Introduction
Cholangiocarcinoma is an aggressive and lethal
disease arising from the epithelial cells in the intra- and
extrahepatic bile ducts. Although the annual incidence of
cholangiocarcinoma is relatively low with 1–2 cases per 100,000 in
western countries (1), it greatly
varies among the population. The incidence is as high as 113 per
100,000 in Southeast Asia, with particularly high rates in
Thailand. This is possibly due to liver fluke infection as an
important risk factor (2). The
cancer is highly resistant to chemotherapy with a high recurrence
rate (3). To date, surgical
resection has been the main treatment option. A five-year survival
rate of only 25–30% has been reported among those who have had
successful resection (4). Thus, an
understanding of tumor biology underlying an aggressive phenotype
of cholangiocarcinoma is crucial for the development of alternative
treatments to improve clinical outcomes.
Inadequate supplies of oxygen as a result of rapid
cell growth and aberrant vasculature formation are commonly found
within most solid tumors (5).
There is accumulating evidence suggesting that hypoxia contributes
to tumor progression and metastasis by activating diverse signaling
pathways (6,7). Tumor hypoxia has been associated with
increased malignancy and poor prognosis (8). Elevated expression of
hypoxia-inducible factor-1α (HIF-1α), a key transcriptional
regulator in hypoxia, is correlated with a low survival rate in a
wide variety of cancers (9).
Numerous in vitro studies have shown that hypoxia can
promote angiogenesis, resistance to chemotherapy and cell invasion
(5–11).
Neoplastic malignancies are usually controlled by
growth factors. Overexpression and overactivation of hepatocyte
growth factor (HGF) receptor (Met) receptor tyrosine kinase (RTK)
is strongly linked to oncogenesis and invasive growth (12,13).
The receptor has a role in cell motility through activation of the
mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase (ERK) and PI3K/Akt pathways as its main
downstream signaling axes upon hepatocyte growth factor (HGF)
binding (14). Met induction by
HGF was reported to have an important role in cholangiocarcinoma
invasion ability (15).
Penacchietti et al (11)
showed that hypoxia can induce Met expression in a number of cancer
cell types, including hepatocarcinoma, lung and ovarian carcinomas.
Moreover, the MAPK/ERK pathway, containing downstream effectors of
Met, regulates multiple cellular functions, including
proliferation, cell motility and apoptosis (16), and hyperactivation of this pathway
is a hallmark of cancer (17). The
ERKs have a role in hypoxic conditions by regulating HIF-1
expression and function (18,19),
which in turn regulates transcription of several genes involved in
proliferation, glycolysis, angiogenesis and cell motility (5). HIF-1 is a heterodimeric basic
helix-loop-helix transcription factor consisting of
oxygen-dependent α- and constitutively expressed β-subunits. During
hypoxia, the subunits dimerize to modulate gene expression.
Although a number of hypoxia-associated proteins
involved in tumor progression have been well characterized in
numerous types of cancers, the hypoxia-mediated pathways in
cholangiocarcinoma remain mostly unknown. The present study was
designed to examine effects of hypoxia on cholangiocarcinoma growth
and its invasive potential, and to investigate the molecular basis
of cancer invasion triggered by chronic hypoxia using a
cholangiocarcinoma cell line.
Materials and methods
Cell culture
The human cholangiocarcinoma cell line RMCCA-1
(established at the Department of Surgery, Rajavithi Hospital,
Bangkok, Thailand) (3), was
cultured in HAM's F12 medium (Gibco-BRL, Life Technology, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS; GE
Healthcare Life Sciences, Logan, UT, USA) and 1%
penicillin/streptomycin at 37°C in a 5% CO2 humidified
atmosphere for normoxic conditions. Incubation under hypoxic
conditions was performed under 1% O2 at 37°C in a
hypoxic chamber (Stem Cell Technologies, Vancouver, BC,
Canada).
Immunofluorescence staining
Cells were plated and cultured on glass cover slips
under hypoxic conditions for 48 h. Cells were then fixed with 4%
paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA), permeabilized
with 1% Triton X-100 (Sigma-Aldrich) and blocked with 1% bovine
serum albumin (United States Biological, Salem, MA, USA). Rabbit
monoclonal antibody against HIF-1α was used (ab51608; 1:100
dilution; Abcam, Cambridge, UK), followed by incubation with Alexa
Fluor® 488-labeled anti-rabbit secondary antibody
(A-11034; 1:200 dilution; Thermo Fisher Scientific, Waltham, MA,
USA).
Cell proliferation assay
Cells were seeded in a 96-well culture plate at a
density of 1,000 cells/well. Cell proliferation was assessed on
days 1, 3 and 5 post-plating using a WST-1 Cell Proliferation Assay
(Roche Diagnostics, Basel, Switzerland) following the
manufacturer's instructions. Cells were incubated in a hypoxic
chamber, which was re-purged every 24 h. The medium was replaced
every other day. Absorbance was measured at 450 nm with a Bio-Rad
680 microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Cell invasion assay
Cells were cultured in normal medium overnight under
hypoxic conditions. On the following day, 5×104 cells
were plated into the upper chamber wells of 24-well Biocoat
Matrigel invasion chambers (8-µm pores) (Becton-Dickinson,
Franklin Lakes, NJ, USA), which were coated with Matrigel and
maintained in 1% FBS F-12 medium under hypoxia or normoxia for 24
h. Non-invaded cells were removed. The invaded cells were fixed
with 25% methanol and stained with crystal violet (Sigma-Aldrich).
Cell numbers were counted in five random ×400 power fields under an
inverted fluorescence light microscope [IX71; Olympus (Thailand)
Co., Ltd., Bangkok, Thailand].
Pathscan RTK signaling antibody array
kit
The PathScan RTK Signaling Antibody Array kit
(#7982; Cell Signaling Technology, Inc., Danvers, MA, USA) was used
according to the manufacturer's instructions to simultaneously
detect phosphorylation of 28 receptor tyrosine kinases and 11
important signaling molecules, including human epidermal growth
factor receptor 2 (HER2) and -3, fibroblast growth factor receptor
2 (FGFR2), insulin receptor (InsR), Met, stem cell factor receptor
(SCFR); ephrin type-A receptor 2 (EphA2), TEK tyrosine kinase,
endothelial (TEK) and S6 ribosomal protein (S6RP). In brief, cells
were washed with ice-cold 1X PBS and lysed in 1X Cell Lysis Buffer
to collect cell lysates of normoxia vs. hypoxia (48 h) samples. The
Array Blocking Buffer was added and lysates were incubated for 15
min at room temperature. The lysates were then added to the assay
wells and incubated for 2 h at room temperature. Subsequently, the
Detection Antibody Cocktail was added to each well and incubated
for 1 h at room temperature followed by an addition of horseradish
peroxidase-linked streptavidin and the slide was incubated for
another 30 min at room temperature. The plates were then covered
with LumiGLO/Peroxide reagent (Cell Signaling Technology, Inc.) and
exposed to film (Kodak, Bangkok, Thailand) for 2–30 sec. The film
was scanned using an Epson E11000XL-PH Photo Scanner (Epson, Suwa,
Japan) and intensity was measured using ImageJ 1.47v (National
Institutes of Health, Bethesda, MD, USA).
Pathscan intracellular signaling array
kit
The array kit (cat. no. 7323; Cell Signaling
Technology, Inc.) was used according to the manufacturer's
instruction. This kit allowed for the detection of phosphorylation
or cleavage of 18 signaling molecules simultaneously, including
ERK, heat shock protein 27 (HSP27), mammalian target or rapamycin
(mTOR), B-cell lymphoma-2-associated death domain (Bad),
c-Jun-N-terminal kinase (JNK), adenosine monophosphate-activated
protein kinase (AMPK) and proline-rich Akt/PKB substrate 40 kDa
(PRAS40). Cell lysates were collected after exposure to hypoxia for
48 h in HAM's F12 medium without fetal bovine serum and processed
as described above.
Inhibition of Met and ERK signaling
Inhibition of Met was performed using small hairpin
(sh)RNA plasmids targeting Met (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) according to the manufacturer's instructions. In
brief, 2×105 cells were plated in a six-well tissue
culture plate in antibiotic-free normal growth medium until the
cells were 70–80% confluent. Cells were then incubated with
Lipofectamine (Invitrogen Life Technologies) together with the
plasmids for 6 h at 37°C prior to replacement with fresh normal
growth medium. Cells were then subjected to an invasion assay as
described above. Block-iT Fluorescent Control (Invitrogen Life
Technologies) was used to confirm transfection efficiency. For
inhibition of ERK, cells were treated with U0126 (10 µg/ml;
Cell Signaling Technology, Inc.) overnight prior to the invasion
assay.
Human cholangiocarcinoma tissue
samples
Cholangiocarcinoma tissue samples used in the
present study were obtained from cholangiocarcinoma patients (n=44)
who underwent surgical resection at Rajavithi Hospital (Bangkok,
Thailand) from 2012–2014 (period of 18 months). The protocol of the
present study was approved by the Ethics Committee of Rajavithi
Hospital (Bangkok, Thailand).
Immunohistochemical staining
Paraffin wax sections of cholangiocarcinoma
specimens were dewaxed in xylene and transferred to various
concentrations of alcohol (100, 85, 70 and 50%. Endogenous
peroxidase activity was blocked with 0.5% hydrogen peroxide
(Sigma-Aldrich) in methanol at room temperature for 10 min, then
the sections were boiled in 10 mM citrate buffer (pH 6.0; Abcam) in
a microwave oven for 10 min (750 W; LG MS-202W; Seoul, South Korea)
for antigen retrieval. Non-specific binding was blocked by
incubation with 3% normal horse serum (Gibco-BRL, Life Technology)
for 20 min. Sections were incubated overnight at 4°C with the
rabbit monoclonal antibody against HIF-1α (ab51608; 1:100 dilution;
Abcam). Biotinylated goat anti-rabbit immunoglobulin G (E0432;
1:500 dilution; Dako, Glostrup, Denmark) was then added followed by
an avidin-biotin-peroxidase conjugate (ABC Elite; Vector
Laboratories, Burlingame, CA, USA) for 30 min at room temperature.
The immunohistochemical reaction was developed with freshly
prepared reagents from a Histofine SAB-PO kit (Nichirei Inc.,
Tokyo, Japan). The slides were then dehydrated with various
concentrations of alcohol (95, 95, 100 then 100%) for 5 min each,
then with xylene. The slides were then mounted. Sections were then
visualized under high-power magnification (×400) using an Olympus
BH2 microscope (field width, 0.5 mm; Olympus, Tokyo, Japan) and
scored using the following three categories based on the percentage
of positively stained cells: 1) negative, <5%; 2) weak, 5–50%;
and 3) strong >50%.
Statistical analysis
All experiments were performed in triplicate and
values are expressed as the mean ± standard deviation. The
student's t-test was used for analysis and P<0.05 was
considered to indicate a statistically significant difference. The
χ2 test was used for statistical analysis of the
association between clinicopathological data and the expression of
HIF-1α.
Results
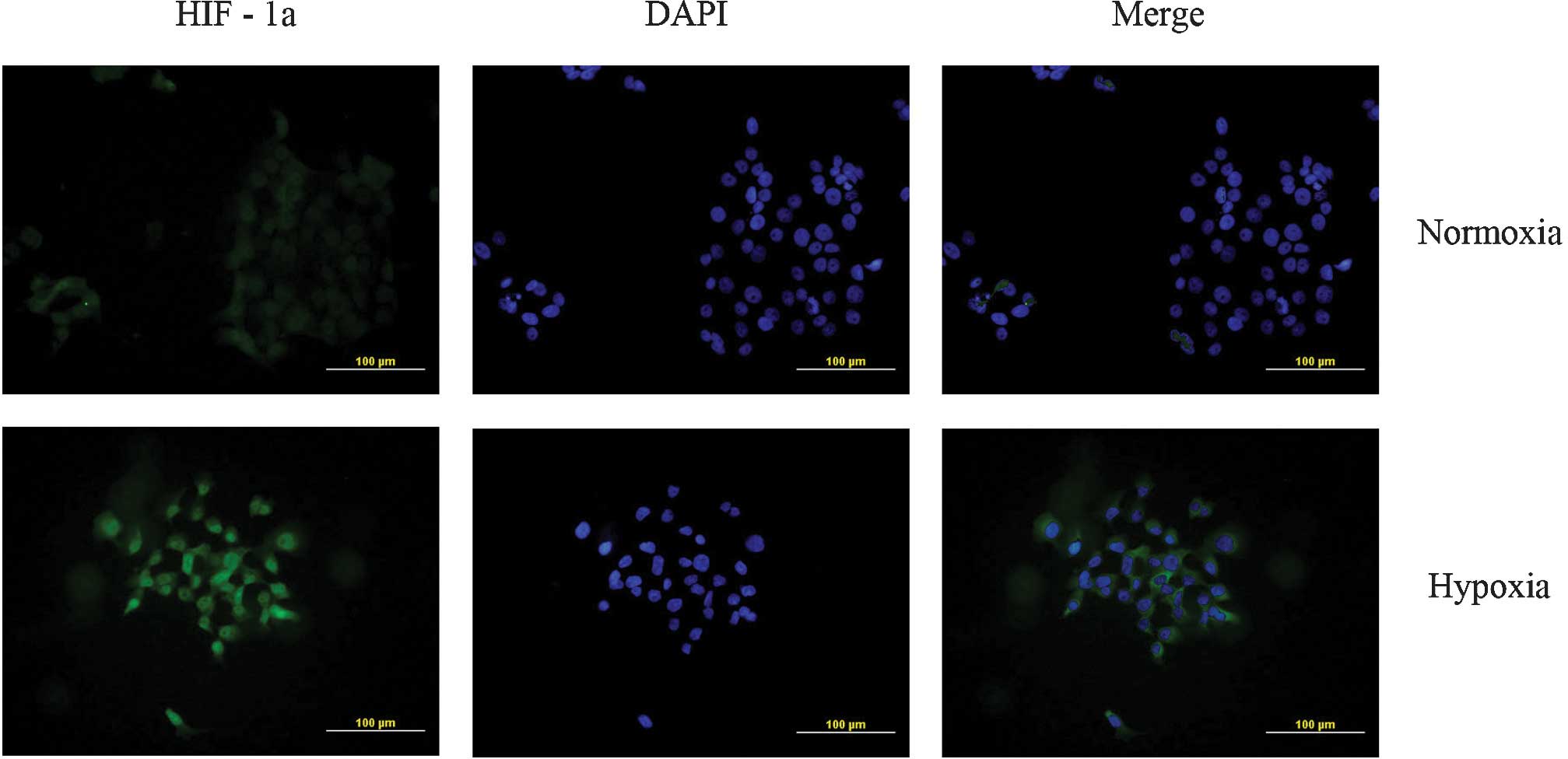
Hypoxia induces HIF-1α protein
expression
Immunofluorescence staining was performed to examine
expression of HIF-1α under hypoxic conditions. An increased HIF-1α
protein expression was observed after hypoxia for 48 h (Fig. 1). HIF-1α expression in the hypoxic
group was detected in the nucleus as well as in the cytoplasm of
RMCCA-1 cells.
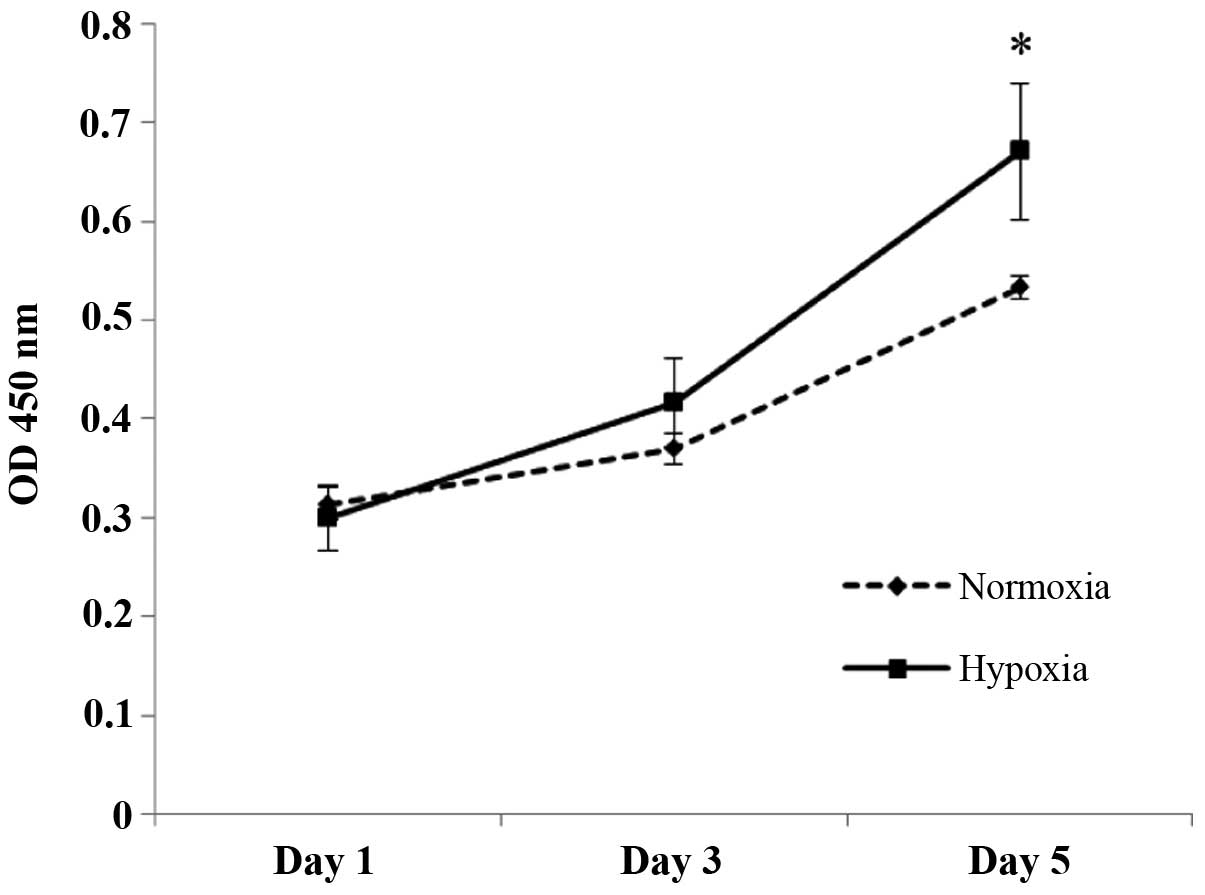
Hypoxia stimulates RMCCA-1
proliferation
Cell proliferation of RMCCA-1 under hypoxic
conditions was investigated over the course of 5 days. The results
showed that hypoxia significantly stimulated RMCCA-1 cell growth
(P<0.05) (Fig. 2). Cells
cultured under hypoxia exhibited a higher growth rate and reached a
statically significant difference compared with that of the
normoxic cells on day 5.
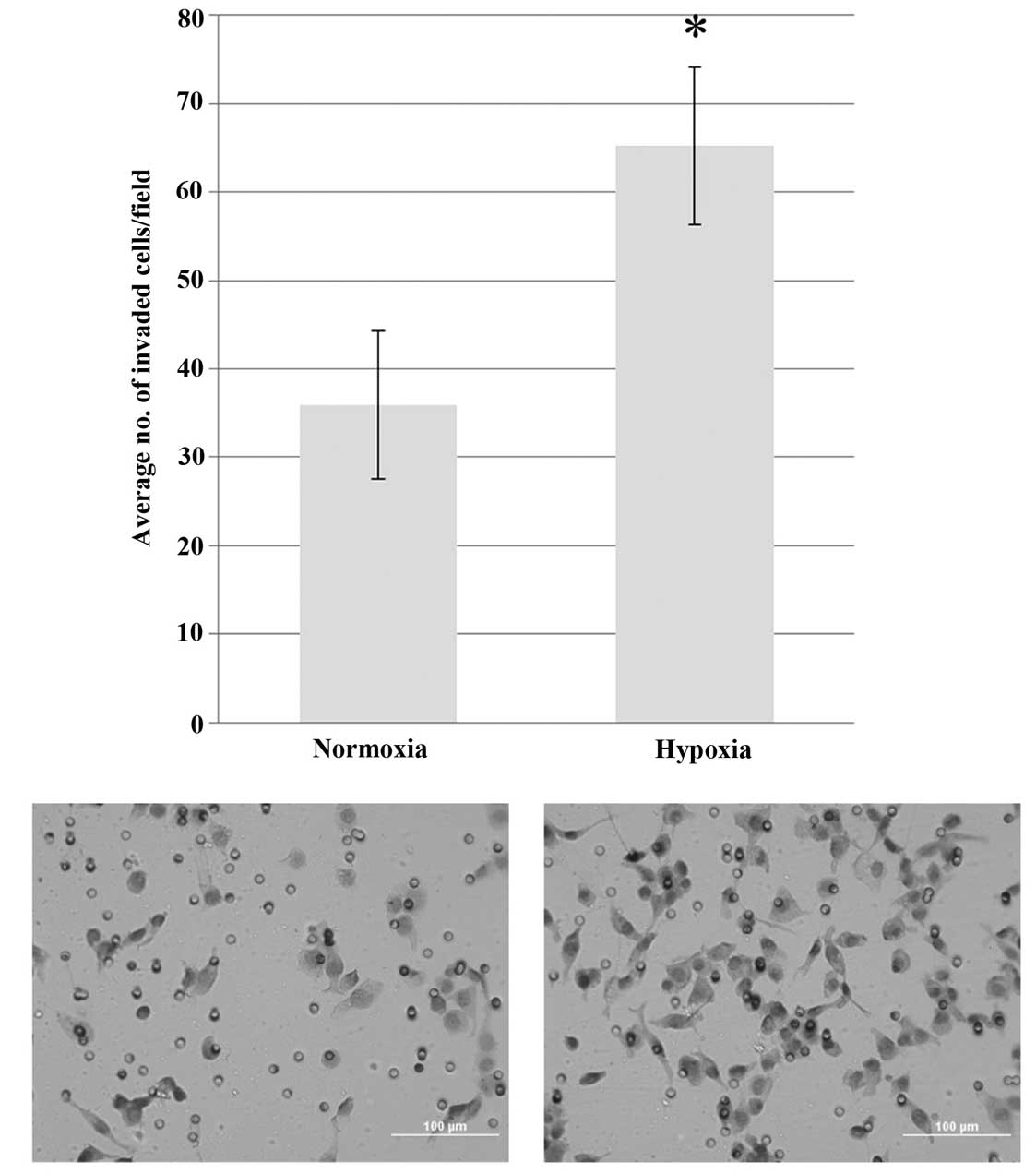
Hypoxia enhances RMCCA-1 invasion
Several studies have reported that hypoxia promotes
cancer cell invasion (5,7). The present study performed a
Transwell invasion assay to examine the effect of hypoxia on the
invasion of RMCCA-1 cells. After 24 h, the number of invaded cells
was significantly increased under hypoxic conditions compared with
that following incubation under normoxic conditions (P<0.05)
(Fig. 3).
Induction of Met under hypoxia
In order to elucidate signaling pathways and
receptors involved in hypoxia, the phosphorylation of various RTKs
and signaling molecules was examined using the Pathscan RTK
signaling antibody array kit. It was found that phosphorylation of
a number of signaling molecules, including HER2, HER3, FGFR4, InsR,
Met/HGFR, c-Kit/SCFR and Tie2/TEK in hypoxic cells was markedly
increased compared with that in normoxic cells (P<0.05)
(Fig. 4). In addition, a
significant decrease in the phosphorylation levels of EphA2 and
S6RP was observed (P<0.05).
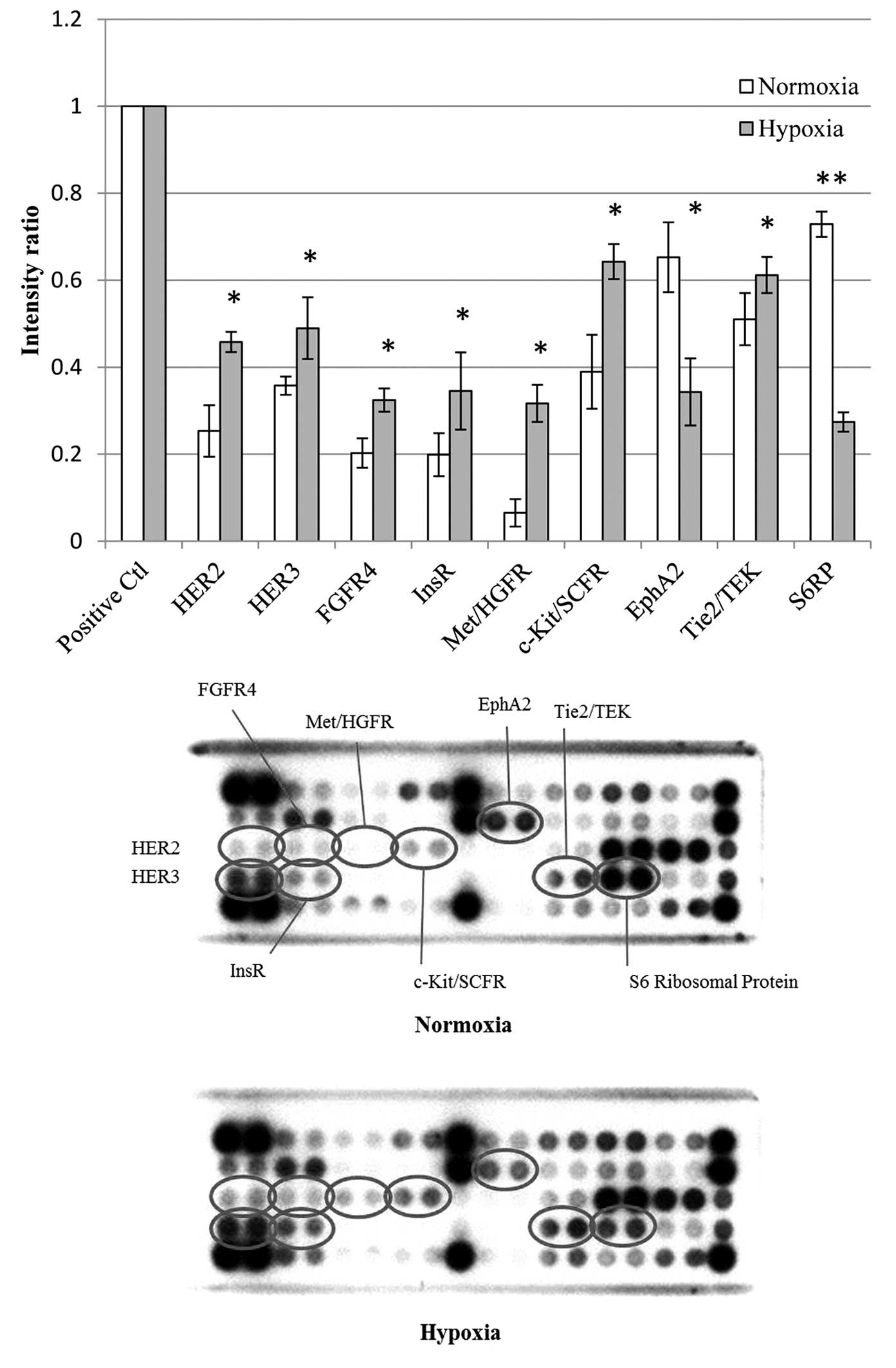 | Figure 4Effects of hypoxia on phosphorylation
of signaling proteins. Lower panel shows a representative pair of
chemiluminescent images produced using the PathScan RTK Signaling
Antibody Array kit. The upper panel shows the pixel intensity ratio
of phosphorylated signaling molecules. *P<0.05,
**P<0.001 for the comparison between hypoxia and
normoxia; values are presented as the mean ± standard deviation.
Ctl, control; HER2, human epidermal growth factor receptor 2;
FGFR2, fibroblast growth factor receptor 2; InsR, insulin receptor;
Met/HGFR, hepatocyte growth factor receptor; SCFR, stem cell factor
receptor; EphA2, ephrin type-A receptor 2; TEK, TEK tyrosine
kinase, endothelial; S6RP, S6 ribosomal protein. |
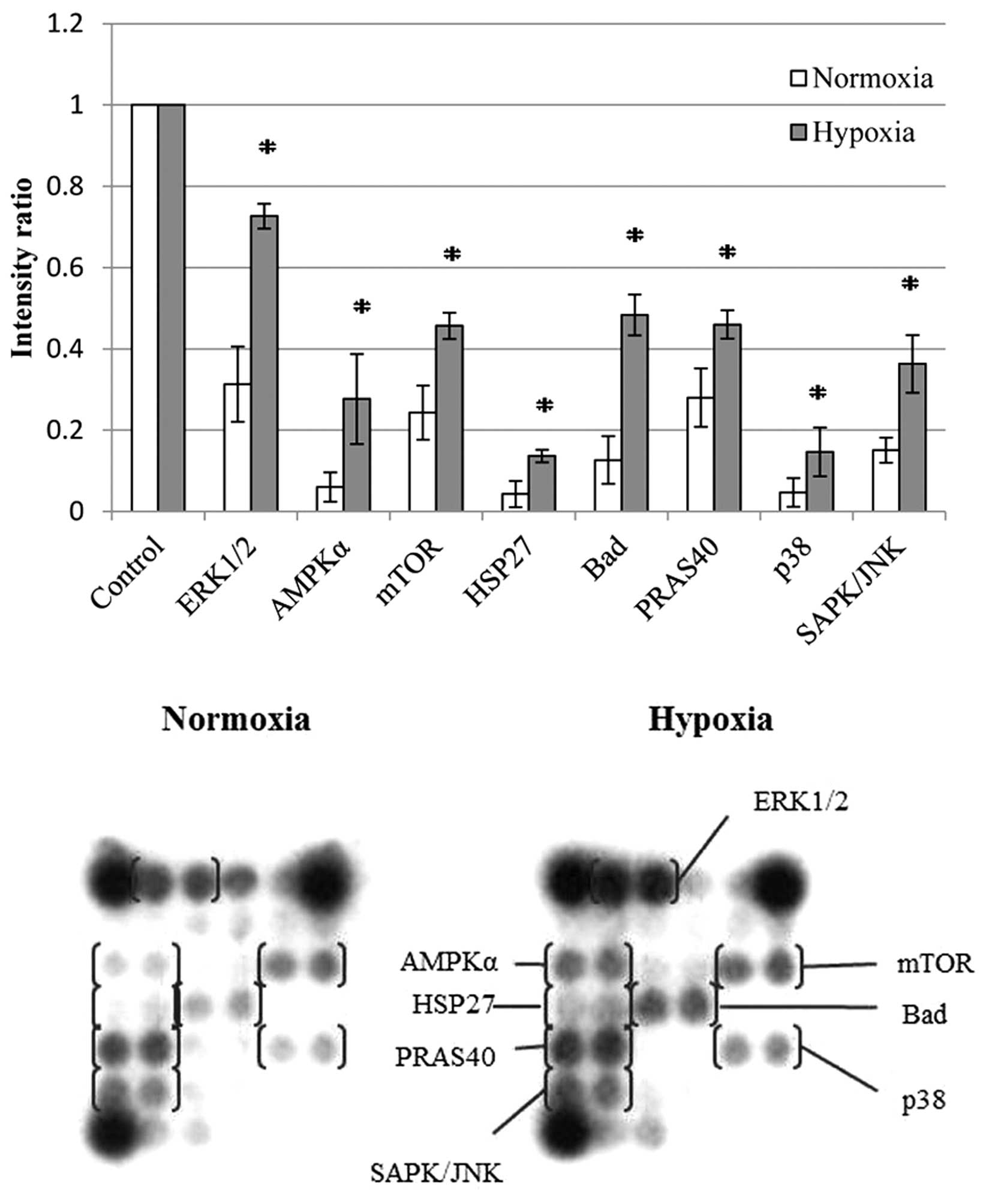
Activation of ERK in hypoxia
The present study further investigated downstream
effectors of Met, which are associated with hypoxia-induced
invasion. The Pathscan intracellular signaling array kit was used
to determine changes in phosphorylation or cleavage of several
intracellular signaling molecules. The results showed an increase
in the phosphorylation levels of ERK1/2, AMPKα, mTOR, HSP27, Bad,
PRAS40, p38 and SAPK/JNK in the hypoxic cells compared with those
in normoxic cells (P<0.05) (Fig.
5).
Inhibition of Met and ERK attenuates
hypoxia-induced invasion
Given that Met exhibited the greatest change in its
phosphorylation levels among the proteins analyzed, the involvement
of Met in hypoxia-induced invasion was assessed by silencing Met
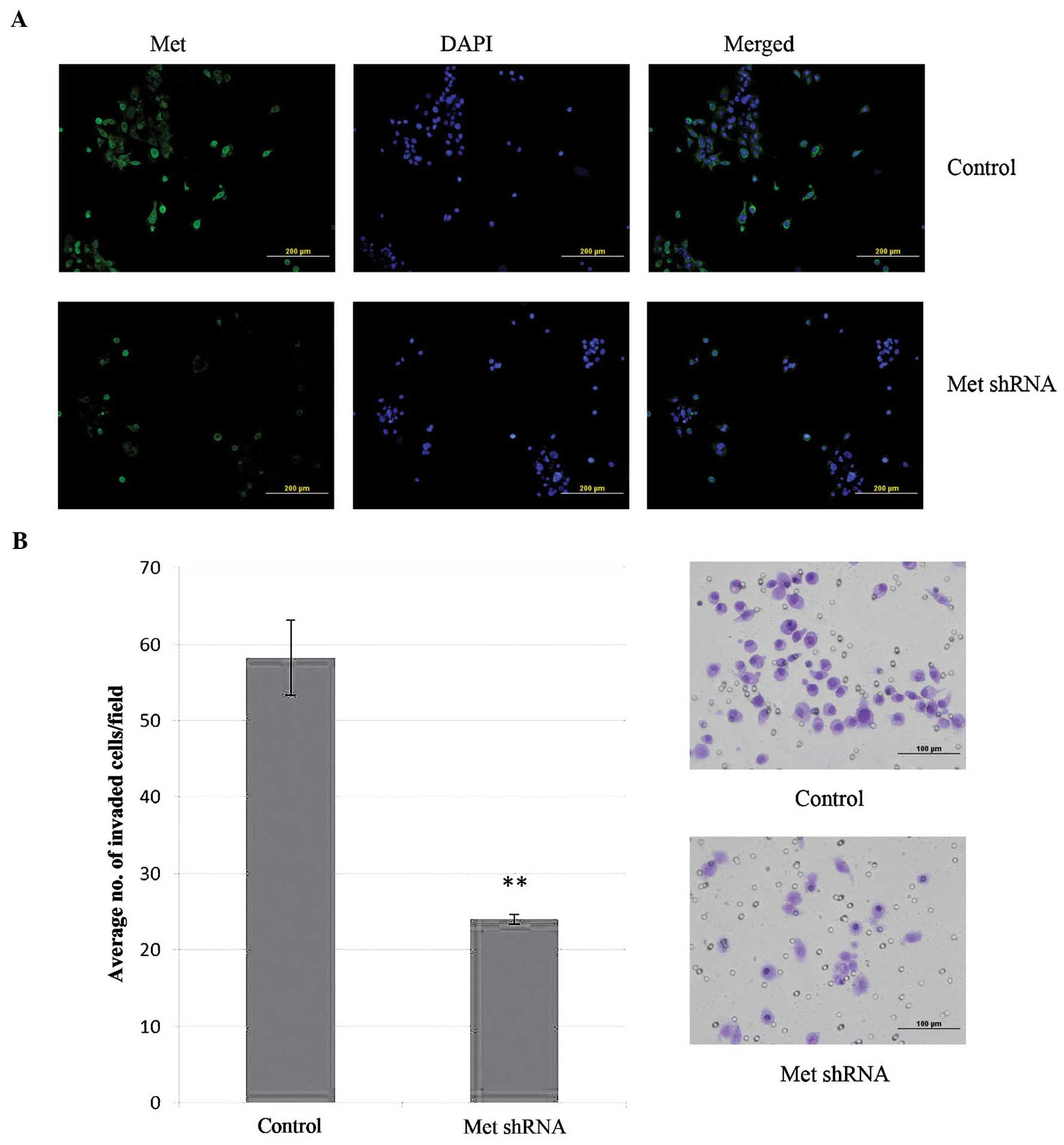
expression in RMCCA-1 cells. Immunofluorescence staining was used
to confirm a decrease in Met expression after transfection
(Fig. 6A). The results showed that
the invasion ability of RMCCA-1 was significantly reduced in cells
treated with shRNA compared to cells treated with control small
interfering RNA (P<0.001) (Fig.
6B). Since ERK is one of the main downstream effectors of Met
and it has been demonstrated to have an important role in
metastasis (15,20), it was hypothesized that Met-induced
invasion of RMCCA-1 cells under hypoxia was mediated through ERK.
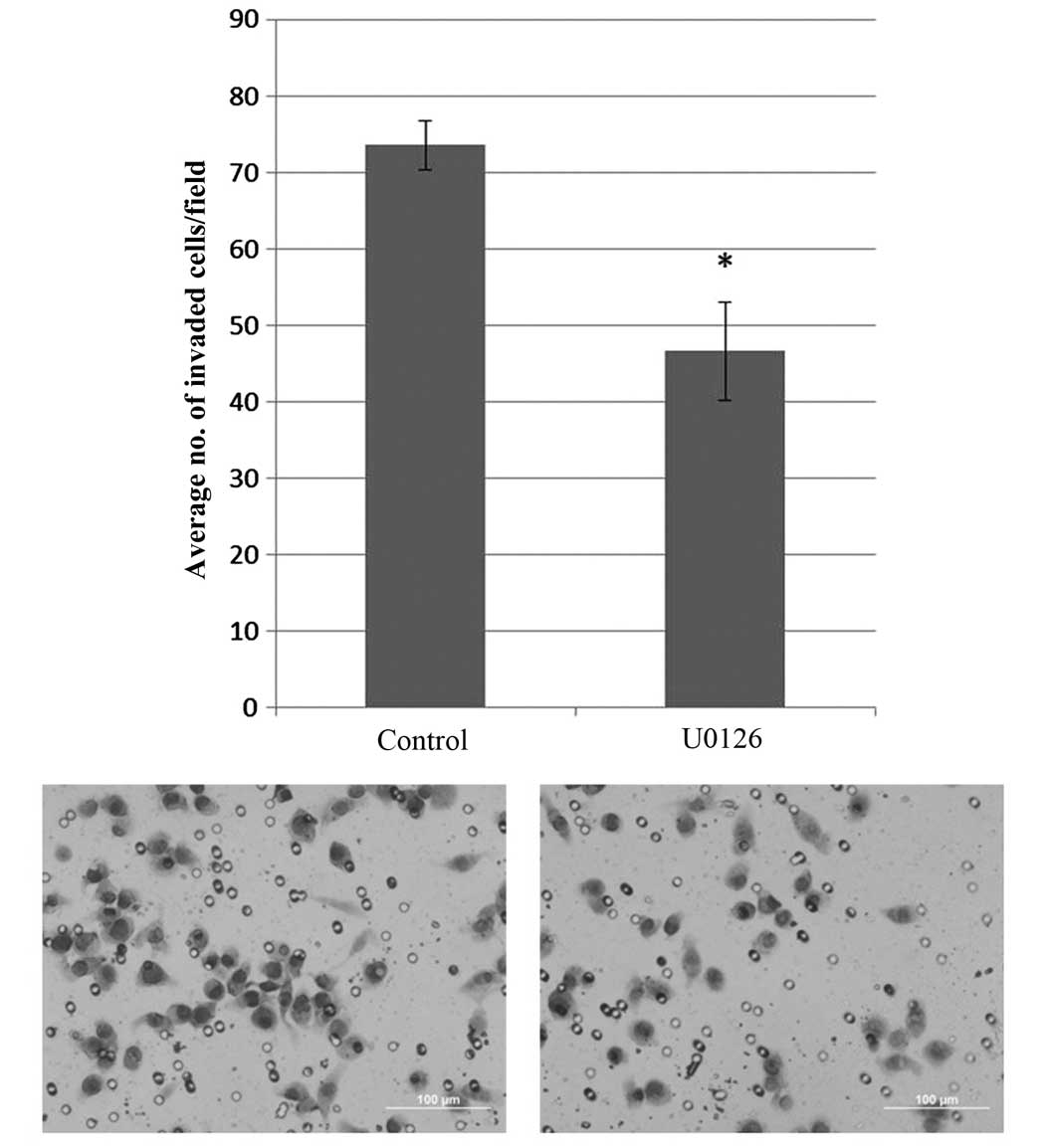
To investigate whether ERK activation was responsible for increased
invasion, U0126 was used to inhibit ERK. A significant decrease in
cell invasion was identified in cells treated with U0126 compared
to that of the control (P<0.05) (Fig. 7). The results therefore suggested
that activation of the Met/ERK pathway is critical for the
hypoxia-induced invasion of RMCCA-1 cells.
HIF-1α expression in
paraffin-embedded cholangiocarcinoma samples
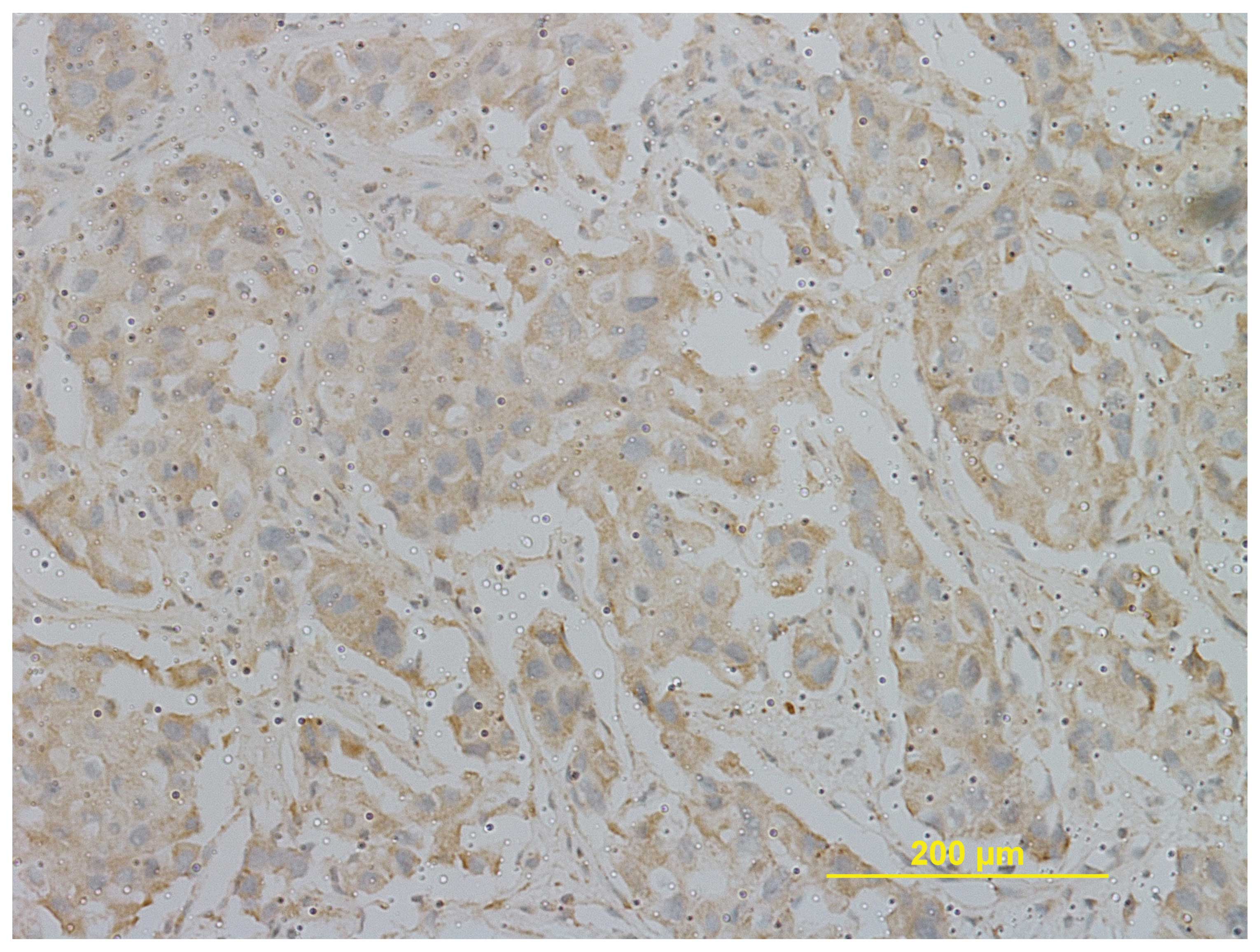
The expression of HIF-1α was determined by
immunohistochemistry in 44 paraffin-embedded cholangio-carcinoma
specimens (Fig. 8). In these
cancerous tissues, HIF-1α-specific signals were localized mainly in
the cytoplasm of cholangiocarcinoma cells but not in normal biliary
epithelial cells. It was found that 66.0% (n=29/44) of the
cholangiocarcinoma specimens were strongly positive for HIF-1α
expression. The expression of HIF in cholangiocarcinoma was
detected in all stages of the disease. However, the expression
levels were not significantly correlated with lymph node or
metastatic status (Table I).
 | Table IAssociation between HIF-1α expression
and clinicopathological features in cholangiocarcinoma samples. |
Table I
Association between HIF-1α expression
and clinicopathological features in cholangiocarcinoma samples.
| | HIF-1α expression
| |
|---|
| Variable | Total (n=44) | Weak | Strong | P-valuea |
|---|
| Age (years) | | | | |
| <60 | 23 | 8 | 15 | 0.919 |
| >60 | 21 | 7 | 14 | |
| Sex | | | | |
| Male | 19 | 7 | 12 | 0.737 |
| Female | 25 | 8 | 17 | |
| TNM stageb | | | | 0.198 |
| I/II | 17 | 8 | 9 | |
| III/IV | 27 | 7 | 20 | |
| Tumor
differentiation | | | | 0.107 |
| Well | 40 | 12 | 28 | |
| Moderate/poor | 4 | 3 | 1 | |
Discussion
The chronic hypoxia within solid tumors provides a
selective pressure for cells with a more aggressive phenotype to
survive. Thus, a better understanding of cellular adaptation to low
oxygen tension is crucial for the development of improved treatment
strategies. The contribution of hypoxia to tumor metastasis has
been reported in several types of cancer, including gliomas
(11), breast cancer (10) and hepatocellular carcinoma
(5). In the present study, an
in vitro experiment demonstrated that hypoxia enhanced the
proliferation and invasion of cholangiocarcinoma. In addition, the
present study suggested that hypoxia-induced invasion is associated
with increased phosphorylation of Met and activation of the
MAPK/ERK signaling pathway. The findings are in agreement with
previous studies suggesting that hypoxia promotes metastatic
progression (5–11). The results of the present study
suggested that the hypoxic environment promotes the malignant
characteristics of cholangiocarcinoma.
The present study first examined the induction of
HIF-1α under hypoxic conditions using immunofluorescence staining
and found higher HIF-1α expression in hypoxic cells, showing HIF-1α
stabilization under hypoxia. The presence of HIF-1α was observed in
the nucleus as well as in the cytoplasm of RMCCA-1, indicating
nuclear translocation for regulating gene transcription.
A cell proliferation assay was then used to identify
the effects of hypoxia on tumor growth. Studies on the association
between hypoxia and cell proliferation have been contradictory in
recent years. Hypoxia was found to inhibit cell proliferation in
certain cell types (5), while
promoting cell growth in others, suggesting a cell-specific
variation in response to hypoxic stress. This is possibly due to
genetic variation among cancer cell populations. The results of the
present study implied that RMCCA-1 cultured under hypoxic
conditions proliferated more efficiently compared to those cultured
under normoxic conditions. These findings were discordant with a
study by Seubwai et al (21), which reported suppressed growth
after exposure of various cholangiocarcinoma cell lines to hypoxia.
The present study reported increased cell proliferation induced by
hypoxia, supporting that hypoxia regulated the expression of genes
involved in glycolysis and accelerated cell growth. Gwak et
al (22) reported that hypoxia
enhanced hepatocellular carcinoma cell growth through induction of
hexokinase II (22). The levels of
HIF-1α were correlated with phosphoinositide-dependent kinase-1,
lactate dehydrogenase A and pyruvate kinase, muscle 2 expression
and inhibition of HIF-1α repressed pancreatic cancer cell growth
(23). However, further studies
are required to identify proteins responsible for accelerated cell
growth following exposure of RMCCA-1 to hypoxia.
Furthermore, the present study found that the
invasion was significantly increased as a result of hypoxic
stimulation. Alterations in phosphorylation/activation of important
signaling molecules were subsequently examined in an attempt to
identify pathways responsible for hypoxia-induced invasion. Hypoxia
is known to mediate various signaling cascades. The antibody array
kit revealed significantly increased phosphorylation of HER2, HER3,
FGFR4, InsR, Met/HGFR, c-Kit/SCFR and Tie2/TEK. The greatest change
in phosphorylation was observed in Met/HGFR. The results of the
intracellular signaling array kit assay further revealed increased
phosphorylation of a number of intracellular signaling nodes,
including ERK1/2, AMPKα, mTOR, HSP27, Bad, PRAS40, p38 and
SAPK/JNK.
A previous study demonstrated an involvement of Met
in cholangiocarcinoma cell lines by showing that inhibition of Met
and ERK activation significantly reduced cell invasion abilities
(15). It was therefore
hypothesized that activation of the ERK pathway by Met may be the
molecular event contributing to the malignant phenotype of RMCCA-1
under hypoxic conditions. To confirm this hypothesis, Met
expression was inhibited using shRNA and ERK expression was blocked
by treatment with U0126, a MAPK kinase (MEK) inhibitor. The results
showed that the increase in cell invasion under hypoxia was
abrogated following blocking of Met or ERK.
Met is a well-known receptor that has a significant
role in tumor progression in various cancers, including
cholangiocarcinoma (24).
Overexpression of this receptor was reported in numerous types of
solid tumor and is associated with malignant disease (25). Stimulation of Met leads to the
activation of multiple downstream signaling pathways with two major
pathways being the MAPK/ERK and the PI3K/Akt signaling axes
(20). In the present study, the
antibody signaling array kits revealed increased phosphorylation of
ERK1/2 but not Akt in the presence of hypoxic stress. ERK is a
downstream signaling molecule activated by MEK in response to
growth stimuli and involved in multiple signaling pathways
regulating cell survival, proliferation and motility (7). The MAPK pathway has been shown to
have a role in HIF-1α phosphorylation to enhance its
transcriptional activity (26). In
addition, there is evidence suggesting that activation of ERK is
associated with hypoxia-induced metastasis (7,27).
A link between hypoxia and Met overexpression in
hypoxia-induced invasion has been demonstrated in a number of
studies (28–31). It was shown that Met expression can
be induced during hypoxia at the transcriptional level in a
HIF-1α-dependent manner (29).
However, the molecular mechanisms of Met activation by hypoxia in
the present study as well as the precise association between tumor
hypoxia and cell invasion in cholangiocarcinoma still remain to be
fully elucidated. In the present study, induction of HIF-1α was
observed when RMCCA-1 cells were exposed to chronic hypoxia. This
may be linked with increased activation of the MAPK cascade, which
in turn results in upregulation of various genes by HIF-1.
In addition, immunohistochemical analysis indicated
that HIF-1α is preferentially expressed in cholangiocarcinoma but
not in the adjacent normal bile duct tissues obtained through
biopsy from cholangiocarcinoma patients. However, the expression
levels of HIF-1α were not significantly correlated with either
tumor differentiation or staging status. This finding is in
contrast to a study on gastric cancer, which found that the
expression of HIF-1α was significantly correlated with the clinical
staging (32). Variations in the
biological features of the tumors and the limited number of
specimens in the present study may account for different
results.
In conclusion, the present study reported that
hypoxic stress enhanced the aggressive phenotype of
cholangiocarcinoma. It was shown that the molecular basis
underlying hypoxia-induced invasion is through the activation of
the Met and the ERK signaling pathway. These findings supported Met
as one of the potential targets in therapeutic approaches to treat
cholangiocarcinoma.
Acknowledgments
The authors would like to thank Ms. Supatip Tujinda
(Department of Immunohistochemical Pathology, Rajavithi Hospital,
Bangkok, Thailand) and Ms. Shunyapat Narong (Division of
Biomolecular Science, Department of Pathology, Rajavithi Hospital)
for their help in the preparation and analysis of the
immunohistochemistry samples.
References
|
1
|
Zogragos GN, Farfaras A, Zagouri F,
Chrysikos D and Karaliotas K: Cholangiocarcinoma: principles and
current trends. Hepatobiliary Pancreat Dis Int. 10:10–20. 2011.
View Article : Google Scholar
|
|
2
|
Razumilava N and Gores GJ: Classification,
diagnosis and management of cholangiocarcinoma. Clin Gastroenterol
Hepatol. 11:13–21. 2013. View Article : Google Scholar
|
|
3
|
Rattanasinganchan P, Leelawat K,
Treepongkaruna SA, Tocharoentanaphol C, Subwongcharoen S,
Suthiphongchai T and Tohtong R: Establishment and characterization
of a cholan-giocarcinoma cell line (RMCCA-1) from a Thai patient.
World J Gastroenterol. 12:6500–6506. 2006.PubMed/NCBI
|
|
4
|
Friman S: Cholangiocarcinoma-current
treatment options. Scand J Surg. 100:30–34. 2011.
|
|
5
|
Miyoshi A, Kitajima Y, Ide T, et al:
Hypoxia accelerates cancer invasion of hepatoma cells by
upregulating MMP expression in an HIF-1α-independent manner. Int J
Oncol. 29:1533–1539. 2006.PubMed/NCBI
|
|
6
|
Yokoi K and Fidler IJ: Hypoxia increases
resistance of human pancreatic cancer cells to apoptosis induced by
gemcitabine. Clin Cancer Res. 10:2299–2306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim JY, Kim YJ, Lee S and Park JH: The
critical role of ERK in death resistance and invasiveness of
hypoxia-selected glioblastoma cells. BMC Cancer. 9:272009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kitajima Y, Ide T, Ohtsuka T and Miyazaki
K: Induction of hepatocyte growth factor activator gene expression
under hypoxia activates the hepatocyte growth factor/c-Met system
via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci.
99:1341–1347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koh MY, Lemos R Jr, Lui X and Powis G: The
hypoxia-associated factor switches cells from HIF-1α- to
HIF-2α-dependent signaling promoting stem cell characteristics,
aggressive tumor growth and invasion. Cancer Res. 71:4015–4027.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu OY, Hou MF, Yang SF, Huang SC and Lee
WY: Cobalt chloride-induced hypoxia modulates the invasive
potential and matrix matelloproteinases of primary and metastatic
breast cancer cells. Anticancer Res. 29:3131–3138. 2009.PubMed/NCBI
|
|
11
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cecchi F, Rabe DC and Bottaro DP:
Targeting the HGF/Met signaling pathway in cancer. Eur J Cancer.
46:1260–1270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Socoteanu MP, Mott F, Alpini G and Frankel
AE: c-Met targeted therapy of cholangiocarcinoma. World J
Gastroenterol. 14:2990–2994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsumura A, Kubota T, Taiyoh H, et al:
HGF regulates VEGF expression via the c-Met receptor downstream
pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int J
Oncol. 42:535–542. 2013.
|
|
15
|
Leelawat K, Leelawat S, Tepaksorn P,
Rattanasinganchan P, Leungchaweng A, Tohtong R and Sobhon P:
Involvement of c-Met/hepatocyte growth factor pathway in
cholangiocarcinoma cell invasion and its therapeutic inhibition
with small interfering RNA specific for c-Met. J Surg Res.
136:78–84. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santarpia L, Lippman SL and El-Naggar AK:
Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.
Expert Opin Ther Targets. 16:103–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katz M, Amit I and Yarden Y: Regulation of
MAPKs by growth factors and receptor tyrosine kinases. Biochim
Biophys Acta. 1773:1161–1176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)α: its protein stability and
biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harris AL: Hypoxia-a key regulatory factor
in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Organ SL and Tsao MS: An overview of the
Met signaling pathway. Ther Adv Med Oncol. 3(Suppl 1): S7–S19.
2011. View Article : Google Scholar
|
|
21
|
Seubwai W, Kraiklang R, Wongkham C and
Wongkham S: Hypoxia enhances aggressiveness of cholangiocarcinoma
cells. Asian Pac J Cancer Prev. 13(Suppl): 53–58. 2012.PubMed/NCBI
|
|
22
|
Gwak GY, Yoon JH, Kim KM, Lee HS, Chung JW
and Gores GJ: Hypoxia stimulates proliferation of human hepatoma
cells through the induction of hexokinase II expression. J Hepatol.
42:358–364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He G, Jiang Y, Zhang B and Wu G: The
effect of HIF-1α on glucose metabolism, growth and apoptosis of
pancreatic cancerous cells. Asia Pac J Clin Nutr. 23:174–180.
2014.
|
|
24
|
Dai R, Li J, Fu J, et al: The tyrosine
kinase c-Met contributes to the pro-tumorigenic function of the p38
kinase in human bile duct cholangiocarcinoma cells. J Biol Chem.
287:39812–39823. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu L, Nilsson MB, Saintigny P, et al:
Epidermal growth factor receptor regulates MET levels and
invasiveness through hypoxia-inducible factor-1α in non-small cell
lung cancer cells. Oncogene. 29:2616–2627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mylonis I, Chachami G, Samiotaka M, et al:
Identification of MAPK phosphorylation sites and their role in the
localization and activity of hypoxia-inducible factor-1α. J Biol
Chem. 281:33095–33106. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee KH, Choi EY, Hyun MS and Kim JR:
Involvement of MAPK pathway in hypoxia-induced up-regulation of
urokinase plasminogen activator receptor in a human prostatic
cancer cell line, PC3MLN4. Exp Mol Med. 36:57–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mazzone M and Comoglio PM: The Met
pathway: master switch and drug target in cancer progression. FASEB
J. 20:1611–1621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eckerich C, Zapf S, Fillbrandt R, Loges S,
Westphat M and Lamszus K: Hypoxia can induce c-Met expression in
glioma cells and enhance SF/HGF-induced cell migration. Int J
Cancer. 121:276–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sullivan R and Graham CH: Hypoxia-driven
selection of the metastatic phenotype. Cancer Metastasis Rev.
26:319–331. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hara S, Nakashiro K, Klosek SK, Ishikawa
T, Shintani S and Hamakawa H: Hypoxia enhances c-Met/HGF receptor
expression and signaling by activating HIF-1alpha in human salivary
gland cancer cells. Oral Oncol. 42:593–598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Li Z, Zhang H, et al: HIF-1α and
HIF-2α correlate with migration and invasion in gastric cancer.
Cancer Biol Ther. 10:376–382. 2010. View Article : Google Scholar : PubMed/NCBI
|