Introduction
Bupivacaine (BPV) is a sodium channel blocker
commonly used as a local anesthetic in order to reduce
postoperative pain. In addition, patients undergoing laparoscopic
surgery may be administered high doses of BPV as a postoperative
analgesic (1). Despite its
widespread usage in clinical settings, BPV may also cause rapid
cardiovascular collapse. Two mechanisms underlying BPV-induced
cardiotoxicity have been proposed: (i) Cardiotoxicity caused by
blockage of the sodium, calcium, and potassium ion channels, or
(ii) cardio-toxicity caused by BPV interference with mitochondrial
energy metabolism (2–4).
The mitochondrial apoptosis pathway is one of the
primary cellular processes that lead to cell death. An important
factor in mitochondrial apoptosis is the mitochondrial permeability
transition pore (mPTP). mPTP is a non-specific pore located in the
inner mitochondrial membrane, which opens in the presence of
elevated calcium concentrations, specifically when these elevated
calcium concentrations are accompanied by high levels of oxidative
stress, and depleted levels of adenine nucleotides (5). Opening of the mPTP causes alterations
in the ionic balance, which in turn leads to destruction of the
inner mitochondrial membrane, and release of cytochrome c
and apoptosis-inducing factors into the cytosol. These factors
result in cell death via caspase-dependent or independent cascade
reactions (6,7).
Elevated levels of mitochondrial calcium are
responsible for the opening of the mPTP (8). However, oxidative stress may induce
mPTP opening by increasing the sensitivity of the pore to calcium
(8). Destabilization of the ion
concentration within the mitochondrial membrane in turn leads to
apoptotic cell death. When cells are under oxidative stress, the
balance between the production and the elimination of reactive
oxygen species (ROS) is deregulated. The levels of ROS within the
cell have a significant role in cell survival. High levels of ROS
induce DNA damage and abnormal protein expression, which ultimately
lead to cell death (9). Another
biological marker of oxidative stress is malondialdehyde (MDA),
which is a product of polyunsaturated fatty acid peroxidation. MDA
has a long half-life and is highly reactive, which allows it to
interact with biomolecules such as nucleic acids and proteins,
often causing irreversibly damage to cellular function (10). The antioxidant defense mechanisms
that eliminate ROS are crucial for protecting the organism against
oxidative damage. Numerous enzymes exhibit antioxidative
properties, including super-oxide dismutase (SOD) and catalase
(CAT) (11). SOD carries out its
protective action by catalyzing the transformation of reactive
oxygen radicals into hydrogen peroxide and oxygen. CAT in turn
reduces hydrogen peroxide into non-toxic water and oxygen (12). The balance between SOD and CAT
activity is crucial to the maintenance of a steady-state between
toxic superoxide radicals and hydrogen peroxide (13).
Until recently, a cardiopulmonary bypass was the
only method shown to be effective in the treatment of refractory
cardiac arrest caused by local anesthetic overdose (14). Previous studies have demonstrated
that treatment with lipid emulsion (LE) attenuates BPV-induced
toxicity in various animal models (15,16),
and in the rat heart (17). Two
hypotheses have been proposed to explain the reversal effects of LE
on BPV-induced toxicity: (i) Lipid sink theory, or (ii) metabolic
theory. The lipid sink theory hypothesizes that the lipophilic
molecules contained within the local anesthetic partition into a
lipemic plasma compartment making them inaccessible to the
surrounding tissue, which in turn leads to an increase in the
concentration of local anesthetic within the plasma (18). However, Litz et al (19) demonstrated that lipid infusion
rapidly reduced serum anesthetic levels, which raises doubt
regarding the “lipid sink” hypothesis. The metabolic theory
hypothesizes that BPV inhibits fatty acid transport within the
inner mitochondrial membrane, and that the lipids prevent the
metabolites from being oxidized (20).
Although there have been numerous successful
clinical reports of LE attenuation of BPV-induced cardiac arrest,
the molecular mechanism underlying the rescue effects of LE has yet
to be elucidated. The present study used H9c2 rat neonatal myoblast
cells to investigate BPV-induced cardiotoxicity, and to elucidate
the mechanisms underlying the rescue effects of LE.
Materials and methods
Drugs and reagents
The 20% LE (C14-24; production batch 13010841) was
purchased from Libang Pharmaceutical Co., Ltd. (Xi'an, China), and
adjusted to a final concentration of 1% using Dulbecco's modified
Eagle's medium (DMEM). The BPV hydrochloride (production batch
121120) was purchased from Wuhu Kangqi Pharmaceutical Co., Ltd.
(Wuhu, China), and adjusted to a final concentration of 1 mM using
complete DMEM medium. Cyclosporin A (CsA) was purchased from
Sigma-Aldrich (St. Louis, MO, USA), and was dissolved in dimethyl
sulfoxide (DMSO) prior to cellular treatment at a final
concentration of 1 μM. Atractyloside (Atr) was purchased
from Nanjing Spring & Autumn Biological Engineering Co., Ltd.
(Nanjing, China). Atr purity (>98%) was determined using high
performance liquid chromatography, and dissolved in DMSO prior to
cellular treatment at a final concentration of 1 μM. Fetal
bovine serum (FBS), DMEM, penicillin, and streptomycin were
purchased from Gibco Life Technologies (Carlsbad, CA, USA). DMSO
and the MTT assay were purchased from Sigma-Aldrich. All primary
and secondary antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The MDA, CAT, and
dichloro-dihydro-flurorescein diacetate (DCFH-DA) ROS detection
kits were obtained from Beyotime Institute of Biotechnology
(Jiangsu, China). The terminal deoxy-nucleotidyl
transferase-mediated biotinylated dUTP nick end labeling (TUNEL)
kit was purchased from Roche Diagnostics Co. (Indianapolis, IN,
USA).
Cell culture
The H9c2 rat neonatal myoblast cells were obtained
from the American Type Culture Collection (Manassas, VA, USA). The
cells were maintained in high glucose DMEM supplemented with 10%
(v/v) heat-inactivated FBS, 100 U/ml penicillin, and 100
μg/ml streptomycin at 37°C in an atmosphere containing 5%
CO2. Once the cells had reached 70-80% confluence, the
media was replaced with DMEM containing 1% LE (LE group), DMEM
containing 1 mM BPV (BPV group), DMEM containing 1 mM BPV and 1% LE
(BPV+LE), or with fresh DMEM (control group).
Cell viability assay
Cell viability was determined using an MTT assay.
Briefly, the H9c2 myoblast cells were seeded into 96-well plates at
a density of 5×103/well. A total of 1 mM BPV was added
to the wells, and the cells were subsequently incubated for 0, 12,
24, and 36 h. A total of 10 μl MTT solution in 5 mg/ml
phosphate-buffered saline (PBS) was then added into each well, and
the cells were incubated at 37°C for a further 4 h. The medium was
discarded, following which 100 μl DMSO was added to each
well, and the plates were gently agitated for 5 min. Optical
density was determined at 590 nm using a Thermo MK3 microplate
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) (21). Cellular absorbance in
the absence of treatment was determined as 100%, indicating cell
survival. Each treatment was repeated four times, and each
experiment was performed in triplicate.
Intracellular ROS assay
ROS levels were measured using the non-fluorescent
DCFH-DA probe as previously described (22). DCFH-DA passively diffuses into
cells where it is deacetylated by esterases in order to form
non-fluorescent 2′-7′-dichlorofluorescein (DCFH). In the presence
of ROS, DCFH reacts to form the fluorescent product
dichlorofluorescein, which remains within the cells. Briefly, the
H9c2 myoblast cells were seeded at a density of
1×105/well into 96-well plates. Following a period of 24
h, the culture wells were treated with various treatment solutions
for 24 h, and the cells were washed three times with
phosphate-buffered saline. DCFH-DA, diluted to a final
concentration of 10 μM in DMEM without FBS, was added to the
cell cultures, which were then incubated for 20 min at 37°C.
Cellular fluorescence was measured at 485 nm excitation and 530 nm
emission using a Safire2 microplate reader (Tecan Schweiz, Uetikon,
Switzerland). An increase in the fluorescence values was determined
as an increase in intracellular ROS, as compared with the
control.
Total SOD (T-SOD) enzymatic activity
assay
T-SOD activity was determined using a WST T-SOD
Assay kit (Dojindo Molecular Technologies, Inc., Kumamoto, Japan),
according to the manufacturer's instructions (23). The T-SOD assay uses highly
water-soluble WST-1 tetrazolium salts that react with superoxide
anions (O2−) in order to produce a water-soluble
formazan dye. The H9c2 cells were seeded at a density of
1×105/well into 96-well plates. Following a period of 24
h the cells were treated with 1 mM BPV, 1% LE, or a combination of
1 mM BPV and 1% LE for 24 h. The cells were then washed three times
with PBS, and the samples were tested, and compared against a
standard curve ranging from 2–200 U/ml. The colorimetric assay is
performed by measuring the levels of formazan produced by the
reaction between WST-1 and O2−. The reaction reduction
rate is linearly correlated to xanthine oxidase activity, which is
inhibited by T-SOD. Cellular absorbance was measured using a
microplate reader (Safire2; Tecan Schweiz AG, Männedorf,
Switzerland) set to 450 nm.
Lipid peroxidation assay
A lipid peroxidation assay was carried out using a
commercial kit according to the manufacturer's instructions, in
order to quantify cellular MDA. Briefly, the H9c2 myoblast cells
were harvested by trypsinization (Gibco Life Technologies), and the
cellular extracts were subsequently prepared by sonication at 950
W, 30% for 10 min, 1 sec with 2 sec interval, in ice-cold buffer
containing 50 mM Tris-HCl pH 7.5, 5 mM EDTA, and 1 mM
dithiothreitol. Following sonication, the cells were centrifuged at
10,000 × g for 20 min in order to remove any debris. The
supernatant was then used to measure MDA levels and protein content
(24). MDA content was quantified
by thiobarbituric acid assay, with 1,1,3,3-tetramethoxypropane as
an external standard. The optical density was measured at 532 nm
using a microplate reader (Safire2; Tecan Schweiz AG), according to
the manufacturer's instructions, and the protein content was
determined using the Bradford method (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China).
CAT specific activity
The H9c2 myoblast cell CAT activity was detected
using a CAT analysis kit (Beyotime Institute of Biotechnology),
according to the manufacturer's instructions (25). Briefly, the cells were treated with
hydrogen peroxide and incubated at 25°C for 1–5 min so that the
hydrogen peroxide could react with CAT. Following treatment, the
remaining hydrogen peroxide was coupled with a substrate prior to
further treatment with peroxidase, in order to produce
N-4-antipyryl-3-chloro-5-sulfonate-benzoquinon emonoimine, which
has a maximum absorption of 520 nm. Cellular absorption was
quantified spectrophotometrically (Safire2; Tecan Schweiz AG). CAT
activity was subsequently calculated from the assay results, and
protein concentration was measured using a Bradford Protein Assay
kit (Beyotime Institute of Biotechnology).
H9c2 Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis assay
The apoptotic rate of the H9c2 cells was detected by
flow cytometry using the Annexin V-FITC/PI double-labeling method.
The cells (1×105 cells/ml) were seeded in 6-well plates,
and were treated with 1 mM BPV and 1% LE, as described above. The
cells were then trypsinized and collected by centrifugation at 160
× g for 5 min. After washing with PBS, the cells were
double-stained with an Annexin V-FITC Apoptosis Detection kit
(eBioscience, San Diego, CA, USA), according to the manufacturer's
instructions. Briefly, the cells were resuspended in Annexin V-FITC
binding buffer, and incubated with Annexin V-FITC (1 μg/ml)
for 15 min at room temperature in the dark, followed by an
incubation with PI (10 μg/ml) for 15 min. Samples were
analyzed using a flow cytometer (FACSCalibur; BD Biosciences,
Mountain View, CA, USA) by two parameter dot-plots. A total of
10,000 cells were recorded in each case.
H9c2 myoblast cell apoptosis assay
H9c2 cell apoptosis was detected using a TUNEL kit
(26). Briefly, the H9c2 myoblast
cells were cultured on cover slips at a density of
1×105/ml for 24 h. Following treatment, the cells were
fixed in 4% paraformaldehyde for 30 min at room temperature. The
cells were then incubated with a methanol solution supplemented
with 0.3% hydrogen peroxide for 30 min at room temperature, prior
to further incubation with a permeabilizing solution supplemented
with 0.1% sodium citrate and 0.1% Triton X-100 (Biosharp, Nanjing,
China) for 2 min at 4°C. The cells were subsequently incubated with
the TUNEL reaction mixture for 60 min at 37°C, and visualized by
inverted fluorescence microscopy (AF6000; Leica Microsystems GmbH,
Wetzlar, Germany). The TUNEL-positive nuclei were counted in four
non-overlapping fields per cover slip, and the total percentage of
TUNEL-positive nuclei was then calculated by determining the ratio
of TUNEL-positive counts to the total number of nuclei, as
determined by DAPI 33342 (Beyotime Institute of Biotechnology)
counterstaining.
Western blot analysis
Proteins were extracted from the H9c2 myoblast cells
using a radioimmunoprecipitation buffer supplemented with 1 mM
phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology). The protein concentrations were determined using
the Bradford Protein Assay kit. The protein samples (50 μg)
were resuspended in sample buffer containing 2% SDS, 2%
β-mercaptoethanol, 50 mmol/l Tris-HCl pH 6.8, 10% glycerol, and
0.05% bromophenol blue. The proteins were separated by 15% SDS-PAGE
and transferred to poly-vinylidene fluoride membranes (Merck
Millipore, Bedford, UK). Following membrane blockage with
Tris-buffered saline containing 0.1% Tween® 20 (TBST)
supplemented with 2.5% bovine serum albumin (Beijing Solarbio
Science & Technology, Co., Ltd., Beijing, China) for 1 h at
room temperature, the membranes were washed twice with TBST prior
to being incubated with the following primary antibodies:
Anti-B-cell lymphoma 2 (Bcl-2; 50E3, cat. no. 2870, 1:1,000),
anti-Bcl-2-associated X protein (Bax; D2E11, cat. no. 5023,
1:1,000), anti-Bcl-2-associated death promoter (Bad; D24A9, cat.
no. 9239, 1:1,000), anti-phosphorylated-Bad (Ser112) (40A9, cat.
no. 5284, 1:1,000), anti-cytochrome c (D18C7, cat. no.
11940, 1:1,000), anti-caspase-9 (cat. no. 9508, 1:1,000),
anti-caspase-3 (cat. no. 9662; 1:1,000), anti-cleaved caspase-3
(cat. no. 9661; 1:1,000), anti-cyclooxygenase-IV (cat. no. 4844;
1:1,000), and anti-β-actin (cat. no. 4970; 1:2,000) overnight at
4°C. The membranes were then washed three times with TBST for 10
min, prior to being incubated for 1 h at room temperature with
horseradish peroxidase-conjugated goat anti-rabbit (cat. no. 7074;
1:3,000) and horse anti-mouse (cat. no. 7076; 1:3,000) secondary
antibodies. Following extensive washing, the proteins were detected
using an enhanced chemiluminescence reagent (Merck Millipore). The
band intensities were quantified using the ChemiDoc™ MP system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the H9c2 cells using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA).
RNA concentration was determined by UV spectrophotometry. cDNA was
reverse transcribed from 1 μg total RNA using ReverTra
Ace-α® kit (Toyobo Co., Ltd., Osaka, Japan). A total of
2 μl template cDNA was used for amplification, RT-qPCR was
carried out using a Step One Plus™ Real-Time PCR system (Applied
Biosystems Life Technologies, Foster City, CA, USA). The RT-qPCR
was performed using Thunderbird SYBR Master Mix (Toyobo Co., Ltd.,
Osaka, Japan), and the following primer sequences were used: Bcl-2
forward, 5′-GGA GGA TTG TGG CCT TCT TTG-3′, and reverse, 5′-CAT CCC
AGC CTC CGT TAT CC-3′ (annealing temperature 60°C); Bax forward,
5′-GGT GAG CGA GGC GGT GAG GAC T-3′, and reverse, 5′-GAG AGG ATG
GCT GGG GAG AC-3′ (annealing temperature 65°C); Bad forward, 5′-TGA
GGA AGA TGA AGG GAT GG-3′, and reverse, 5′-GCT TTG TCG CAT CTG TGT
TG-3′ (annealing temperature 60°C); caspase-9 forward, 5′-AGC TGG
CCC AGT GTG AAT AC-3′, and reverse, 5′-GCT CCC ACC TCA GTC AAC
TC-3′ (annealing temperature 60°C); cytochrome c forward,
5′-CTT GGG CTA GAG AGC GGG A-3′, and reverse, 5′-GGT ATC CTC TCC
CCA GGT GAT-3′ (annealing temperature 60°C); GAPDH forward, 5′-GAT
CCC GCT AAC ATC AAA TG-3′, and reverse, 5′-GAG GGA GTT GTC ATA TTT
CTC-3′ (annealing temperature 60°C). All of the primers were
designed and synthesized by GenScript Co., Ltd. (Nanjing, China).
The RT-qPCR cycling conditions were as follows: 95°C for 10 min,
followed by 95°C for 15 sec, the respective primer annealing
temperatures for 30 sec, and 72°C for 30 sec, and 95°C for 15 sec
for 40 cycles. GAPDH expression was used as an internal control.
2−ΔΔCT was calculated for every sample in order to
determine the mRNA expression levels, which were normalized to
GAPDH (27).
Measurement of mitochondrial membrane
potential (ΔΨm)
The mitochondria of H9c2 cells were stained with
JC-1. A total of 1×106 cells were incubated with 10
μg/ml JC-1 (Nanjing Keygene Biotech Co., Ltd., Nanjing,
China) dissolved in DMEM without FBS at 37°C for 30 min, then
washed three times with PBS. The fluorescence of the cells was
measured at an excitation wavelength of 507 nm and an emission
wavelength of 529 nm using a Safire2 microplate reader (Tecan
Schweiz AG).
Statistical analysis
Statistical analyses were performed using SPSS 10.0
(SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean ±
standard deviation. A one-way analysis of variance was performed
when >2 groups were compared, and when statistically significant
a Student-Newman-Keuls test was used to investigate the differences
between individual groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
BPV exhibits inhibitory effects on H9c2
myoblast cell proliferation
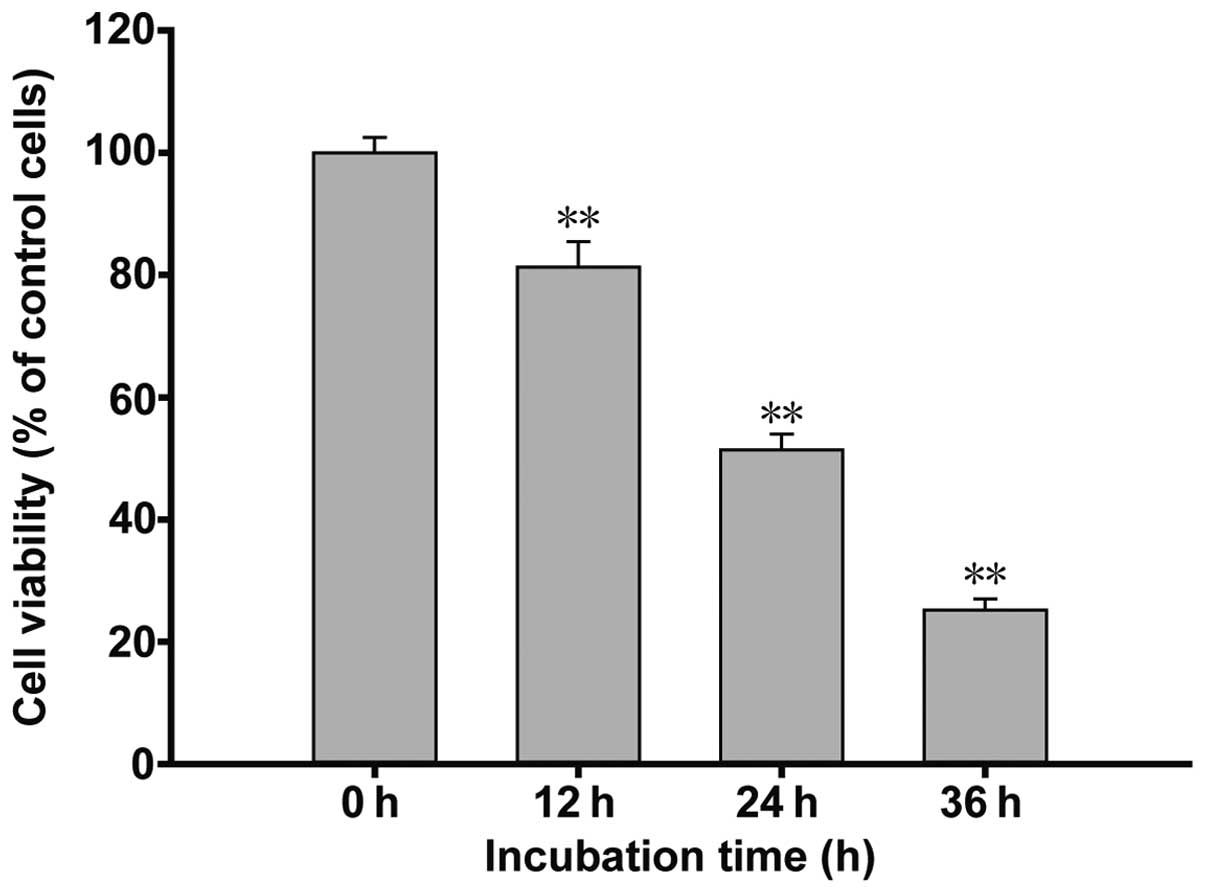
The results of the cell viability assay are shown in
Fig. 1. The H9c2 myoblast cells
were treated with 1 mM BPV over a time course of 12, 24 and 36 h.
The cell viability levels at the 12 h time point decreased to
81.32±2.92%, as compared with the control cells. At 24 h, an
increase in BPV-induced inhibition of cell proliferation was
observed (51.23±2.15%), whereas at 36 h, cell viability was further
reduced to 24.57±1.58%. These results suggest that BPV exerted
inhibitory effects on H9c2 myoblast cell growth in a time-dependent
manner.
LE treatment exhibits rescue effects on
BPV-induced H9c2 myoblast cell apoptosis
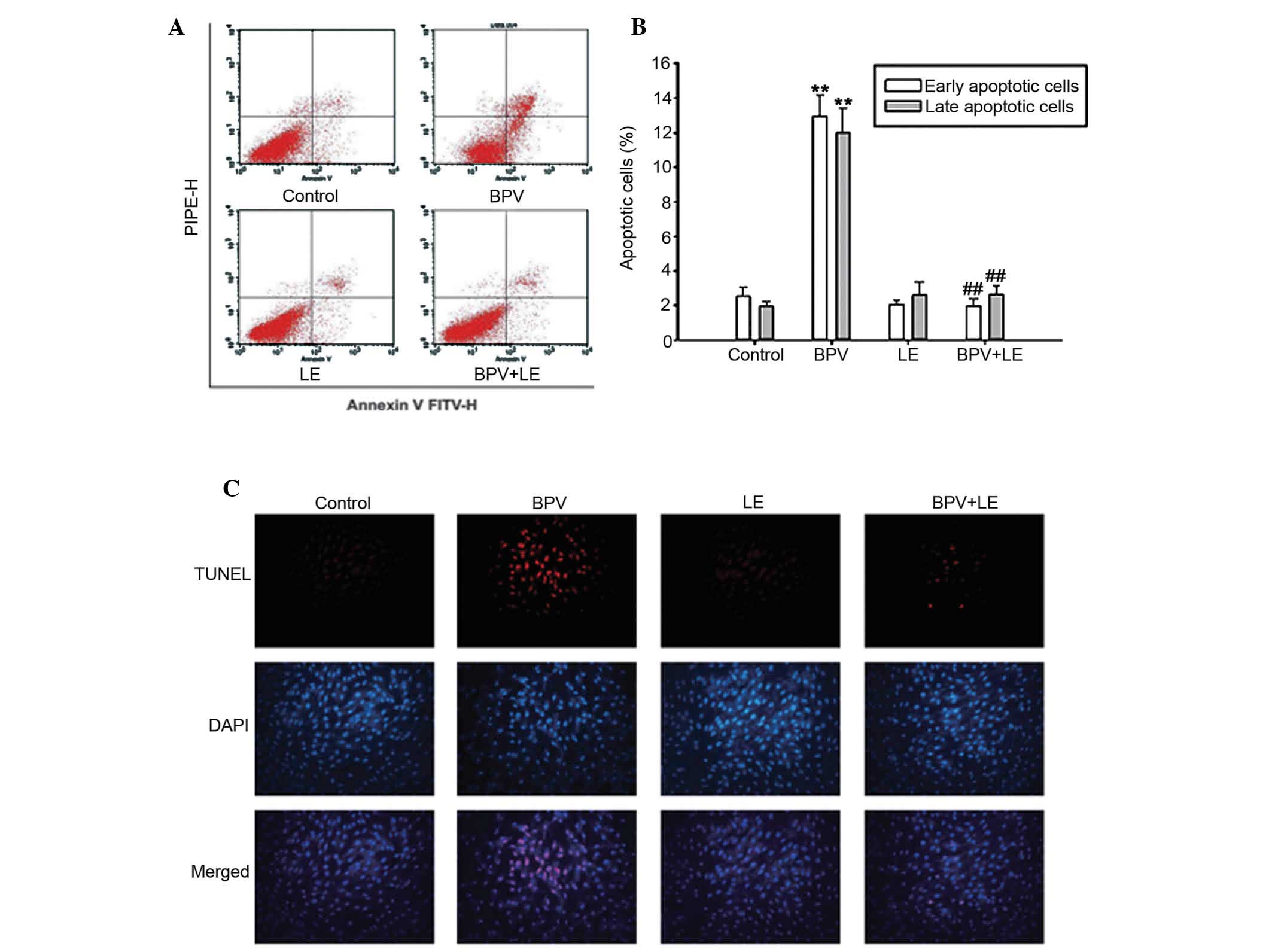
BPV-induced apoptosis of the H9c2 myoblast cells was
investigated using Annexin V/PI staining assay. As shown in
Fig. 2A and B, the apoptotic rate
of the H9c2 myoblast cells was measured for 24 h following cellular
treatment with BPV or LE alone, and following co-treatment with BPV
and LE. The percentage of apoptotic cells was 4.54±0.20% for the
untreated control group, whereas the cells treated with 1 mM BPV
exhibited 24.66±2.57% apoptosis, including both early- and
late-stage apoptotic cells. When the H9c2 myoblast cells were
treated with 1 mM BPV combined with 1% LE, LE was able to attenuate
the apoptotic effects induced by BPV treatment, and the co-treated
cell apoptotic levels resembled those of the control group. The
addition of LE to the BPV-treated group reduced the apoptotic rate
to a normal level (5.07±0.41%). A TUNEL assay was used in order to
investigate the apoptotic cells. As shown in Fig. 2C, the H9c2 myoblast cells exhibited
extensive apoptosis following BPV treatment. However, LE treatment
lowered BPV-induced apoptosis. These results suggest that
co-treatment with BPV and LE may protect H9c2 myoblast cells from
apoptosis.
LE treatment reverses the effects of BPV
on the Bcl-2 and caspase families
Bcl-2 and caspase are two protein families that have
a crucial role in cell apoptosis. In the present study, western
blotting and RT-qPCR were used to investigate the changes in
protein and mRNA expression levels of Bcl-2 and caspase. The
results of the western blot analysis revealed that the protein
expression levels of Bax, cleaved caspase 9, and cleaved caspase 3
were elevated in the BPV-treated cells, whereas the protein
expression levels of Bcl-2 and phosphorylated-Bad were
downregulated. However, co-treatment of the H9c2 myoblast cells
with LE and BPV resulted in opposing results (Fig. 3A and B). The downregulated
expression levels of Bcl-2 and phosphorylated-Bad were restored to
normal, and similarly the expression levels of cleaved caspase-3
and -9 were reduced to that of the control sample. RT-qPCR was
performed in order to verify whether Bcl-2 and Bax were
transcriptionally regulated. The results of the RT-qPCR confirmed
that the mRNA expression levels of Bcl-2 and Bax were also
regulated by BPV and LE in a similar manner to the protein
expression levels (Fig 3C). In
addition, the ratio of Bax/Bcl-2 doubled following BPV treatment,
indicating that the apoptotic levels has increased. This ratio
returned to normal in the BPV and LE co-treated H9c2 myoblast cells
(Fig. 3C and D). These results
indicate that BPV and LE may regulate apoptosis via Bcl-2 and
caspase at both the protein and mRNA level.
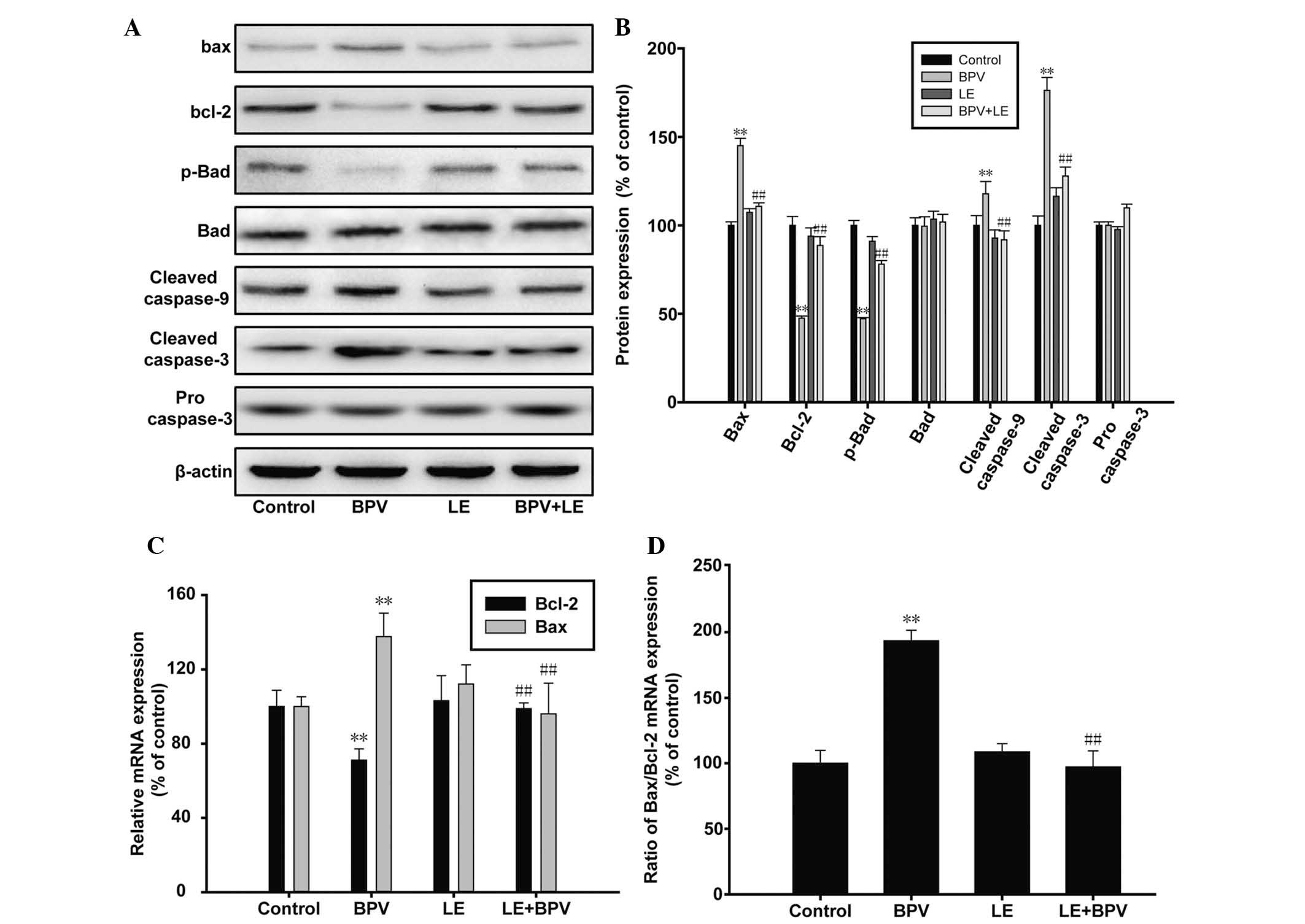 | Figure 3Effects of bupivacaine (BPV) on
B-cell lymphoma 2 (Bcl-2) and caspase. (A and B) The H9c2 rat
myoblast cells were treated with 1 mM BPV or 1% lipid emulsion (LE)
alone, or co-treated with both BPV and LE for 24 h, prior to the
collection of the cell lysate. Western blot analysis was performed
using antibodies targeting Bcl-2, Bcl-2-associated X protein (Bax),
phosphorylated (p)-Bcl-2-associated death promoter (Bad), Bad,
cleaved caspase 3, cleaved caspase 9 and procasepase 3. (C and D)
The mRNA expression levels of Bcl-2 and Bax in the H9c2 myoblast
cells were quantified using reverse transcription-quantitative
polymerase chain reaction. The data are presented as the mean ±
standard deviation, and all experiments were performed in
tripliacate. **P<0.01, vs. the control group;
##P<0.01, vs. the 1 mM BPV treatment group. |
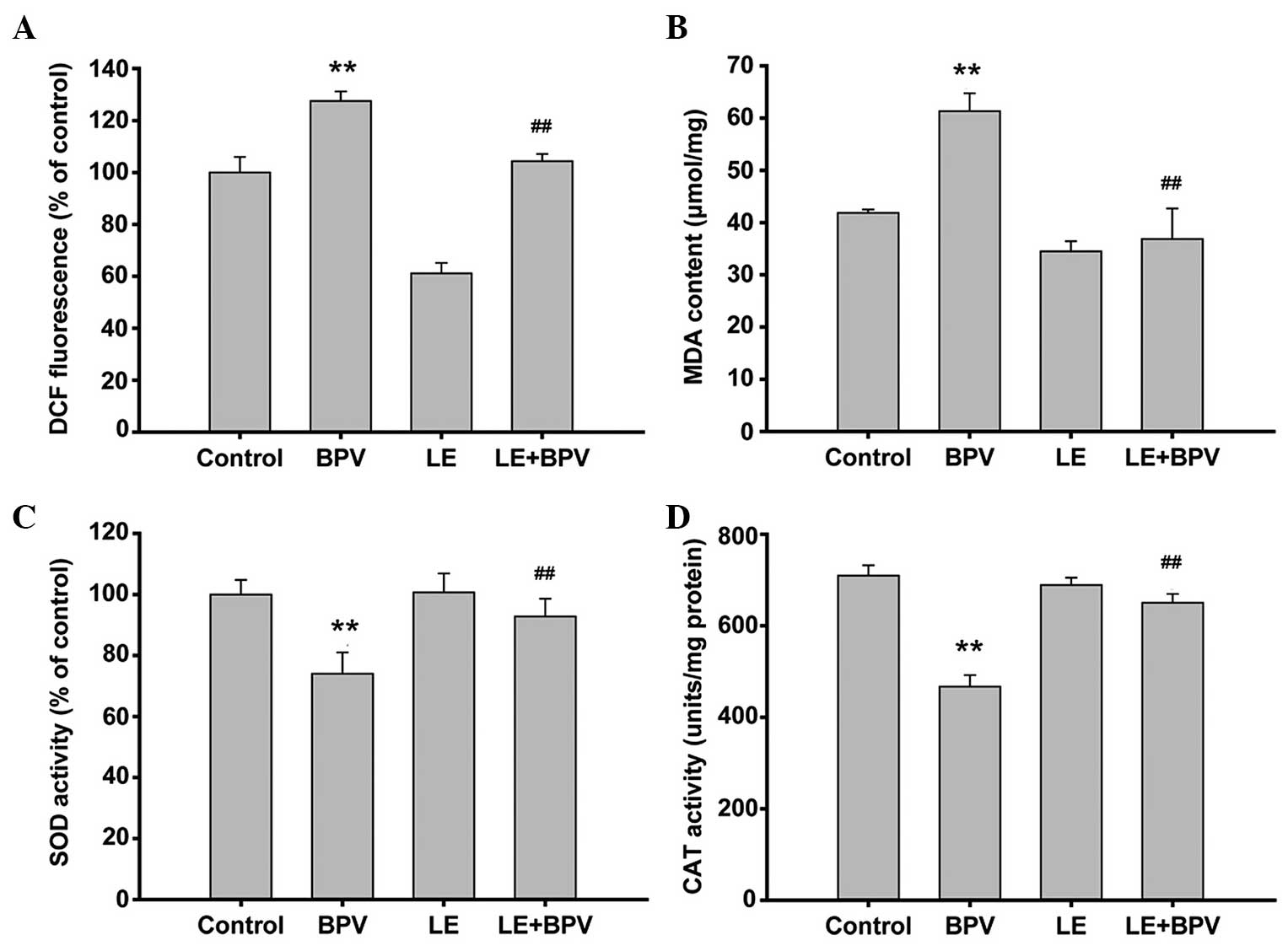
LE treatment attenuates BPV-triggered
oxidative stress in H9c2 myoblast cells
Oxidative stress is caused by disturbance to the
oxidation and antioxidation balance, leading to apoptotic cell
death (28). In order to
investigate whether oxidative stress mediated the effects of BPV
and LE, the activity levels of various enzymes were monitored. ROS
levels, the accumulation of which may lead to mitochondrial
dysfunction, were detected using a DCFH-DA probe. The results
indicated that a rapid production of ROS occurred following H9c2
myoblast cell exposure to BPV. However, BPV-induced ROS production
was reversed by LE treatment (127.54±3.68% in the BPV group, vs.
104.33±2.77% in the BPV+LE group, Fig.
4A). MDA is a marker for lipid peroxidation, which was used as
a further indicator of endogenous ROS levels. The BPV-treated cell
samples contained elevated levels of MDA (61.33±3.43
μmol/mg), as compared with the control group (41.86±0.64
μmol/mg) (Fig. 4B). These
results suggest that BPV treatment causes apoptotic cell death by
increasing cellular ROS levels. In order to further verify this
hypothesis, the activity levels of SOD were investigated. SOD
antioxidant activity was measured by WST-1. The activity levels of
SOD were significantly reduced following BPV treatment (Fig. 4C). In addition, co-treatment of the
H9c2 myoblast cells with LE restored SOD activity levels to those
of the control (74.03±6.23% in the BPV-treated sample, vs.
92.83±5.81% in the BPV+LE co-treated sample; Fig. 4C). CAT activity, which is another
key indicator of endogenous ROS levels, was measured in order to
assess the levels of antioxidation within the H9c2 myoblast cells.
As shown in Fig. 4D, the CAT
activity levels exhibited a similar trend to SOD activity levels.
CAT activity was reduced in the BPV-treated cells, whereas
co-treatment with LE restored CAT activity levels to those of the
control group (709.74±22.40 U/mg in the control group, 467.03±25.39
U/mg in the BPV-treated group, and 650.47±19.430 U/mg in the BPV+LE
co-treated group). These results suggest that BPV-induced toxicity
is attributable to an increase in the oxidative stress levels of
H9c2 myoblast cells. Furthermore, co-treatment with BPV and LE
reversed the observed increase in cellular oxidation, bringing the
levels of endogenous ROS back to normal.
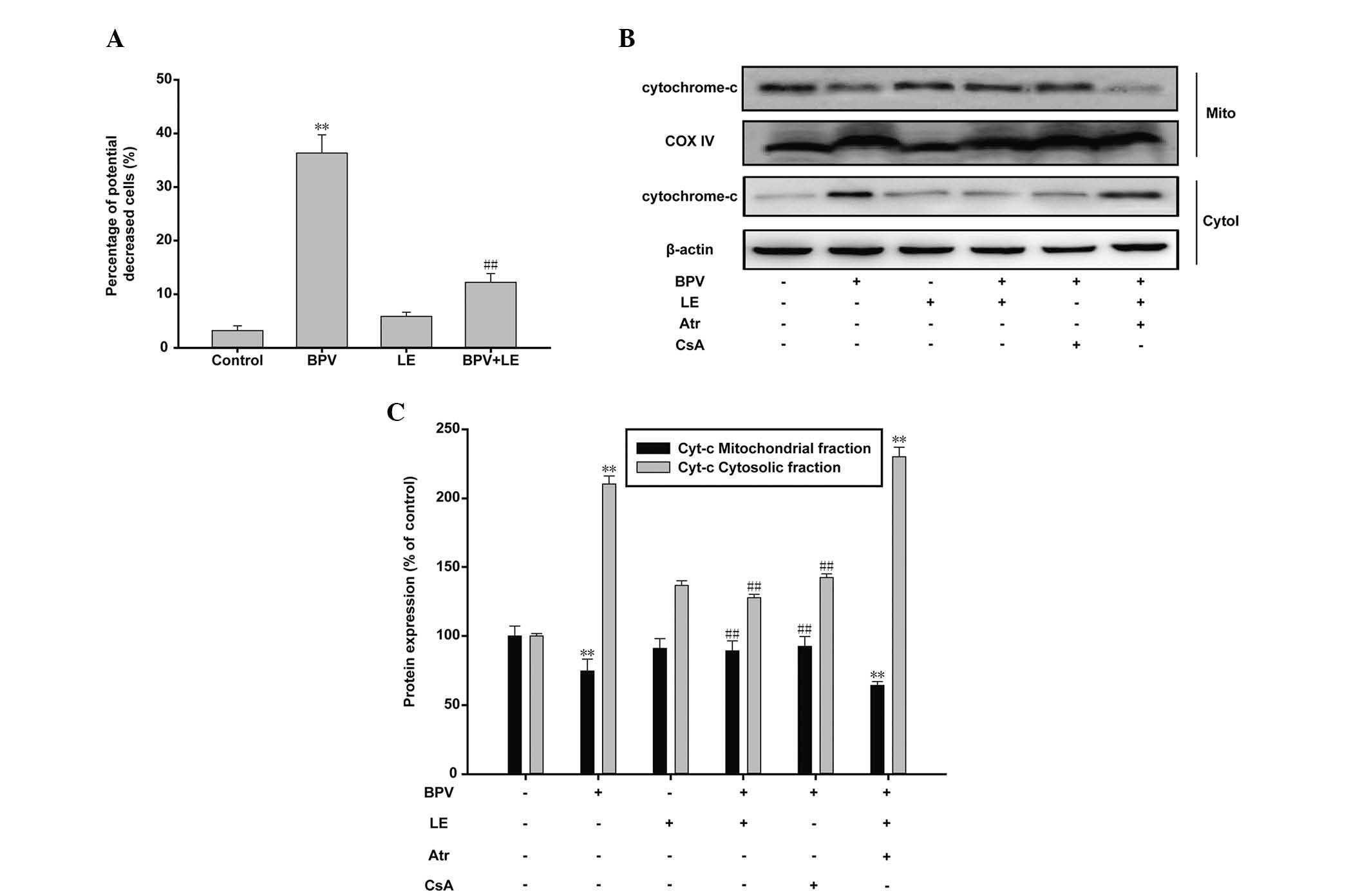
LE reverses BPV-induced apoptosis by
promoting mitochondrial survival
Two major pathways trigger apoptotic cell death: The
death receptor-induced extrinsic pathway, and the mitochondrial
apoptosome-mediated apoptotic intrinsic pathway (29). The mitochondrial membrane potential
(ΔΨ)-sensitive dye JC-1 was used in order to determine through
which pathway BPV and LE affect H9c2 myoblast cell survival. When
mitochondrial ΔΨ is high, JC-1 accumulates in the mitochondrial
matrix, and forms red fluorescent JC-1 aggregates. However, when
mitochondrial ΔΨ is low, JC-1 becomes a monomer and emits green
fluorescence. The ratio of green to red fluorescence indicates the
percentage of mitochondrial depolarization. Flow cytometric
analysis revealed that the H9c2 myoblast cells became more
susceptible to mitochondrial membrane depolarization following BPV
treatment, as compared with those in the control group (Fig. 5A). The percentage of green
fluorescence increased from 3.21±0.91% to 36.34±3.40% following BPV
treatment, indicating a decrease in mitochondrial ΔΨ, and
mitochondrial dysfunction. The percentage of JC-1 monomers also
decreased significantly in the cells co-treated with BPV and LE
(12.22±1.63%). mPTP opening promotes uncontrolled ion flow between
the mitochondrial membranes, which leads to mitochondrial swelling
and rupture, followed by the release of cytochrome c
(30). Therefore, the levels of
cytochrome c reflect mPTP state, which provides an
indication of mitochondrial viability. In the present study,
cytochrome c release within the mitochondria was quantified
via immunoblotting. The results demonstrated that treatment with
BPV significantly reduced mitochondrial expression levels of
cytochrome c, whereas BPV treatment significantly increased
cytosolic cytochrome c expression levels (Fig. 5B and C). The addition of LE to the
BPV-treated cells reduced the cytosolic expression levels of
cytochrome c similar to those observed in the control cells.
CsA treatment also inhibited cytochrome c release in the
BPV-treated cells. Cellular treatment with Atr eliminated the
reversal effects induced by LE treatment. These results strongly
support the hypothesis that LE counteracts the cellular toxicity
induced by treatment with BPV. Furthermore, the results of the
present study suggest that the molecular mechanism underlying the
protective effects of LE on H9c2 myoblast cells is modulated via
the mPTP.
Discussion
Previous studies have demonstrated that BPV exhibits
cellular toxicity both in vitro and in vivo (15–17).
However, LE treatment is able to reverse BPV-induced
cardiotoxicity. Chen et al (31) recently reported that BPV-induced
asystole may be reversed by LE in rat hearts. Rosenblatt et
al (32) also reported the
successful clinical use of LE to resuscitate patients following
BPV-associated cardiac arrest. However, the mechanism underlying
BPV toxicity and the protective effects of LE have yet to be
elucidated.
In the present study, the MTT assay demonstrated
that BPV inhibited cellular proliferation in a time-dependent
manner. The results of Annexin V/PI staining indicated that BPV
induced cellular apoptosis, and the results of the TUNEL assay
further confirmed this observation. The present study successfully
demonstrated that LE exhibited protective effects on H9c2 myoblast
cells, and rescued them from BPV-induced apoptosis.
Oxidative stress is closely associated with
apoptosis. Previous studies have demonstrated that ROS and
oxidative stress have an important role in the onset of apoptosis.
Antioxidants and thiol reductants, such as N-acetylcysteine, as
well as the overexpression of manganese-SOD are able to prevent or
delay apoptosis (33). The present
study investigated changes in the activity levels of ROS and
antioxidants, and demonstrated that treatment with BPV caused
oxidative stress. Incubation of the H9c2 myoblast cells with BPV
for 24 h resulted in elevated levels of ROS and MDA, and reduced
levels of SOD and CAT. Treatment with LE counteracted BPV-induced
cell death caused by ROS-induced cellular damage. The results of
the present study suggested that BPV-induced cellular apoptosis may
be closely associated with oxidative stress. Furthermore, the
rescue effects observed following LE treatment are likely
attributable to an increase in the levels of antioxidants.
The present study investigated the effects of
various apoptosis-associated proteins, and revealed that BPV and LE
influence the expression levels of Bcl-2 and caspase family
proteins. The results of the present study showed that treatment
with BPV increased the protein expression levels of Bax, and
inhibited the protein expression levels of Bcl-2. RT-qPCR further
demonstrated that the mRNA expression levels of Bax and Bcl-2 were
affected. In addition, treatment with BPV activated caspases-9 and
-3. The upregulation of apoptotic proteins following BPV treatment
was rescued by LE treatment. The ratio of Bax/Bcl-2 is
representative of the severity of mitochondrial outer membrane
permeabilization, which is a critical factor of the intrinsic
apoptotic pathway (34–36). These results support the hypothesis
that the mitochondria are critical in cellular apoptosis.
Furthermore, mitochondria have been implicated in the generation of
ROS (37). These previous results
are concordant with the results of the present study, and suggest
that BPV-induced toxicity directly affects mitochondrial
survival.
In order to further confirm this hypothesis, and to
elucidate the molecular mechanism underlying the rescue effects
induced by LE treatment, a series of experiments were carried out.
The results of JC-1 staining indicated that treatment with BPV
affected H9c2 myoblast cell survival by initiating mitochondrial
dysfunction. Release of cytochrome c from the mitochondria
into the cytosol also occurred following BPV treatment, and was
determined to be a key marker of apoptosis. Mitochondrial function
and the release of cytochrome c were inversely correlated to
LE treatment. mPTP, which is a non-specific pore located in the
inner mitochondrial membrane, is responsible for the release of
cytochrome c. The effects of BPV and LE on mPTP are
associated with the levels of cytochrome c release. In
addition, the effects of CsA and Atr, an mPTP inhibitor and
activator respectively, on the release of cytochrome c were
compared with that of BPV and LE. The results demonstrated that
mPTP has a significant role in the mechanism underlying the
protective effects of LE on BPV-induced toxicity. The results of
the present study confirmed the involvement of the mitochondria in
BPV-induced toxicity, and the important role of mPTP in the
mechanism underlying the protective effects of LE treatment.
The present study demonstrated the potential
clinical applications of LE as a treatment for BPV-induced
toxicity. Furthermore, the present study revealed that the
mechanism underlying BPV-induced toxicity is associated with the
mitochondria, via the alteration of mPTP sensitivity. In
conclusion, LE may protect H9c2 myoblast cells from BPV-induced
toxicity through restoration of mitochondrial function.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81360284),
and from the Natural Science Foundation of Ningxia (grant no.
NZ13148).
References
|
1
|
Malhotra N, Chanana C, Roy KK, Kumar S,
Rewari V and Sharma JB: To compare the efficacy of two doses of
intraperitoneal bupivacaine for pain relief after operative
laparoscopy in gynecology. Arch Gynecol Obstet. 276:323–326. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clarkson CW and Hondeghem LM: Mechanism
for bupivacaine depression of cardiac conduction: Fast block of
sodium channels during the action potential with slow recovery from
block during diastole. Anesthesiology. 62:396–405. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch C III: Depression of myocardial
contractility in vitro by bupi-vacaine, etidocaine, and lidocaine.
Anesth Analg. 65:551–559. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castle NA: Bupivacaine inhibits the
transient outward K+ current but not the inward
rectifier in rat ventricular myocytes. J Pharmacol Exp Ther.
255:1038–1046. 1990.PubMed/NCBI
|
|
5
|
Halestrap AP, McStay GP and Clarke SJ: The
permeability transition pore complex: Another view. Biochimie.
84:153–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Halestrap AP, Clarke SJ and Khaliulin I:
The role of mitochondria in protection of the heart by
preconditioning. Biochim Biophys Acta. 1767:1007–1031. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bernardi P, Krauskopf A, Basso E,
Petronilli V, Blachly-Dyson E, Di Lisa F and Forte MA: The
mitochondrial permeability transition from in vitro artifact to
disease target. FEBS J. 273:2077–2099. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Halestrap AP, Clarke SJ and Javadov SA:
Mitochondrial permeability transition pore opening during
myocardial reperfusion - a target for cardioprotection. Cardiovasc
Res. 61:372–385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stone JR and Yang S: Hydrogen peroxide: A
signaling messenger. Antioxid Redox Signal. 8:243–270. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Del Rio D, Stewart AJ and Pellegrini N: A
review of recent studies on malondialdehyde as toxic molecule and
biological marker of oxidative stress. Nutr Metab Cardiovasc Dis.
15:316–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu JQ, Kosten TR and Zhang XY: Free
radicals, antioxidant defense systems, and schizophrenia. Prog
Neuropsychopharmacol Biol Psychiatry. 46:200–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gałecka E, Jacewicz R, Mrowicka M,
Florkowski A and Gałecki P: Antioxidative enzymes - structure,
properties, functions. Pol Merkur Lekarski. 25:266–268. 2008.In
Polish.
|
|
13
|
Bowler C, Slooten L, Vandenbranden S, De
Rycke R, Botterman J, Sybesma C, Van Montagu M and Inzé D:
Manganese superoxide dismutase can reduce cellular damage mediated
by oxygen radicals in transgenic plants. EMBO J. 10:1723–1732.
1991.PubMed/NCBI
|
|
14
|
Long WB, Rosenblum S and Grady IP:
Successful resuscitation of bupivacaine-induced cardiac arrest
using cardiopulmonary bypass. Anesth Analg. 69:403–406. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weinberg G, Ripper R, Feinstein DL and
Hoffman W: Lipid emulsion infusion rescues dogs from
bupivacaine-induced cardiac toxicity. Reg Anesth Pain Med.
28:198–202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weinberg GL, VadeBoncouer T, Ramaraju GA,
Garcia-Amaro MF and Cwik MJ: Pretreatment or resuscitation with a
lipid infusion shifts the dose-response to bupivacaine-induced
asystole in rats. Anesthesiology. 88:1071–1075. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weinberg GL, Ripper R, Murphy P, Edelman
LP, Hoffman W, Strichartz G and Feinstein DH: Lipid infusion
accelerates removal of bupivacaine and recovery from bupivacaine
toxicity in the isolated rat heart. Reg Anesth Pain Med.
31:296–303. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weinberg GL: Lipid infusion therapy:
Translation to clinical practice. Anesth Analg. 106:1340–1342.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Litz RJ, Roessel T, Heller AR and Stehr
SN: Reversal of central nervous system and cardiac toxicity after
local anesthetic intoxication by lipid emulsion injection. Anesth
Analg. 106:1575–1577. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weinberg GL, Palmer JW, VadeBoncouer TR,
Zuechner MB, Edelman G and Hoppel CL: Bupivacaine inhibits
acylcarnitine exchange in cardiac mitochondria. Anesthesiology.
92:523–528. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Daoud A, Song J, Xiao F and Shang J:
B-9-3, a novel β-carboline derivative exhibits anti-cancer activity
via induction of apoptosis and inhibition of cell migration in
vitro. Eur J Pharmacol. 724:219–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian YY, An LJ, Jiang L, Duan YL, Chen J
and Jiang B: Catalpol protects dopaminergic neurons from
LPS-induced neurotoxicity in mesencephalic neuron-glia cultures.
Life Sci. 80:193–199. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martin RC, Liu Q, Wo JM, Ray MB and Li Y:
Chemoprevention of carcinogenic progression to esophageal
adenocarcinoma by the manganese superoxide dismutase
supplementation. Clin Cancer Res. 13:5176–5182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qian J, Jiang F, Wang B, Yu Y, Zhang X,
Yin Z and Liu C: Ophiopogonin D prevents
H2O2-induced injury in primary human
umbilical vein endothelial cells. J Ethnopharmacol. 128:438–445.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He Z, Yu S, Mei G, Zheng M, Wang M, Dai Y,
Tang B and Li N: Maternally transmitted milk containing recombinant
human catalase provides protection against oxidation for mouse
offspring during lactation. Free Radic Biol Med. 45:1135–1142.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Yan M, Yu A, Mao H and Zhang J:
Inhibitory effects of β-tricalciumphosphate wear particles on
osteocytes via apoptotic response and Akt inactivation. Toxicology.
297:57–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Sies H and Cadenas E: Oxidative stress:
Damage to intact cells and organs. Philos Trans R Soc Lond B Biol
Sci. 311:617–631. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu W and Kavanagh JJ: Anticancer therapy
targeting the apoptotic pathway. Lancet Oncol. 4:721–729. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Halestrap A: Biochemistry: A pore way to
die. Nature. 434:578–579. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Y, Xia Y, Liu L, Shi T, Wang Q, Chen
L, Papdimos TJ and Xu X: Lipid emulsion reverses
bupivacaine-induced asystole in isolated rat hearts:
Concentration-response and time-response relationships.
Anesthesiology. 113:1320–1325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosenblatt MA, Abel M, Fischer GW,
Itzkovich CJ and Eisenkraft JB: Successful use of a 20% lipid
emulsion to resuscitate a patient after a presumed
bupivacaine-related cardiac arrest. Anesthesiology. 105:217–218.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kannan K and Jain SK: Oxidative stress and
apoptosis. Pathophysiology. 7:153–163. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Osorio LM, De Santiago A,
Aguilar-Santelises M, Mellstedt H and Jondal M: CD6 ligation
modulates the Bcl-2/Bax ratio and protects chronic lymphocytic
leukemia B cells from apoptosis induced by anti-IgM. Blood.
89:2833–2841. 1997.PubMed/NCBI
|
|
35
|
Del Poeta G, Venditti A, Del Principe MI,
Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A,
Mazzone C, et al: Amount of spontaneous apoptosis detected by
Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML).
Blood. 101:2125–2131. 2003. View Article : Google Scholar
|
|
36
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kulbacka J, Bar J, Chwilkowska A, Dumanska
M, Drag-Zalesinska M, Wysoka T, Stach H, Becharz I, Lugowski M,
Markinkowska A, et al: Oxidative modulation of marcaine and
lekoptin in H9C2 rat myoblasts. Acta Pharmacol Sin. 30:184–192.
2009. View Article : Google Scholar : PubMed/NCBI
|