-1/Bcl-2
signaling pathway contributes to autophagy activation. (A) Cerebral
cortical neurons were treated with 20 μmol/l Cd for various
durations and cell lysates were analyzed by western blot with the
indicated antibodies. β-actin was used as a loading control. (B)
Indicated proteins from A were semi-quantified using Image Lab
software. Values are expressed as the mean ± standard deviation of
results from three independent experiments. *P<0.05,
**P<0.01 vs. control group. (C) Cortical neurons were
harvested following 6 h of treatment with (or without) 20
μmol/l Cd in the presence or absence of 20 μmol/l LY.
Cell lysates were analyzed by immunoblot assay with indicative
antibodies with β-actin used as a loading control. (D) The optical
densities of the respective protein bands in C were analyzed using
Image Lab and normalized to the loading control (β-actin). Values
are expressed as the mean ± standard deviation of date from three
independent experiments. ##P<0.01 compared to the Cd
group. (E) Immunofluorescence analysis. Cortical neurons were
treated for 6 h with (or without) 20 μmol/l Cd in the
presence or absence of 20 μmol/l LY and incubated with
anti-LC3, FITC-conjugated secondary antibody, and then DAPI to
stain the nuclei (scale bar, 10 μm). (F) Quantified cerebral
cortical neurons with punctate FITC-LC3 from E; values are
expressed as mean ± standard deviation. #P<0.05
compared to the Cd group. Bcl-2, B-cell lymphoma 2; PI3K,
phosphoinositide 3 kinase; LC-3, microtubule-associated protein
1A/1B-light chain 3; Ly, PI3K inhibitor LY294002; Con, control;
FITC, fluorescein isothiocyanate. |
To further explore the association between the class
III PI3K/beclin-1/Bcl-2 pathway and autophagy, LY, a PI3K
inhibitor, was applied during Cd treatment. The results
demonstrated that a concentration of 20 μmol/l LY
significantly decreased the expression of beclin-1, class III PI3K
and LC3-II and promoted Bcl-2 protein expression in Cd-treated
cerebral cortical neurons (Fig. 4C and
D). Concomitantly, autophagocytosis was also obviously
decreased after treatment with Cd and LY compared to that following
treatment with Cd alone (Fig.
4C–F). These results indicated that the decreased expression of
beclin-1 and class III PI3K or the increased expression of Bcl-2 by
a PI3K inhibitor inhibits autophagy in Cd-injured cerebral cortical
neurons.
Discussion
Cd is a neurotoxic metal known to be a major
environmental contaminant. The potential impact of Cd on the
nervous system has long been recognized (22). A recent study by our group
demonstrated that exposure of PC-12 cells to Cd stimulates
autophagy, which can delay the occurrence of apoptosis (23). The present study provided further
evidence that Cd can induce cytoprotective autophagy by activating
the class III PI3K/beclin-1/Bcl-2 signaling pathway in rat cerebral
cortical neurons.
Recent reports demonstrated that the autophagy
process is important for the cellular response to oxidative stress,
as it helps to conserve cellular energy as well as to limit damaged
proteins and organelles that amplify toxic signals (24–26);
therefore, the present study hypothesized that Cd-induced autophagy
is a housekeeping process that protects neurons from damage at the
early stage of Cd exposure. This hypothesis was verified by the
observed CQ-mediated inhibition of autophagy and decrease in cell
viability, as well as the RAP-induced activation of autophagy and
recovery of cell viability. These results indicated that cerebral
cortical neurons can adapt to handle Cd-induced stress through the
removal and recycling of damaged proteins and organelles. Similar
results were also found following Cd treatment in hematopoietic
stem cells (27), lung epithelial
fibroblasts (28), rat kidney
NRK-52E cells (29) and proximal
convoluted tubule cells (30), but
not in mesangial cells (31) and
skin epidermal cells (32) treated
with Cd. The differences in autophagy induction by Cd in these
studies may be due to differential Cd concentration and duration of
exposure as well as variability in the metabolic state and cell
type studied. In the present study, autophagy did not completely
protect cells from damage caused by Cd treatment as demonstrated by
the MTT assay. Other processes that may be involved in the
Cd-induced reduction in cell viability are damage to DNA-repairing
systems (33) and activation of
P53, which leads to cell cycle arrest and the induction of cell
death (34). In the late stages of
autophagy, the features of apoptosis, including chromatin
condensation and cytoplasmic vacuolization, were observed using
TEM, indicating that Cd causes autophagy as well as apoptosis,
depending on the CD concentration and duration of treatment. It may
be postulated that upon initial exposure to Cd, autophagy is
induced, but at later stages, when the damage becomes irreversible,
apoptosis takes over. Further studies are required to investigate
the association between Cd treatment, autophagy and apoptosis.
Various signaling pathways are thought to be
involved in regulating autophagy induced by Cd (28,32,35),
but the molecular mechanisms that govern Cd-induced autophagy have
yet to be fully elucidated. This inspired the present study to
investigate which signaling pathways are responsible for Cd-induced
autophagy in cerebral cortical neurons. In the present study, it
was demonstrated that class III PI3K/beclin-1/Bcl-2 signaling has a
major role in regulating Cd-induced autophagy. Of note, the
decrease of Bcl-2 expression preceded the elevation of beclin-1,
suggesting that a decrease in Bcl-2 facilitates the release of
beclin-1, allowing it to form a complex with class III PI3K, which
is required for regulating autophagy (36,37).
The present study further confirmed that disrupting Bcl-2/beclin-1
interactions may be a mechanism used by cells to promote autophagy
and cell survival during conditions of stress (38,39).
However, the results showed that after a rapid decline during the
first hours of Cd treatment, Bcl-2 levels began to rise again after
longer durations of Cd treatment. One of the possible explanations
for this is that cells initiate compensatory mechanisms under
stressful conditions (6). Further
studies are required to elucidate the detailed mechanism of how
Bcl-2 regulates Cd-induced autophagy.
In conclusion, the results of the present study
suggested that Cd induces a decrease in cell viability and
initiates autophagy (in the early stages) in rat cerebral cortical
neurons. The results also suggested that autophagy may have an
important protective role in Cd-induced cytotoxicity by activating
class III PI3K/beclin-1/Bcl-2 signaling pathways. These results
provided essential preliminary data for the further assessment of
Cd-induced neurotoxicity.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 31302058, 31101866 and
31172373), a Project Funded by the Priority Academic Program
Development of Jiangsu Higher Education Institutions (PAPD) and the
Joint Foundation of Guizhou Province [LKB (2013) 02].
References
|
1
|
Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ and
Chiou HL: Dioscin-induced autophagy mitigates cell apoptosis
through modulation of PI3K/Akt and ERK and JNK signaling pathways
in human lung cancer cell lines. Arch Toxicol. 87:1927–1937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang M, Jeong CW, Ku JH, Kwak C and Kim
HH: Inhibition of autophagy potentiates atorvastatin-induced
apoptotic cell death in human bladder cancer cells in vitro. Int J
Mol Sci. 15:8106–8121. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang R, Zeh H, Lotze M and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex-at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maiuri MC, Le Toumelin G, Criollo A, et
al: Functional and physical interaction between Bcl-XL and a
BH3-like domain in Beclin-1. EMBO J. 26:2527–2539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He C and Levine B: The beclin 1
interactome. Curr Opin Cell Biol. 22:140–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Muneer S, Hakeem KR, Mohamed R and Lee JH:
Cadmium toxicity induced alterations in the root proteome of green
gram in contrasting response towards iron supplement. Int J Mol
Sci. 15:6343–6355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Łukawski K, Nieradko B and
Sieklucka-Dziuba M: Effects of cadmium on memory processes in mice
exposed to transient cerebral oligemia. Neurotoxicol Teratol.
27:575–584. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Curtis JT, Hood AN, Chen Y, Cobb GP and
Wallace DR: Chronic metals ingestion by prairie voles produces
sex-specific deficits in social behavior: An animal model of
autism. Behav Brain Res. 213:42–49. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mascagni R, Consonni D, Bregante G,
Chiappino G and Toffoletto F: Olfactory function in workers exposed
to moderate airborne cadmium levels. Neurotoxicology. 24:717–724.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fernández-Pérez B, Caride A, Cabaleiro T
and Lafuente A: Cadmium effects on 24 h changes in glutamate,
aspartate, glutamine, GABA and taurine content of rat striatum. J
Trace Elem Med Biol. 24:212–218. 2010. View Article : Google Scholar
|
|
14
|
Minami A, Takeda A, Nishibaba D, Takefuta
S and Oku N: Cadmium toxicity in synaptic neurotransmission in the
brain. Brain Res. 894:336–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
López E, Figueroa S, Oset-Gasque MJ and
González MP: Apoptosis and necrosis: Two distinct events induced by
cadmium in cortical neurons in culture. Brit J Pharmacol.
138:901–911. 2003. View Article : Google Scholar
|
|
16
|
Yuan Y, Jiang CY, Xu H, et al:
Cadmium-induced apoptosis in primary rat cerebral cortical neurons
culture is mediated by a calcium signaling pathway. PloS One.
8:e643302013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan Y, Bian JC, Zhong LX, Zhang Y, Sun Y
and Liu ZP: Oxidative stress and apoptotic changes of rat cerebral
cortical neurons exposed to cadmium in vitro. Biomed Environ Sci.
25:172–181. 2012.PubMed/NCBI
|
|
18
|
Chiarelli R and Roccheri MC: Heavy metals
and metalloids as autophagy inducing agents: Focus on cadmium and
arsenic. Cells. 1:597–616. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zaidi AU, McDonough JS, Klocke BJ, et al:
Chloroquine-induced neuronal cell death is p53 and Bcl-2
family-dependent but caspase-independent. J Neuropathol Exp Neurol.
60:937–945. 2001.PubMed/NCBI
|
|
20
|
Geng Y, Kohli L, Klocke BJ and Roth KA:
Chloroquine-induced autophagic vacuole accumulation and cell death
in glioma cells is p53 independent. Neuro Oncol. 12:473–481.
2010.PubMed/NCBI
|
|
21
|
Klionsky DJ, Abeliovich H, Agostinis P, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SM, Park JG, Baek WK, et al: Cadmium
specifically induces MKP-1 expression via the glutathione
depletion-mediated p38 MAPK activation in C6 glioma cells. Neurosci
Lett. 440:289–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Q, Zhu J, Zhang K, et al: Induction
of cytoprotective autophagy in PC-12 cells by cadmium. Biochem
Biophys Res Commun. 438:186–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jisun L, Samantha G and Jianhua Z:
Autophagy, mitochondria and oxidative stress: Cross-talk and redox
signalling. Biochem J. 441:523–540. 2012. View Article : Google Scholar
|
|
25
|
Benavides GA, Liang Q, Dodson M,
Darley-Usmar V and Zhang J: Inhibition of autophagy and glycolysis
by nitric oxide during hypoxia-reoxygenation impairs cellular
bioenergetics and promotes cell death in primary neurons. Free
Radic Bio Med. 65:1215–1228. 2013. View Article : Google Scholar
|
|
26
|
Duan WJ, Liu FL, He RR, et al: Autophagy
is involved in the effects of resveratrol on prevention of
splenocyte apoptosis caused by oxidative stress in restrained mice.
Mol Nutr Food Res. 57:1145–1157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Gioacchino M, Petrarca C, Perrone A,
Martino S, Esposito DL, Lotti LV and Mariani-Costantini R:
Autophagy in hematopoietic stem/progenitor cells exposed to heavy
metals. Autophagy. 4:537–539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lim SC, Hahm KS, Lee SH and Oh SH:
Autophagy involvement in cadmium resistance through induction of
multidrug resistance-associated protein and counterbalance of
endoplasmic reticulum stress WI38 lung epithelial fibroblast cells.
Toxicology. 276:18–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kato H, Katoh R and Kitamura M: Dual
regulation of cadmium-induced apoptosis by mtorc1 through selective
induction of IRE1 branches in unfolded protein response. PloS One.
8:e643442013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chargui A, Zekri S, Jacquillet G, et al:
Cadmium-induced autophagy in rat kidney: an early biomarker of
subtoxic exposure. Toxicol Sci. 121:31–42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang SH, Shih YL, Ko WC, Wei YH and Shih
CM: Cadmium-induced autophagy and apoptosis are mediated by a
calcium signaling pathway. Cell Mol Life Sci. 65:3640–3652. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Son YO, Wang X, Hitron JA, et al: Cadmium
induces autophagy through ROS-dependent activation of the LKB1-AMPK
signaling in skin epidermal cells. Toxicol Appl Pharmacol.
255:287–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giaginis C, Gatzidou E and Theocharis S:
DNA repair systems as targets of cadmium toxicity. Toxicol Appl
Pharmacol. 213:282–290. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu ZM, Chen GG, Vlantis A, Tse G, Shum C
and Van Hasselt C: Calcium-mediated activation of PI3K and p53
leads to apoptosis in thyroid carcinoma cells. Cell Mol Life Sci.
64:1428–1436. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang LY, Wu KH, Chiu WT, Wang SH and Shih
CM: The cadmium-induced death of mesangial cells results in
nephrotoxicity. Autophagy. 5:571–572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Simonsen A and Tooze SA: Coordination of
membrane events during autophagy by multiple class III PI3-kinase
complexes. J Cell Biol. 186:773–782. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fung C, Lock R, Gao S, Salas E and Debnath
J: Induction of autophagy during extracellular matrix detachment
promotes cell survival. Mol Biol Cell. 19:797–806. 2008. View Article : Google Scholar :
|