Introduction
Liver cancer is one of the most common types of
cancer and is the third most frequent cause of cancer-associated
mortality. Hepatocellular carcinoma (HCC) is the major histological
subtype among types of primary liver cancer, and it is estimated
that >600,000 cases of HCC are newly diagnosed each year
worldwide, >50% of which are in China (1,2).
Despite advances in diagnosis and multimodality therapies for HCC,
the prognosis of the disease remains poor, with a 5-year overall
survival rate of <14% (3).
The environmental risk factors for HCC include
hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, food
aflatoxin contaminants, alcohol intake and smoking (2). HBV infection is the dominant global
attributable risk factor, accounting for >80% of the cases of
HCC in China (2). HBV is small
enveloped DNA virus, belonging to the hepadnavirus family, which is
capable of integrating into and altering the host genome. A total
of four overlapping open reading frames, encoding for the surface
protein, core protein, polymerase protein and X protein (HBX), have
been identified in the small 3.2 kb DNA of HBV (4,5). HBX
is the regulatory protein of the virus, and is frequently detected
in the tumor tissues of patients with HBV-associated HCC (4,5). HBX
transgenic mice have been reported to exhibit a positive
correlation between the expression levels of HBX and the
development of HCC (6,7). Consuming food containing aflatoxin is
another high environmental risk factor for HCC. Aflatoxin B1 is the
most abundant form of aflatoxin, produced by the fungus
Aspergillus flavus, which contaminates food, particularly
under hot and humid conditions (8–10).
The metabolites of Aflatoxin B1 can form a DNA adduct at the third
base of codon 249 in the TP53 gene, inducing a G>T transversion
and an R249S mutation, which is the most frequently altered type of
TP53 in HCC tissues (8–10).
Alterations in multiple signaling pathways and
patterns of host gene expression have been documented following HBV
infections. Somatic mutations, which trigger oncogenes or
inactivate tumor suppressor genes, contribute to the development of
HCC. Previous studies using several strategies have demonstrated
that certain genetic alterations occur in HCC cells, including
TP53, CTNNB1, Axin1 and ARID1A high frequency mutation genes and
Kras and APC low frequency mutation genes (11–13).
The development of next generation sequencing (NGS) has led to an
increase in the number of mutations identified in HCC tissues.
Huang et al (14)
investigated the spectrum of molecular aberrations in liver cancer
using whole exome sequencing, and identified 356 candidate somatic
single nucleotide variants (SNVs) within 347 genes. Despite efforts
to improve the diagnosis and treatment of HCC, the molecular
mechanisms leading to the development and progression of
HBV-associated HCC remain to be fully elucidated. In addition,
investigations focus predominantly on HCC samples from southern and
eastern China, while molecular data from HCC samples in western
China remains limited. Due to the different environmental factors,
patients with HCC in western China may exhibit characteristic
patterns of gene alterations.
In the present study, targeted sequencing of 372
genes was performed using an NGS platform, in order to investigate
the mutation spectrum of HCC samples from western China. The
investigation aimed to provide resources for understanding the
molecular alterations associated with the development of HCC.
Materials and methods
Tissue samples
A total of 12 patients from western China, who had
undergone surgery for HCC at Tangdu Hospital, Fourth Military
Medical University (Xi'an, China) between December 2010 and
September 2012, were included in the present study. The patients'
cancerous and adjacent non-cancerous tissues (minimum, 3 cm) were
collected and analyzed in the present study. Both cancerous and
non-cancerous samples were flash frozen in liquid nitrogen
following surgery and then stored at −80°C, until DNA extraction.
The size of tissue samples used for DNA extraction was ~0.2×0.2×0.2
cm. The majority of these patients were chronic HBV carriers. The
study population consisted of 11 males and 1 female, with a median
age of 51 years old (range 33–65 years). All the HCC cases were
histopathologically confirmed. Cirrhosis and capsules were
confirmed by histochemical analysis using a light microscope
(CDM965; Shanghai Tuming Optical Instrument Co., Ltd., Shanghai,
China). Information regarding family history, smoking history, and
drinking history was acquired by questionnaire.
The experiments were performed with the
understanding and written consent of each patient, and the
investigation was performed in accordance with The Code of Ethics
of the World Medical Association (Declaration of Helsinki), printed
in the British Medical Journal (1964) (15). The present study was also approved
by the Ethics Committee of Tangdu Hospital, Fourth Military Medical
University.
Target enrichment of genomic DNA and
sequencing
Genomic DNA was extracted using a QIAamp DNA Mini
kit (cat. no. 51306), according to the manufacturer's instructions
(Qiagen, Dusseldorf, Germany). The targeted regions included all
exons of 372 genes. The targeted genes were selected from the
Catalogue of Somatic Mutations in Cancer (Cosmic; http://www.sanger.ac.uk/genetics/CGP/cosmic) database
using the 2011 updated edition. Illumina TruSeq technology was used
to capture the exon region of 372 genes, using a TruSeq Custom
Enrichment Trial kit, according to the manufacturer's instructions
(Illumina, Inc., San Diego, CA, USA). An Illumina TruSeq DNA Sample
Prep kit was used for TruSeq sample preparation, and the samples
were then sequenced using the Illumina HiSeq platform at Shanghai
ZhangJiang Translational Medicine Research Center (Shanghai,
China).
Data analysis
For each patient, DNA was extracted from both tumor
tissues and non tumor tissues, and was sequenced at the same time.
The SNVs and insertions/deletions were identified using a Genome
Analyzer Toolkit (GATK, version 3.0; Broad Institute, Cambridge,
MA, USA). Coverage represents the number of times that each
nucleotide is sequenced at the NGS platform. Effective rate
represents the ratio of clean data (following removal of low
quality and contaminated data) to raw data. To identify somatic
mutations, the sequence variants of the non tumor tissues were
subtracted from those of the tumor tissues. The Database for
Annotation, Visualization and Integrated Discovery (DAVID;
http://david.abcc.ncifcrf.gov/) was used
for pathway analysis.
Results
Sample characteristics
The study population comprised of 11 males and 1
female. Of these, 11 patients were chronic HBV carriers and 1
patient was not an HBV carrier. A total of 11 patients had liver
cirrhosis and 1 patient had no cirrhosis, three patients had a
history of smoking and seven patients had a history of drinking.
All the HCC tumors were accompanied by tumor capsules, and tumor
cell invasion was observed by microscopy. The patient
characteristics are described in Table
I.
 | Table ISummary of hepatocellular carcinoma
samples. |
Table I
Summary of hepatocellular carcinoma
samples.
| Sample ID | Number of
mutations | Age (years) | Gender | HBV | Cirrhosis | Metastsis | Smoking | Drinking | AFP (ng/ml) | NCCN staging | T | N | M | Single or
multiple | Tumor size (mm) | Capsule |
|---|
| H1 | 7 | 33 | Male | + | + | − | − | + | 9.7 | I | T1 | N0 | M0 | − | 8×6×6 | + |
| H2 | 6 | 42 | Male | + | + | − | − | + | 15766 | I | T1 | N0 | M0 | − | 10×8×8 | + |
| H3 | 5 | 47 | Male | + | + | − | + | + | 7.9 | II | T1 | N0 | M0 | − | 5×5×4 | + |
| H4 | 8 | 54 | Male | + | + | + | − | − | 6568 | IVa | T3 | N1 | M0 | + | 8×6×5 | + |
| H5 | 5 | 55 | Male | + | + | + | − | − | 2.9 | IVa | T3 | N1 | M0 | − | 10×7×6 | + |
| H6 | 7 | 49 | Male | + | + | − | + | − | 81575 | IIIc | T4 | N0 | M0 | − | 7×6×6 | + |
| H7 | 10 | 65 | Male | − | + | − | − | + | 81.1 | I | T1 | N0 | M0 | − | 5×5×5 | + |
| H8 | 9 | 35 | Male | + | − | − | − | − | 71060 | IIIa | T3 | N0 | M0 | + | 8×6×6 | + |
| H9 | 8 | 60 | Female | + | + | − | − | − | 898.4 | IVb | T3 | N0 | M1 | + | 6×5×5 | + |
| H10 | 3 | 58 | Male | + | + | − | − | + | 6.2 | IIIa | T3 | N0 | M0 | − | 6×6×6 | + |
| H11 | 6 | 53 | Male | + | + | − | + | + | 7.1 | I | T1 | N0 | M0 | − | 4×4×3 | + |
| H12 | 7 | 35 | Male | + | + | − | − | + | 121001 | IIIc | T4 | N0 | M0 | − | 9×8×7 | + |
Sequencing profiles
The genomic DNA obtained from 12 patients with HCC
was screened for somatic mutations in 372 genes. On average, 0.197
Gb sequencing data was obtained for each sample. The average
coverage of each base in the target regions was ~40× per tumor
sample, and the effective rate for each sample was >86%.
Mutation profiles
In total, 81 non-synonymous somatic mutations were
identified in 62 different genes. Of the somatic mutations, 80 were
heterozygous and 1 was homozygous. A total of 77 SNVs were observed
in 58 genes from 12 patients and four deletion variations,
including one frameshift mutation and three non-frameshift
mutations, were identified in four genes from three patients. Point
mutations identified included 67 missense mutations and 10 nonsense
mutations. The HCC mutation spectrum was dominated by C>T and
C>A transitions, and the median number of mutations in each
tumor sample was seven (range, 3–10). Due to the small sample
number, no significant association was observed between the
mutation spectrum and clinical features. Deletion was observed in
MAML2, NN1, RET and MLLT3 genes, and the deletion of MAML2 resulted
in a frameshift alteration. No insertion mutations were observed in
the present study.
Recurrent mutations
The two most frequently mutated genes in the present
study were TP53 and RUNX1, with frequencies of 5/12 and 3/12,
respectively. The mutations of TP53 were missense or nonsense and
located in exons 2, 3 and 4, and four mutations were present in the
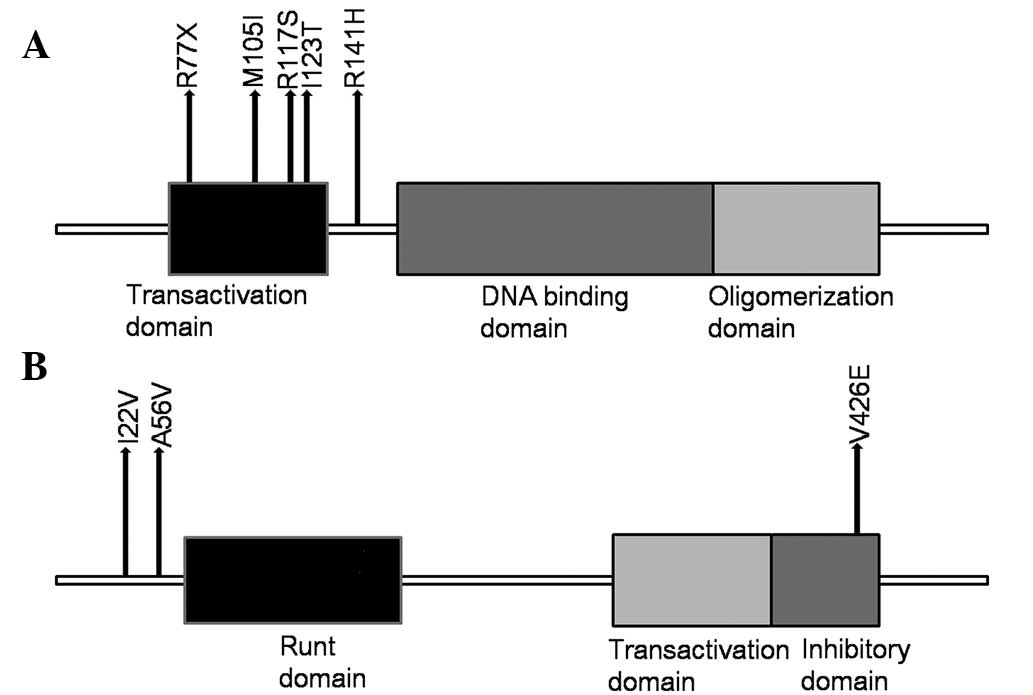
transactivation domain (Fig. 1A).
The mutations of RUNX1 were all missense, located in exons 1, 3 and
6, one of which was in the inhibitory domain (Fig. 1B). Additionally, 9 other genes,
which were recurrently mutated in at least 2/12 tumors were
identified (Table II). The H7
tumor exhibited two mutations in the FLCN gene, resulting in
c.G268T:p.A90S and c.G1536T:p.M512I, while H8 had two mutations in
the RECQL4 genes, resulting in c.T274C:p.S92P and
c.G187T:p.E63X.
| Table IIRecurrent mutations in hepatocellular
carcinoma samples. |
Table II
Recurrent mutations in hepatocellular
carcinoma samples.
| Gene | Mutation
frequency | Mutation | Sample |
|---|
| TP53 | 5/12 |
c.G422A:p.R141H, | H12 |
| |
c.T368C:p.I123T, | H8 |
| |
c.A229T:p.R77X, | H9 |
| |
c.G315A:p.M105I, | H4 |
| |
c.G351T:p.R117S | H5 |
| RUNX1 | 3/12 |
c.T1277A:p.V426E, | H3 |
| |
c.C167T:p.A56V, | H7 |
| | c.A64G:p.I22V | H4 |
| JAK | 2/12 |
c.C3124T:p.R1042W, | H1 |
| |
c.G3205T:p.E1069X | H3 |
| CREBBP | 2/12 |
c.G2103C:p.M701I, | H7 |
| |
c.G6291T:p.Q2097H | H9 |
| FLCN | 2/12 |
c.G1536T:p.M512I, | H7 |
| | c.G268T:p.A90S | |
| HOXC13 | 2/12 | c.C71A:p.A24E, | H6 |
| |
c.A425T:p.Q142L | H5 |
| KRAS | 2/12 |
c.G507T:p.K169N, | H5 |
| | c.G38A:p.G13D | H2 |
| MECOM | 2/12 |
c.A197T:p.N66I, | H7 |
| |
c.A725G:p.K242R | H9 |
| NF1 | 2/12 |
c.A7382T:p.D2461V, | H12 |
| |
c.G1716T:p.E572D | H6 |
| NOTCH1 | 2/12 |
c.G236A:p.R79H, | H8 |
| |
c.C7397T:p.T2466M | H1 |
| PDE4DIP | 2/12 |
c.G4111A:p.V1371I, | H3 |
| |
c.G512A:p.R171K | H11 |
| USP6 | 2/12 |
c.G1573A:p.V525I, | H9 |
| | c.C215G:p.T72R | H11 |
| RECQL4 | 2/12 |
c.T274C:p.S92P, | H8 |
| | c.G187T:p.E63X | H8 |
Pathways
Pathway analysis was performed for genes containing
mutations in each tumor using the DAVID database. A total of 77
altered genes were analyzed. Following the exclusion of
disease-associated pathways, including pathways in cancer, six
pathways were found to be significantly involved: The Janus kinase
(JAK)-signal transducer and activator of transcription (STAT)
signaling pathway (hsa04630), neurotrophin pathway (hsa04722),
apoptosis pathway (hsa04210), focal adhesion signaling pathway
(hsa04510), notch signaling pathway (hsa04330) and p53 signaling
pathway (hsa04115). Among these pathways, the JAK-STAT signaling
pathway was the most frequently involved, with six patients (50%)
exhibiting possible altered function in this pathway.
Discussion
Multiple genetic events accumulate during the
progression of HCC development. In the present study, the profile
of genetic alterations in HCC was analyzed using a NGS platform. In
total, 372 genes from 12 pairs of normal and tumor tissue specimens
were sequenced and a total of 81 non-silent somatic point mutations
were found.
The samples used in the present study were from
patients from western China, therefore, the profiling of the
mutation spectrum performed was, to a certain extent, different
from previous studies (8–10,13,14).
The two most commonly mutated genes observed in the present study
were TP53 and RUNX1. The TP53 gene, termed 'the guardian of the
genome' has been reported to be the most frequently altered gene in
various types of cancer, including HCC (16). In the present study, the mutation
frequency of TP53 was ~ 41.7%. However, the distribution of
mutations was different from that observed in previous studies
(8–10,13,14).
The p53 protein can be divided into three main domains, the
transactivation domain (exons 2 and 3), the DNA binding domain
(exons 5–8) and the oligomerization domain (exons 9 and 10). The
transactivation domain, at the N terminus of the protein, is rich
in serine and threonine and is able to induce protein activation.
The DNA binding domain recognizes and binds a consensus sequence in
the promoter sequence of genes, which is regulated by p53 at the
transcriptional level (17,18).
The oligomerization domain, at the C terminus of the p53 protein,
assists in the formation of an active tetramer (17,18).
Previous studies (8–10) have reported that the majority of
p53 mutations are localized in the DNA binding domain, and that
R249 was the most common site for this, which has been observed in
>30% of HCC cases in geographical areas of high HCC incidence.
The R249S mutation is associated with the induction of AFB1
(8–10). AFB1 is the metabolite of
aflatoxin-producing fungi, which is able to form a DNA adduct at
the third base of codon 249 in the TP53 gene, inducing a G>T
transversion and an R249S mutation (8,9). In
an area with a hot and humid climate, aflatoxin-producing fungi are
able to grow and can contaminate food, including corn and peanuts,
therefore the AFB1-inducing mutation R249S is commonly observed
(8–10). In the present study, all the
mutations identified were in exons 2, 3 and 4, which is possibly
due to the fact that the samples were from patients from western
China, facing different environmental factors and, thus, exhibiting
a different mutation spectrum for p53. RUNX1 is located on
chromosome 21q22 and contains an 138 amino acid Runt homology
domain, which is necessary for DNA consensus sequence recognition
and interaction with other co-factors (19). RUNX1 is able to increase or inhibit
transcriptional activity of target genes. Its roles include
regulating cell differentiation, growth and survival (20). A previous study (21) reported that RUNX1 was one of the
most common targets of chromosomal rearrangement in leukemia, and
was dysregulated in certain types of solid tumor. Miyagawa et
al (22) reported that RUNX1
was downregulated in HCC tissue, however, to the best of our
knowledge no mutation of RUNX1 in HCC has been previously reported.
In the present study, three RUNX1 mutations were identified,
suggesting its importance in liver carcinogenesis. In addition, the
present study identified mutations in several cancer genes, which
had not been previously linked to HCC, including JAK3. JAK3 is a
member of the non-receptor tyrosine kinase family, the members of
which are able to bind to various cell surface receptors and are
important in cytokine-induced signal transduction (23,24).
Previous studies (24,25) have reported that JAK3, unlike the
JAK1, JAK2 or Tyk2 members of the non-receptor tyrosine kinase
family, is primarily expressed in cells of a hematopoietic lineage,
and mutations of JAK3 have been identified in leukemic patients and
cell lines. In the present study, two non-synonymous mutations for
JAK3 were identified in patients with HCC. This, to the best of our
knowledge, is the first time that mutations of JAK3 have been
reported in HCC, suggesting a novel role of JAK3.
In conclusion, the present study identified several
novel genes involved in HCC using NGS. The results of the present
study provide a resources for understanding the molecular
alterations underlying the development of HCC, however, further
investigations, with larger sample sizes, are required to fully
examine genetic alteration in HCC development.
Acknowledgments
The authors would like to thank Shanghai ZhangJiang
Translational Medicine Research Center for their assistance with
data analysis.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Center MM and Jemal A: International
trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers
Prev. 20:2362–2368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim CM, Koike K, Saito I, Miyamura T and
Jay G: HBx gene of hepatitis B virus induces liver cancer in
transgenic mice. Nature. 351:317–320. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koike K, Moriya K, Iino S, et al:
High-level expression of hepatitis B virus HBx gene and
hepatocarcinogenesis in transgenic mice. Hepatology. 19:810–819.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bouchard MJ and Schneider RJ: The
enigmatic X gene of hepatitis B virus. J Virol. 78:12725–12734.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seeger C and Mason WS: Hepatitis B virus
biology. Microbiol Mol Biol Rev. 64:51–68. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gouas D, Shi H and Hainaut P: The
aflatoxin-induced TP53 mutation at codon 249 (R249S): Biomarker of
exposure, early detection and target for therapy. Cancer Lett.
286:29–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang
NJ and Harris CC: Mutational hotspot in the p53 gene in human
hepatocellular carcinomas. Nature. 350:427–428. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen HM and Ong CN: Mutations of the p53
tumor suppressor gene and ras oncogenes in aflatoxin
hepatocarcinogenesis. Mutat Res. 366:23–44. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li M, Zhao H, Zhang X, et al: Inactivating
mutations of the chromatin remodeling gene ARID2 in hepatocellular
carcinoma. Nat Genet. 43:828–829. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boyault S, Rickman DS, de Reyniès A, et
al: Transcriptome classification of HCC is related to gene
alterations and to new therapeutic targets. Hepatology. 45:42–52.
2007. View Article : Google Scholar
|
|
13
|
Zender L, Villanueva A, Tovar V, Sia D,
Chiang DY and Llovet JM: Cancer gene discovery in hepatocellular
carcinoma. J Hepatol. 52:921–929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang J, Deng Q, Wang Q, et al: Exome
sequencing of hepatitis B virus-associated hepatocellular
carcinoma. Nat Genet. 44:1117–1121. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gandevia B and Tovell A: Declaration of
Helsinki. Med J Aust. 2:320–321. 1964.PubMed/NCBI
|
|
16
|
Levrero M, De Laurenzi V, Costanzo A, Gong
J, Wang IY and Melino G: The p53/p63/p73 family of transcription
factors: Overlapping and distinct functions. J Cell Sci.
113:1661–1670. 2000.PubMed/NCBI
|
|
17
|
Laptenko O and Prives C: Transcriptional
regulation by p53: One protein, many possibilities. Cell Death
Differ. 13:951–961. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho Y, Gorina S, Jeffrey PD and Pavletich
NP: Crystal structure of a p53 tumor suppressor-DNA complex:
Understanding tumorigenic mutations. Science. 265:346–355. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rossetti S and Sacchi N: RUNX1: A MicroRNA
hub in normal and malignant hematopoiesis. Int J Mol Sci.
14:1566–1588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blyth K, Cameron ER and Neil JC: The RUNX
genes: Gain or loss of function in cancer. Nat Rev Cancer.
5:376–387. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meyers S, Downing JR and Hiebert SW:
Identification of AML-1 and the (8; 21) translocation protein
(AML-1/ETO) as sequence-specific DNA-binding proteins: The runt
homology domain is required for DNA binding and protein-protein
interactions. Mol Cell Biol. 13:6336–6345. 1993.PubMed/NCBI
|
|
22
|
Miyagawa K, Sakakura C, Nakashima S, et
al: Down-regulation of RUNX1, RUNX3 and CBFbeta in hepatocellular
carcinomas in an early stage of hepatocarcinogenesis. Anticancer
Res. 26:3633–3643. 2006.PubMed/NCBI
|
|
23
|
Yamaoka K, Saharinen P, Pesu M, Holt VE
III, Silvennoinen O and O'Shea JJ: The Janus kinases (Jaks). Genome
Bio1. 5:2532004. View Article : Google Scholar
|
|
24
|
Pesu M, Laurence A, Kishore N, Zwillich
SH, Chan G and O'Shea JJ: Therapeutic targeting of Janus kinases.
Immunol Rev. 223:132–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elliott NE, Cleveland SM, Grann V, Janik
J, Waldmann TA and Davé UP: FERM domain mutations induce gain of
function in JAK3 in adult T-cell leukemia/lymphoma. Blood.
118:3911–3921. 2011. View Article : Google Scholar : PubMed/NCBI
|