Introduction
Osteosarcoma (OS) is the most common primary bone
malignancy in children and young adults (1,2). In
spite of extensive advancements in diagnostic methods and surgical
techniques in recent years, the five-year survival rate of
osteosarcoma patients remains poor at 25–40% due to the high
frequency of pulmonary metastasis (3). Therefore, an improved understanding
of the mechanisms underlying osteosarcoma development and
progression is urgently required to optimize therapeutic options or
develop novel effective therapies.
MicroRNAs (miRNAs or miRs) are a class of highly
conserved short non-coding small RNAs, usually 18–25 nucleotides in
length, which inhibit translation and cleave mRNA by base-pairing
to the 3′ untranslated region of the target genes (4–8). In
mammals, miRNAs were predicted to regulate the activity of >50%
of all protein-coding genes (9).
Due to their widespread regulating functions, miRNAs are able to
regulate a variety of biological processes, including cell
proliferation, differentiation, migration, metabolism and apoptosis
(10). For cancers, homeostatic
expression of miRNAs is disrupted, which results in aberrant gene
expression in tumor initiation, development and metastasis. An
increasing number of studies has pointed out that multiple miRNAs
are involved in carcinogenesis and tumor progression in OS
(11–15). For example, miR-124 was reported to
function as a tumor oncogene by downregulating putative tumor
suppressor 1 (11). miR-145 exerts
tumor-suppressive effects in osteosarcoma genesis through
suppression of Rho-associated, coiled-coil containing protein
kinase 1 (12). A recent miRNA
microarray analysis by Lulla et al (13) identified 22 differentially
expressed miRNAs in osteosarcomas and found that miR-210, a key
factor in the hypoxic response, was significantly elevated in
osteosarcoma tissues compared with that in the corresponding
controls. Subsequently, Cai et al (14) further confirmed this result, and
found that miR-210 expression was significantly increased in
osteosarcoma tissues compared with that in corresponding
non-cancerous bone tissues, and that miR-210 upregulation more
frequently occurred in osteosarcoma tissues with large tumor size,
poor response to pre-operative chemotherapy and positive
metastasis. In addition, miR-210 has been demonstrated to be
upregulated in all cell types under hypoxic conditions (15). However, the precise role of miR-210
in OS cells remains elusive at present.
Therefore, the aim of the present study was to
identify the detailed roles of miR-210 in OS cells in vitro
and in vivo in order to determine its utility in OS therapy.
The effect of downregulation of miR-210 on clonogenicity, migration
and invasion, as well as apoptosis and cell cycle were examined
in vitro. In addition, the effect of miR-210 on tumor growth
in nude mice was assessed. The findings of the present study
implied that miR-210 is a potential therapeutic target for OS.
Materials and methods
Cell lines and cell culture
The human osteosarcoma cell lines MG63, Saos-2, U2OS
and the human osteoblast cell line h-OB were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA). Cells
were grown in Dulbecco's modified Eagle's medium (DMEM; HyClone, GE
Healthcare, Little Chalfont, UK) supplemented with 10% fetal bovine
serum (FBS; Gibco-BRL, Invitrogen Life Technologies, Carlsbad, CA,
USA), 2 mM glutamine, 100 U/ml penicillin and 100 mg/ml
streptomycin (Sigma-Aldrich, St Louis, MO, USA) at 37°C in a 5%
CO2 atmosphere.
miR NA transfection
miR-210 inhibitor (5′-TCAGCCGCT GTCACACGCACAG-3′) or
negative control miRNA (5′-GTG TAACACGTCTATACGCCCA-3′) (Applied
Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) were
transiently transfected into MG63 cells in six-well plates using
Lipofectamine 2000 (Invitrogen Life Technologies) according to the
manufacturer's instructions at a concentration of 100 nM.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Transfected cells were harvested by centrifugation
at 1,000 × g for 5 min at room temperature after 48 h, and total
RNA enriched with small RNAs was isolated using the mirVana miRNA
Isolation kit (Ambion, Life Technologies, Thermo Fisher Scientific)
according to manufacturer's instructions. Concentration and purity
of RNA were determined using a GeneQuant RNA/DNA Calculator (GE
Healthcare Life Sciences, Uppsala, Sweden), and miRNA was reversely
transcribed using the One Step Prime script miRNA cDNA Synthesis
kit (Qiagen, Hilden, Germany) and then quantified by real-time
RT-PCR using SYBR Premix Ex Taq (Takara, Dalian, China). All PCR
reactions were monitored using an ABI 7900 Fast system (Applied
Biosystems). The primers of miR-210 (CUGUGCGUGUGACAGCGGCUGA) and
RNU6B (CGCAAGGAUGACACGCAAAUUCGUGAA GCGUUCCAUAUUUUU) were purchased
from Applied Biosystems. The expression levels of RNU6B were used
as an internal control. PCR was performed at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The
relative quantified miRNA levels were calculated as the fold change
after normalization to U6 RNA using the equation 2−∆∆Ct
with the Rotor-Gene 6000 Series Software 1.7 (Qiagen, Hilden,
Germany). miR-210 levels in the negative control cells were
compared with those of untransfected cells and non-significant
changes <5% were observed. All experiments were repeated three
times to reduce curve-derived variance.
Cell prolifration and colony formation
assay
Cell viability was examined using an MTT assay. In
brief, MG63 cells were seeded into 96-well plates at a density of
2,000 cells/well containing 100 µl culture medium and
cultured overnight, and cells were then transfected with 100 nM
miR-210 inhibitor or negative control. Non-transfected cells
(blank) were used as a control. Fresh complete medium was changed
for cells at 24 h post-transfection except for those to be tested
for viability at this time-point. Every 24-h interval, 20 µl
5 mg/ml MTT (Sigma-Aldrich) was added into each corresponding test
well and incubated for 4 h in a humidified incubator. The
supernatant was then discarded and 200 µl dimethylsulfoxide
(DMSO; Sigma-Aldrich) was added to each well to dissolve the
formazan. The optical density (OD) was evaluated by measuring the
absorbance at a test wavelength of 490 nm and a reference
wavelength of 630 nm using a SpectraMax M3 ELISA spectrophotometer
(Molecular Devices, LLC, Sunnyvale, CA, USA). Wells without cells
(DMSO alone) were used as a control to normalize the results. Each
test was performed daily for five consecutive days and repeated in
eight wells. The experiments were repeated three times
independently.
For the colony formation assay, cells were seeded in
six-well plates at a low density (1×103 cells/well) and
cultured overnight. Cells were then transfected with 100 nM miR-210
inhibitor or negative control and cultured for 7 days. Cells were
then fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 min and
counted after staining with 1% crystal violet (Sigma-Aldrich). The
experiments were performed in triplicate wells for at least three
times.
Cell cycle analysis by flow
cytometry
24 h prior to transfection, MG63 cells were seeded
into six-well plates at a density of 1×105 cells/well.
Cells were then transfected with 100 nM miR-210 inhibitor or
corresponding negative control. 48 h after transfection, the
transfected MG63 cells in the logarythmic growth phase were stained
with the DNA-binding dye propidium iodide (PI; 50 mg/ml;
Sigma-Aldrich) and RNase (1.0 mg/ml; Sigma-Aldrich) for 30 min at
37°C in the dark and examined by fluorescence-activated
cell-sorting (FACS) using a flow cytometer (FACScan; BD
Biosciences, Franklin Lakes, NJ, USA), and DNA histograms were
analyzed with CellQuest v 3.3 software (BD Biosciences).
Cell apoptosis assay
MG63 cells were seeded into six-well plates at a
density of 1×105 cells/well and cultured overnight.
Then, cells were transfected with 100 nM miR-210 inhibitor or
corresponding negative control. 48 h after transfection, MG63 cells
were incubated with Annexin V/PI (Sigma-Aldrich) for 15 min at room
temperature in the dark. The cells were then analyzed by flow
cytometry with fluorescence-activated cell-sorting (FACS) flow
cytometer (FACScan; BD Biosciences). Each test was repeated in
triplicate. Experiments were performed in triplicate.
In addition, caspase-3, caspase-8 and caspase-9
activity were tested using Caspase Colorimetric Protease Assay kits
(EMD Millipore, Billerica, MA, USA) as an additional indicator of
apoptosis.
Caspase activity
The activity of caspase-3, caspase-8 and caspase-9
was determined by caspase colorimetric protease assay kits (EMD
Millipore) according to the manufacturer's instructions. In brief,
MG63 cells were seeded into six-well plates at a density of
1×105 cells/well and cultured overnight. Cells were then
transfected with 100 nM miR-210 inhibitor or corresponding negative
control and cultured for 48 h. The cells were washed twice with
ice-cold phosphate-buffered saline (PBS; Sigma-Aldrich) and
harvested by centrifugation at 800 ×g for 5 min. The cell pellets
were then lysed in 150 µl buffer provided in the kit.
Protein concentrations of lysates were measured by the Lowry
method. An aliquot of lysates (80 µl) was incubated with 10
µl substrate of each caspase at 37°C for 1 h. Samples were
analyzed using a Multiskan Spectrum microplate reader (Thermo
Fisher Scientific) at 405 nm. The relative caspase activity of the
negative control group was referred to as 100.
Wound-healing assays
MG63 cells were treated with miR-210 inhibitor or
corresponding negative control when cells were grown to 80–90%
confluence in 24-well plates. At 24 h following transfection,
linear scratch wounds were created on the confluent cell monolayers
with a 100-µl pipette tip. To stop cells from entering the
cell cycle prior to wounding, cells were maintained in serum-free
medium. To visualize migrating cells and wound healing, images were
captured at 0 and 24 h using an IX51 inverted microscope (Olympus,
Tokyo, Japan). Five random fields of view were selected from each
well and the cells in three wells of each group were
determined.
Cell invasion assay
For the cell invasion assay, cells transfected with
miR-210 inhibitor or corresponding negative control were harvested
and re-suspended in serum-free DMEM. Aliquots of cells
(5×104) were placed in chambers coated with 150 mg
Matrigel (BD Biosciences), which were fitted into 24-well plates
and incubated for 24 h in DMEM with 10% FBS. Cells remaining on the
upper surface of the membranes were removed, whereas those adhering
to the lower surface were fixed with ice-cold methanol
(Sigma-Aldrich) and stained with 1% crystal violet. Stained cells
were visualized and counted under the IX51 inverted microscope
(Olympus). Experiments were performed in triplicate.
Western blot analysis
At 48 h post-transfection, MG63 cells were washed
with pre-chilled PBS and then subjected to lysis in a
radioimmunoprecipitation assay buffer containing 0.5% SDS and 3%
proteinase inhibitor cocktail (Sigma-Aldrich) on ice for 30 min.
The lysate was then centrifuged at 14,000 ×g for 20 min, and the
supernatant was harvested as total proteins for subsequent
analysis. The total concentration of protein was measured using a
bicinchoninic acid protein assay kit (Boster, Beijing, China).
Equal amounts of protein lysates (20 µg per lane) was
separated by 8–15% SDS-PAGE (Sigma-Aldrich) and then
electrotransferred onto nitrocellulose membranes (Invitrogen Life
Technologies). The membranes were blocked with Tris-buffered saline
containing Tween 20 (TBST; Sigma-Aldrich) with 5% non-fat milk
powder for 2 h and incubated with the following primary antibodies:
Rabbit monoclonal anti-human anti-matrix metalloproteinase (MMP)-2
(#13132; 1:1,000; Cell Signaling Technology, Danvers, MA, USA);
mouse monoclonal anti-human anti-MMP-9 (sc-12759; 1:2,000; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA); rabbit polyclonal
anti-human anti-vascular endothelial growth factor (VEGF; #2445;
1:2,000; Cell Signaling Technology) and mouse monoclonal anti-human
anti-β-actin (sc-47778; 1:10,000; Santa Cruz Biotechnology)
overnight at 4°C. After three washes with TBST, the membranes were
incubated with polyclonal goat anti-mouse (sc-2005) or goat
anti-rabbit (sc-2004) horseradish peroxidase-conjugated secondary
antibodies (1:10,000; Santa Cruz Biotechnology, Inc.) for 2 h at
room temperature. Blots were washed three times with TBST again,
and proteins were detected by enhanced chemilumi-nescence (ECL;
ECL-Plus; Thermo Fisher Scientific). β-actin was used as a loading
control. The optical density of protein fragments was quantified by
Quantity One v.4.6.1 software (Bio-Rad, Billerica, MA, USA).
Tumor growth in vivo
Logarithmically growing MG63 cells transfected with
miR-210 inhibitor or negative control were harvested and
re-suspended in PBS, and aliquots of 2×106 cells
inoculated subcutaneously into five-week-old male BALB/c nude mice
(HFK Bio-Technology, Co., Ltd; Beijing, China), respectively. The
mice (n=10 per group) were maintained in specific pathogen-free
conditions with free access to food and water, and were maintained
at normal 12 h light/dark cycles. After 30 days, mice were
sacrificed by cervical dislocation, followed by surgical excision
of tumors. Tumors were weighed and their volumes were calculated as
0.5236 × width2 × length. All animal experiments were
performed following the standards of animal care as outlined in the
Guide for the Care and Use of Experimental Animals of Dalian
Medical University Jilin University (Dalian, China), following a
protocol approved by the Ethics Committees of the Disease Model
Research Center, Dalian Medical University (Dalian, China).
Statistical analysis
All values are expressed as the mean ± standard
deviation. Student's t test and one-way analysis of variance
were used to determine statistical significance. All statistical
tests were two-sided. P<0.05 was considered to indicate a
statistically significant difference between values. Statistical
analyses were performed using SPSS 16.0 statistics software (SPSS
Inc, Chicago, IL, USA) and GraphPad Prism version 5.01 (GraphPad
Software, La Jolla, CA, USA) for Windows®.
Results
miR-210 is upregulated in human
osteosarcoma cell lines
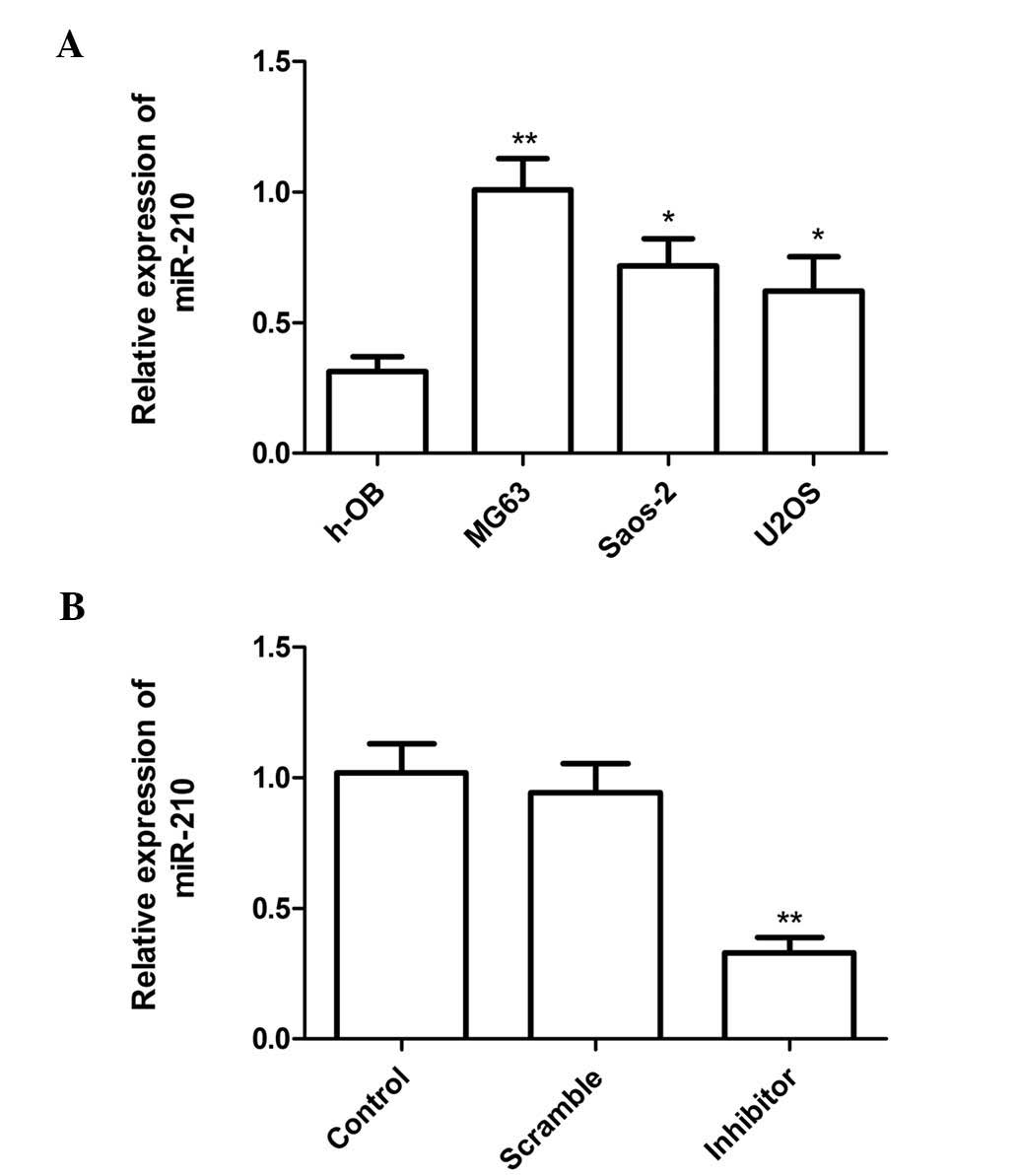
To elucidate the role of miR-210 in human
osteosarcoma, the expression levels of miR-210 in three human
osteosarcoma cell lines, U2OS, Saos-2 and MG63, as well as in the
human osteoblast cell line h-OB were determined using RT-qPCR. The
h-OB cell line was used as a control. The results showed that
osteosarcoma cell lines (U2OS, Saos-2 and MG-63) exhibited higher
miR-210 expression as compared with that in the human osteoblast
cell line (h-OB) (Fig. 1A). These
results indicated that miR-210 is significantly upregulated in
osteosarcoma cell lines.
To investigate the function of miR-210, miR-210
inhibitor or corresponding scrambled control were transfected into
MG63 cells (which had the highest expression levels of miR-210). At
24 h after transfection, miR-210 expression was evaluated by
RT-qPCR. The results showed that miR-210 significantly decreased
the expression of miR-210 compared with that in the scrambled group
and the blank group. In addition, no statistically significant
difference in miR-210 expression was found between the scrambled
and blank control groups (Fig.
1B).
miR-210 inhibition suppresses cell
proliferation and colony formation of MG63 cells
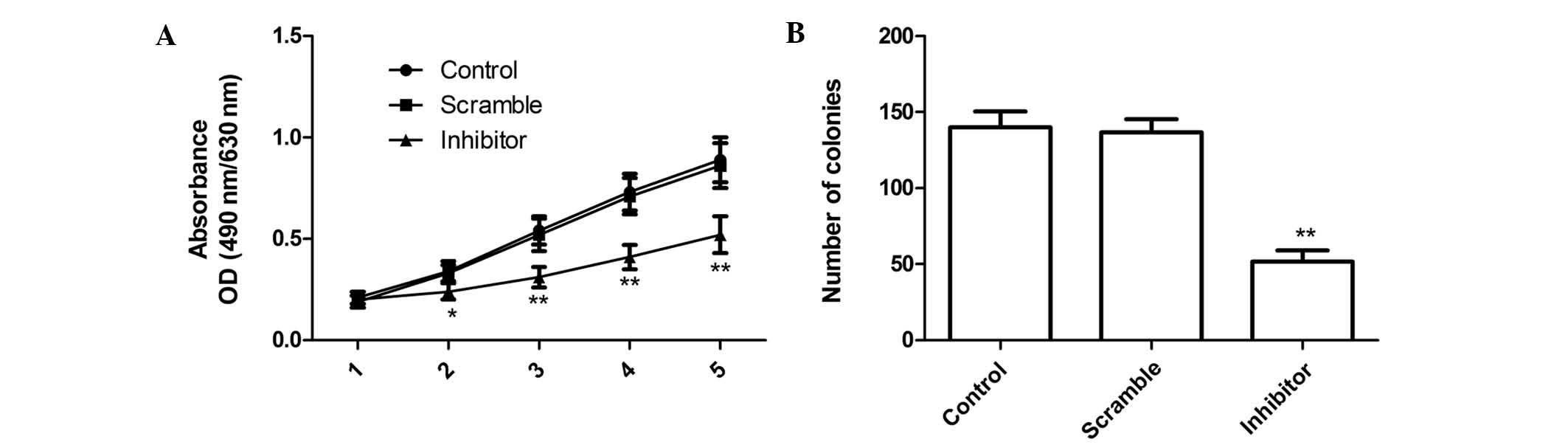
The proliferation rates of MG63 cells were
determined via MTT assay following transfection with miR-210
inhibitor. The cell proliferation curve is depicted in Fig. 2A. The results demonstrated that the
viability of MG63 cells was markedly decreased by transfection with
miR-210 inhibitor compared with that in the scrambled and the blank
group, and the suppressive effect of miR-210 inhibitor on cell
proliferation was observed from day 2 onwards, becoming more
obvious on days 4 and 5 (P<0.05; Fig. 2A). No statistically significant
difference was observed between the proliferation rate of the blank
and that of the scrambled group (Fig.
2A).
In addition, the present study determined the effect
of miR-210 on cell colony formation of MG63 cells. The results
showed that miR-210 inhibitor significantly inhibited cell colony
formation compared to that in the blank and scrambled group
(P<0.05; Fig. 2B). No
statistically significant difference was observed rate between the
proliferation rate in the blank and that of the scrambled group.
These findings suggested that the downregulation miR-210 greatly
inhibited cell proliferation and colony formation in MG63
cells.
miR-210 inhibition induces G1 arrest and
cell apoptosis in MG63 cells
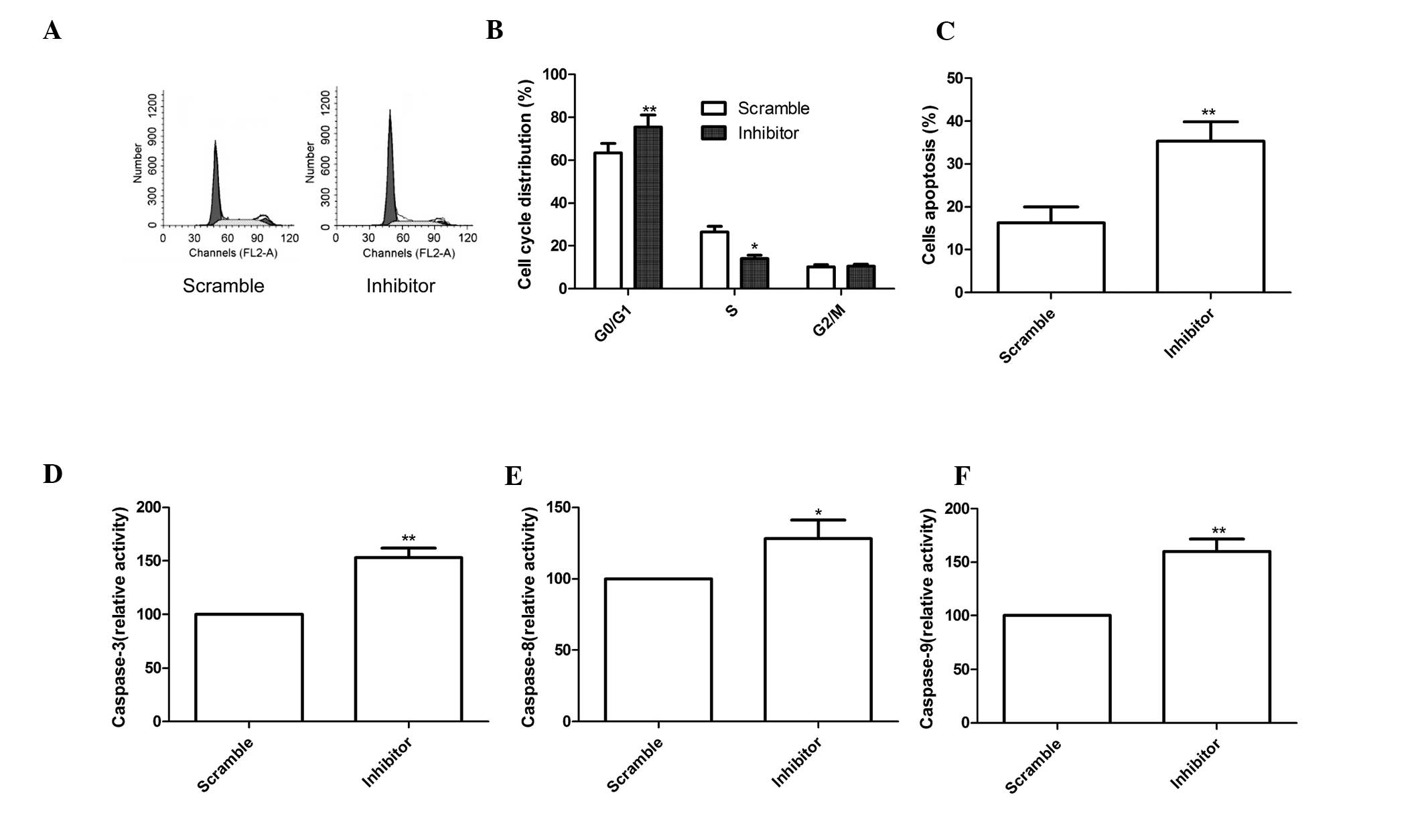
To determine the effects of miR-210 on the cell
cycle, FACScan flow cytometry assays were performed. The results
demonstrated that miR-210 inhibitor was able to increase the
percentage of cells in G1 phase, while decreasing the percentage of
cells in S phase compared with that in the scramble group
(P<0.05, Fig. 3A and B).
Next, the role of miR-210 in MG63-cell apopto sis
was assessed using Annexin V/PI staining and flow cytometric
analysis. The results showed that the miR-210 inhibitor
significantly induced cell apoptosis compared with that in the
scrambled group (P<0.01; Fig.
3C).
Finally, the effects of miR-210 on caspase-3,
caspase-8 and caspase-9 activity were assessed using ELISA. As
shown in Fig. 3D–F, caspase-3,
caspase-8 and caspase-9 activity in the miR-210 inhibitor group
were significantly increased compared with those in the scrambled
group (all P<0.01).
MiR-210 inhibition reduces cell migration
and invasion in MG63 cells
To ascertain the inhibitory effect of miR-210
inhibitor on cell motility in MG63 cells, scratch wound migration
assays were performed. MG63 cells transfected with miR-210
inhibitor migrated at a significantly decreased rate compared with
that of cells transfected with the negative control (P<0.01;
Fig. 4A and B).
 | Figure 4miR-210 inhibitor reduced cell
migration and invasion of MG63 cells. (A) Cell migration was
determined using the wound-healing assay, magnification, ×200. (B)
Quantified numbers of migrated cells. (C) Cell invasion was
determined by a Matrigel Transwell assay, magnification, ×200. (D)
Quantified number of invaded cells. (E) Western blot analysis of
VEGF, MMP-2 and MMP-9 protein expression after transfection with
miR-210 inhibitor and corresponding scrambled control. β-actin was
used as an internal control. (F) Relative quantification of VEGF,
MMP-2 and MMP-9 protein by densitometric analysis.
*P<0.05; **P<0.01, vs. the negative
control. All values are expressed as the mean ± standard deviation.
VEGF, vascular endothelial growth factor; MMP, matrix
metalloproteinase; miR, microRNA. |
In addition, the ability of miR-210 to affect the
invasiveness of MG63 cells was further investigated using the
Transwell assay. The results showed that miR-210 inhibition
significantly inhibited cell invasion compared with that in the
scrambled group (P<0.01; Fig. 4C
and D).
To determine the potential mechanism of the effect
of miR-210 on cell migration and invasion, the levels of the cell
invasion-associated proteins VEGF, MMP-2 and MMP-9 were assessed
using western blot analysis. As shown in Fig. 4E and F, VEGF, MMP-2 and MMP-9
protein expression were significantly decreased in the miR-210
inhibitor group compared with that in the scrambled group
(P<0.05).
miR-210 inhibitor suppresses tumor growth
in a nude mouse model
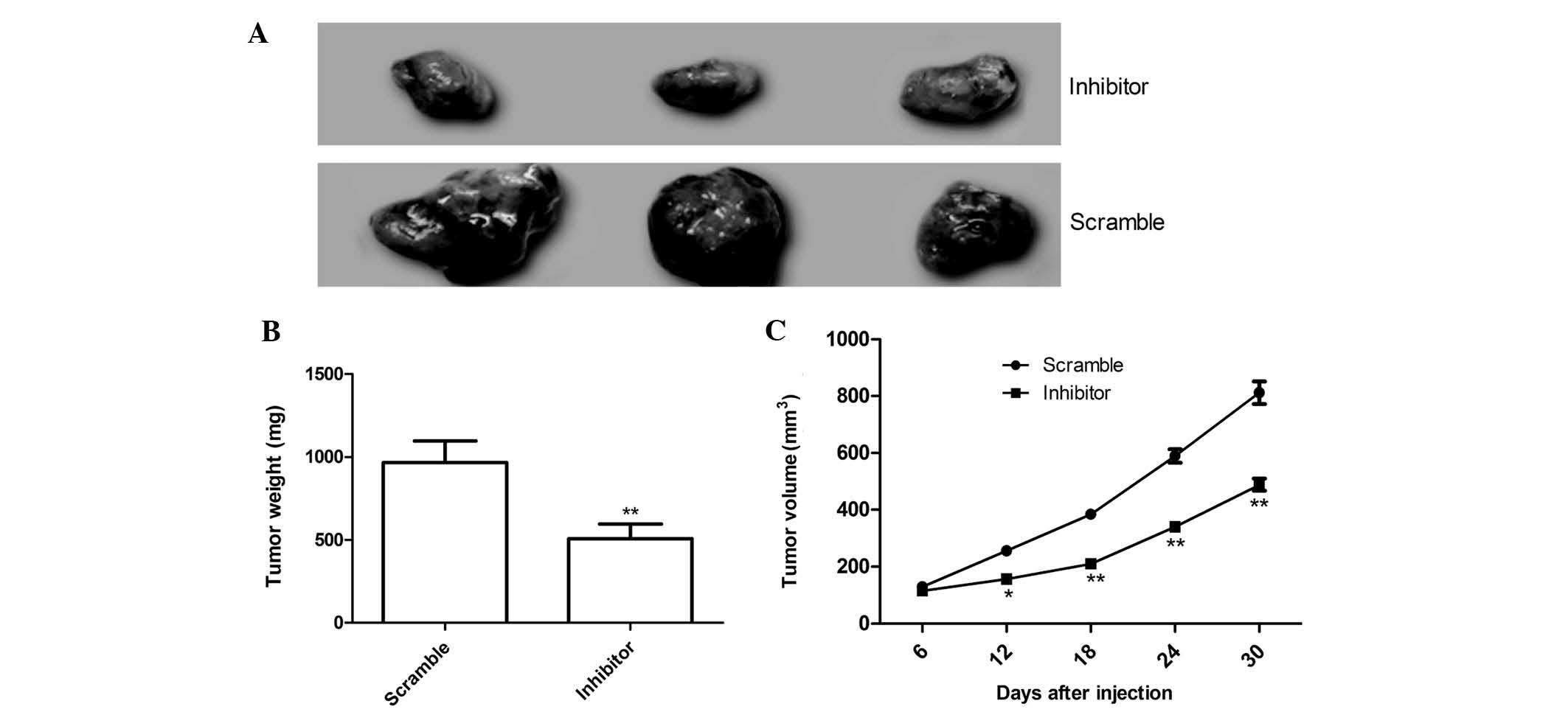
To further determine whether miR-210 affects OS
tumor growth in vivo, MG63 cells engineered to stably
over-express miR-210 inhibitor or the scrambled sequence were
subcutaneously inoculated into nude mice (n=10), respectively, and
the tumor growth was measured over 30 days. As shown in Fig. 5A, tumors in the miR-210 inhibitor
group were significantly smaller than tumors in the negative
control group. The average volume and weight of the tumors in
miR-210 inhibitor group were significantly reduced compared with
those in the scrambled group (P<0.05; Fig. 5B and C). These data collectively
indicated that miR-210 was able to suppress OS tumor growth in
vivo.
Discussion
The results of the present study showed that miR-210
was increased in human osteosarcoma cell lines compared with that
in h-OB osteoblasts cells. Furthermore, the results indicated that
downregulation miR-210 inhibited cell migration and invasion of
MG63 cells. In addition, downregulation miR-210 suppressed tumor
growth of OS in nude mice. To the best our knowledge, the present
study was the first to show that miR-210 has a crucial role in OS
development and progression.
miR-210, an intronic miRNA, is located within the
genomic loci of transcript AK123483 (16). miR-210 has been reported to be
involved in the regulation of a hypoxia-inducible factor-1
(HIF-1)-dependent (17,18), HIF-2-dependent (17) and HIF-independent (19) pathways. miR-210 has also been
identified a variety of functionally important targets involved in
a wide range of physiological processes, including cell cycle
regulation, differentiation, DNA damage repair, cell survival,
angiogenesis as well as metabolism (20,21).
miR-210 has been shown to be overexpressed in a variety of
malignancies, including pancreatic cancer (22), head and neck squamous cell
carcinomas (23), breast cancer
(24), lung cancer (25), renal cell cancer (26), non-small lung cancer (27) and osteosarcoma (14). Most studies have reported that
miR-210 may act as an oncogenic miRNA and is associated with poor
prognosis in a number of human epithelial cancer types (27–31).
However, certain studies have indicated that miR-210 expression is
lost during tumorigenesis and that the miRNA exerts a
tumor-suppressor effect on human epithelial ovarian and esophageal
squamous cell carcinoma (32,33).
These findings suggested that miR-210 may have important roles
during tumorigenesis and cancer progression and may exert various
effects on different cancer types. Regarding OS, previous studies
demonstrated that miR-210 was upregulated in OS tissue and an OS
cell line (13,14). Of note, miR-210 was found to be a
positive regulator of osteoblastic differentiation through the
inhibition of activin A receptor type 1B (34). In line with the results of previous
studies, the present study found that miR-210 was increased in
human osteosarcoma cell lines, and that downregulation of miR-210
inhibited tumor growth of osteosarcoma in vitro and in
vivo. The present results, together with those of other
studies, suggested that miR-210 may act as an oncogenic miRNA and
have crucial roles in OS tumorigenesis.
With the development of bioinformatic miRNA target
prediction tools as well as the improvement of experimental
approaches, numerous diverse targets of miR-210 have been
identified, including E2F3, NPTX1, RAD52, ACVR1B, MNT, CASP8AP2,
FGFRL1 and HOXA1 (15,35–37).
It has been shown that miR-210 targets >35 genes (36,37).
The present study demonstrated that miR-210 is involved in OS cell
proliferation, clonogenicity, migration and invasion, as well as
cell apoptosis via targeting multiple genes.
In conclusion, the results of the present study
provided novel evidence that downregulation of miR-210 can inhibit
cell proliferation, clonogenicity, migration, invasion, as well as
induced G1 arrest and cell apoptosis in vitro, as well as
suppress tumor growth in vivo. The findings therefore
implied that miR-210 may be a potential therapeutic target for
osteosarcoma.
References
|
1
|
Ottaviani G, Robert RS, Huh WW, Palla S
and Jaffe N: Sociooccupational and physical outcomes more than 20
years after the diagnosis of osteosarcoma in children and
adolescents: limb salvage versus amputation. Cancer. 119:3727–3736.
2013.PubMed/NCBI
|
|
2
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
3
|
Yang J and Zhang W: New molecular insights
into osteosarcoma targeted therapy. Curr Opin Oncol. 25:398–406.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim VN, Han J and Siomi MC: Biogenesis of
small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Erhard F, Haas J, Lieber D, et al:
Widespread context dependency of microRNA-mediated regulation.
Genome Res. 24:906–919. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Almeida MI, Reis RM and Calin GA: MicroRNA
history: discovery, recent applications and next frontiers. Mutat
Res. 717:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Z and Wang T: miR-214 promotes the
proliferation and invasion of osteosarcoma cells through direct
suppression of LZTS1. Biochem Biophys Res Commun. 449:190–195.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li E, Zhang J, Yuan T and Ma B: miR-145
inhibits osteosarcoma cells proliferation and invasion by targeting
ROCK1. Tumour Biol. 35:7645–7650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lulla RR, Costa FF, Bischof JM and Chou
PM: Identification of differentially expressed microRNAs in
osteosarcoma. Sarcoma. 2011:7326902011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cai H, Lin L, Cai H, Tang M and Wang Z:
Prognostic evaluation of microRNA-210 expression in pediatric
osteosarcoma. Med Oncol. 30:4992013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fasanaro P, Greco S, Lorenzi M, et al: An
integrated approach for experimental target identification of
hypoxia-induced miR-210. J Biol Chem. 284:35134–35143. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giannakakis A, Sandaltzopoulos R, Greshock
J, et al: miR-210 links hypoxia with cell cycle regulation and is
deleted in human epithelial ovarian cancer. Cancer Biol Ther.
7:255–264. 2008. View Article : Google Scholar
|
|
17
|
McCormick RI, Blick C, Ragoussis J, et al:
miR-210 is a target of hypoxia-inducible factors 1 and 2 in renal
cancer, regulates ISCU and correlates with good prognosis. Br J
Cancer. 108:1133–1142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang X, Ding L, Bennewith KL, Tong RT,
Welford SM, Ang KK, Story M, Le QT and Giaccia AJ:
Hypoxia-inducible mir-210 regulates normoxic gene expression
involved in tumor initiation. Mol Cell. 35:856–867. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takikawa T, Masamune A, Hamada S, Nakano
E, Yoshida N and Shimosegawa T: miR-210 regulates the interaction
between pancreatic cancer cells and stellate cells. Biochem Biophys
Res Commun. 437:433–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Devlin C, Greco S, Martelli F and Ivan M:
miR-210: More than a silent player in hypoxia. IUBMB Life.
63:94–100. 2011.PubMed/NCBI
|
|
21
|
Chan SY and Loscalzo J: MicroRNA-210: a
unique and pleiotropic hypoxamir. Cell Cycle. 9:1072–1083. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Greither T, Grochola LF, Udelnow A,
Lautenschlager C, Wurl P and Taubert H: Elevated expression of
microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated
with poorer survival. Int J Cancer. 126:73–80. 2010. View Article : Google Scholar
|
|
23
|
Gee HE, Camps C, Buffa FM, et al:
hsa-mir-210 is a marker of tumor hypoxia and a prognostic factor in
head and neck cancer. Cancer. 116:2148–2158. 2010.PubMed/NCBI
|
|
24
|
Foekens JA, Sieuwerts AM, Smid M, et al:
Four miRNAs associated with aggressiveness of lymph node-negative,
estrogen receptor-positive human breast cancer. Proc Natl Acad Sci
USA. 105:13021–13026. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Puissegur MP, Mazure NM, Bertero T, et al:
miR-210 is overexpressed in late stages of lung cancer and mediates
mitochondrial alterations associated with modulation of HIF-1
activity. Cell Death Differ. 18:465–478. 2011. View Article : Google Scholar :
|
|
26
|
Juan D, Alexe G, Antes T, et al:
Identification of a microRNA panel for clear-cell kidney cancer.
Urology. 75:835–841. 2010. View Article : Google Scholar
|
|
27
|
Eilertsen M, Andersen S, Al-Saad S, et al:
Positive prognostic impact of miR-210 in non-small cell lung
cancer. Lung Cancer. 83:272–278. 2014. View Article : Google Scholar
|
|
28
|
Ying Q, Liang L, Guo W, et al:
Hypoxia-inducible microRNA-210 augments the metastatic potential of
tumor cells by targeting vacuole membrane protein 1 in
hepatocellular carcinoma. Hepatology. 54:2064–2075. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong L, Yang J, Han Y, Lu Q, Cao J and
Syed L: High expression of miR-210 predicts poor survival in
patients with breast cancer: a meta-analysis. Gene. 507:135–138.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wotschofsky Z, Busch J, Jung M, et al:
Diagnostic and prognostic potential of differentially expressed
miRNAs between metastatic and non-metastatic renal cell carcinoma
at the time of nephrectomy. Clin Chim Acta. 416:5–10. 2013.
View Article : Google Scholar
|
|
31
|
Qu A, Du L, Yang Y, et al:
Hypoxia-inducible MiR-210 is an independent prognostic factor and
contributes to metastasis in colorectal cancer. PLoS One.
9:e909522014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Giannakakis A, Sandaltzopoulos R, Greshock
J, et al: miR-210 links hypoxia with cell cycle regulation and is
deleted in human epithelial ovarian cancer. Cancer Biol Ther.
7:255–264. 2008. View Article : Google Scholar
|
|
33
|
Tsuchiya S: The role of microRNA-210 in
esophageal squamous cell carcinoma. Yakugaku Zasshi. 132:1069–1073.
2012.In Japanese. View Article : Google Scholar
|
|
34
|
Mizuno Y, Tokuzawa Y, Ninomiya Y, et al:
miR-210 promotes osteoblastic differentiation through inhibition of
AcvR1b. FEBS Lett. 583:2263–2268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan YC, Banerjee J, Choi SY and Sen CK:
miR-210: the master hypoxamir. Microcirculation. 19:215–223. 2012.
View Article : Google Scholar :
|
|
36
|
Mei Y, Gao C, Wang K, et al: Effect of
microRNA-210 on prognosis and response to chemotherapeutic drugs in
pediatric acute lymphoblastic leukemia. Cancer Sci. 105:463–472.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qin Q, Furong W and Baosheng L: Multiple
functions of hypoxia-regulated miR-210 in cancer. J Exp Clin Cancer
Res. 33:502014. View Article : Google Scholar : PubMed/NCBI
|