Introduction
Pokeweed antiviral protein (PAP) is an
N-glycosidase of the ribosome-inactivating protein (RIP)
superfamily, a widely-distributed family of toxins found in plants,
fungi and bacteria (1,2). RIPs function by irreversibly
depurinating the sarcin-ricin loop (SRL), a highly conserved
sequence found in the RNA of all large ribosomal subunits (3). Depurination prevents ribosomes from
completing the translocation step of the elongation cycle due to
the inability to bind elongation factor 2 (4), leading to a ribotoxic stress response
and RIP-induced apoptosis (5). In
addition to targeting the SRL, PAP can depurinate a wide variety of
RNA substrates, including viral RNA (6) and eukaryotic mRNA (7,8). The
strong antiviral activity of PAP exists against a wide range of
viruses, including human immunodeficiency virus-1, influenza,
herpes, polio and human T-cell leukemia virus I (9,10).
Investigations into the phytotherapeutic and prophylactic usage of
PAP in this context have shown promise (11,12).
Based on their structure, RIPs are traditionally
classified as either type I or type II. Type I RIPs, including PAP,
are enzymatically active ~30 kDa monomeric toxins. By contrast,
type II RIPs, including ricin and Shiga toxin, possess a dimeric
quaternary structure with an active A-subunit (homologous to type I
RIPs) and a larger lectin B-subunit that attaches to
galactose-containing surface receptors of target cells. The
mechanisms of cell entry and intracellular trafficking of type II
RIPs have been investigated in detail, although less is known about
these processes in type I RIPs, including PAP (5). Due to the absence of the cell-binding
B subunit, type I RIPs are less toxic compared with type II RIPs;
however, cellular uptake and cytotoxicity can be markedly enhanced
through conjugation with monoclonal antibodies, lectins, hormones
or growth factors (2,13).
Several studies on American pokeweed (Phytolacca
americana) have revealed differential PAP gene expression at
various developmental stages, with PAP-I, PAP-II, PAP-S1/PAP-S2 and
PAP-R representing the different isozymes that appear in spring
leaves, summer leaves, seeds and roots, respectively. In
vivo toxicity studies in a preclinical mouse model revealed
that the natively-isolated seed isoform, PAP-S1, has far greater
toxicity than PAP-I and the A-subunit of ricin (14). The crystal structures of PAP-I and
PAP-S1 from Phytolacca americana have been determined
(15–17), although significantly less is known
about the tissue distribution, biochemical properties and
structural features of potentially therapeutic PAPs from other
species. To address this deficiency, the present study combined
protein sequencing, mass spectrometry and X-ray crystallography to
elucidate the complete covalent and 1.7 Å X-ray crystal structure
of a PAP homolog isolated from seeds of Asian pokeweed
(Phytolacca acinosa). The sequence and structure of this
RIP, termed PAP-S1aci, was compared to other known RIPs
including the American pokeweed seed homolog (Phytolacca
americana). An unusual type of post-translational carbohydrate
modification identified in PAP-S1aci is discussed within
the context of type-I RIP cytotoxicity, and more generally, is
compared to well-established eukaryotic N-glycosylation patterns
and pathways. Finally, the potential application of these findings
in the development of specific RIP-based therapies is presented
herein.
Materials and methods
Purification of PAP-S1aci for
primary structure determination
The two isoenzymes of PAP-Saci
were isolated from mature seeds of P. acinosa as previously
described (18). Unless otherwise
indicated, all chemicals were purchased from Carl Roth GmbH &
Co. KG (Karlsruhe, Germany). The individual isoenzymes were
subsequently separated by reversed phase high performance liquid
chromatography (HPLC) on a Vydac C4 column (4.6×250 mm, 5
μm; Vydac; Grace Davison Discovery Sciences, Lokeren,
Belgium) equilibrated with 0.1% trifluoroacetic acid (TFA) and
eluted with an acetonitrile gradient (0–60% acetonitrile; 1 ml/min;
60 min; A280 nm). The higher molecular weight isoenzyme
(PAP-S1aci) eluted first at ~35% acetonitrile and
the two major peaks were individually collected and dried under a
vacuum. The PAP-S1aci and
PAP-S2aci fractions, which were ~95% pure as
determined by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), were transferred onto polyvinylidene
fluoride (PVDF) membranes, and the area corresponding to
PAP-S1aci was extracted for N-terminal
sequencing. The PAP-S1aci elution fraction
produced an N-terminal sequence identical to the blotted sample and
was therefore used for subsequent sequencing experiments.
Preparation of PAP-S1aci
cleavage peptides
For cyanogen bromide (CNBr) cleavage, the dried
PAP-S1aci sample (5 mg) was dissolved in 400
μl of 70% formic acid containing 400 μg of CNBr,
sealed under nitrogen and incubated for 24 h at room temperature in
the dark. The mixture was desiccated under a vacuum and the residue
was dissolved in 500 μl of 0.1% TFA and immediately
separated by reversed phase HPLC on a Vydac C4 column as described
above except that elution was monitored at 230 nm. A total of eight
fractions were collected and desiccated. For further enzymatic
digestion, the CNBr fragments were dissolved in 50 mM
ethylmorpholine-acetate (pH 8.0) and digested with Arg-C sequencing
grade Arg-C proteinase (Boehringer Ingelheim Deutschland GmbH,
Ingelheim am Rhein, Germany; substrate:enzyme ratio 100:1) for 18 h
at 37°C. Digestion was terminated by desiccation and the peptides
(totaling 16 individual peaks) were separated as described
above.
Automated Edman degradation
N-terminal sequencing of proteins and peptides was
performed on an automatic LF3600D Protein Sequencer (Beckman
Coulter, Miami, FL, USA). Cysteine residues were modified by
acrylamide and detected as cys-S-β-propionamide (19). For sequencing of the C-terminal
peptide, the arginine peptide R12 prepared from the CNBr fragment
M6 was immobilized to a Sequelon AA membrane (Millipore
Corporation, Billerica, MA, USA) using carbodiimide
condensation.
In-gel protein digestion
Selected spots were excised from Coomassie gels and
the gel plugs were minced, washed with water and soaked in 100 mM
ethylmorpholine acetate buffer (pH 8.5) in 50% acetonitrile.
Following complete gel destaining in a sonication bath, the gel
sections were repeatedly washed with water and dehydrated in
acetonitrile. The supernatant was removed and the gel was partially
dried under a vacuum. Gel sections were swollen in a digestion
buffer containing 50 mM ethylmorpholine acetate (Sigma-Aldrich,
Munich, Germany), pH 8.0, 1 mM CaCl2, 10% acetonitrile
and sequencing grade trypsin (Roche Diagnostics, Mannheim, Germany;
trypsin:protein ratio=1:75). Following overnight digestion
(agitation at 37°C) the resulting peptides were extracted from the
gel by increasing the acetonitrile concentration to 50% and
addition of TFA to a final concentration of 1%. Following
sonication for 15 min, the liquid phase containing the extracted
peptides was dried and the peptides were redissolved in aqueous 1%
TFA and desalted on ZipTip microcolumns (Millipore Corporation,
Hesse, Germany).
Matrix-assisted laser
desorption/ionization mass spectrometry (MALDI-MS)
A saturated solution of α-cyano-4-hydroxycinnamic
acid (Sigma-Aldrich) in aqueous 50% acetonitrile/0.2% TFA was used
as a MALDI matrix. A total of 0.5 μl of a 1:1 sample:matrix
mixture was placed on the sample target and dried at ambient
temperature. Positive ion MALDI mass spectra were measured on a
Bruker BIFLEX reflectron time-of-flight mass spectrometer
(Bruker-Franzen, Bremen, Germany) equipped with a SCOUT 26 sample
inlet, a gridless delayed extraction ion source and a nitrogen
laser (337 nm; Laser Science, Cambridge, MA, USA). The ion
acceleration voltage and reflectron voltage were set to 19 and 20
kV, respectively. Spectra were calibrated externally using the
monoisotopic [M+H]+ ion of α-cyano-4-hydroxycinnamic
acid and a peptide standard (angiotensin II; Bachem, Bubendorf,
Switzerland).
Tandem mass spectrometry
Peptides were loaded onto a capillary column (0.18
mm inner diameter) packed with 10 cm of reversed-phase resin (MAGIC
C-18, 200 Å, 5 μm; Michrom Bioresources, Auburn, CA, USA)
and separated using a 5–40% gradient of acetonitrile in 0.5% acetic
acid (50 min; 2 μl/min). The column was interfaced with an
LCQ DECA ion trap mass spectrometer (ThermoQuest Corp., San Jose,
CA, USA) set to acquire a full MS spectrum over the mass range
300–1,800 a.m.u. followed by full MS/MS scans of the two most
intense ions from the preceding MS scan. Dynamic exclusion was set
to two repeat counts with a repeat duration of 0.5 min and a 3 min
exclusion duration window. The spray voltage was held at 1.5 kV and
the tube lens potential was −2 V. The heated capillary was kept at
175°C with a voltage of 13 V. Spectra were interpreted manually
and/or with the Sequest™ v. B22 software (Thermo Finnigan, San
Jose, CA, USA) and MS/MS MASCOT error tolerant search engine
(http://www.matrixscience.com) (20).
Crystallization and X-ray structure
determination
Protein preparation, crystallization and preliminary
phasing have been described previously (18). In brief, multiple rounds of ion
exchange and gel filtration chromatography on P. acinosa
seed homogenate yielded an approximate equimolar mixture of
PAP-S1aci and PAP-S2aci.
Crystallization of PAP-S1aci was achieved at 289
K by hanging-drop vapor diffusion of a solution containing 10 mg/ml
of the isoenzyme mixture, 21–22% polyethylene glycol (PEG) 4 K, 0.2
M sodium citrate and 100 mM phosphate buffer (pH 7.2) over
reservoirs containing 42–44% PEG 4K and 100 mM phosphate buffer (pH
7.2). An X-ray diffraction dataset to 1.7 Å was collected at 100 K
at the joint University of Hamburg-IMB Jena-EMBL Beamline X13 at
DESY (Hamburg, Germany). The crystals belonged to space group I222
with unit cell constants a=78.63, b=84.19 and c=90.88 Å and one
monomer per asymmetric unit. The structure was solved by molecular
replacement with the atomic coordinates of P. americana
PAP-I (15; Protein Data Bank code 1PAF) as a search model.
Preliminary X-ray sequencing and model rebuilding were conducted
prior to determination of the full protein sequence. The
preliminary X-ray sequenced model had correct residue assignments
at 246 of 261 positions (with five errors due to Asn/Asp and
Gln/Glu side-chain ambiguities). Final model building into
σA-weighted 2Fo-Fc and
Fo-Fc maps (21) was conducted with the molecular
graphics program Crystallographic Object-Oriented Toolkit
(Coot) (22).
Crystallographic refinement was performed in CNS 1.1 (23) using anisotropic bulk solvent
correction, positional and isotropic atomic B-factor protocols. The
final PAP-S1aci model, which consists of 261
amino-acid residues, 1 N-linked GlcNAc and 464 water
molecules, exhibits good geometry as determined by MolProbity
(http://molprobity.biochem.duke.edu/)
(24). An exception is the pre-Pro
residue Val173, which violates the pre-Pro Ramachandran plot
despite clear electron density supporting its refined main-chain
conformation (see Results and Discussion). Final R and
Rfree values are 16.9 and 21.1% for data in the
29.7–1.7 Å range (with 5% of the observed data reserved for
calculation of Rfree). Final model statistics are
presented in Table I. Secondary
structure assignments were performed with the program DSSP
(25). Structural superpositions
were performed with LSQKAB (26),
as implemented in the CCP4 suite software (http://www.ccp4.ac.uk) (27). Crystal contact analyses and surface
area calculations were conducted with the CCP4 programs CONTACT and
AREAIMOL, respectively. Molecular figures were generated with PyMOL
(28). The refined coordinates of
PAP-S1aci have been deposited in the RCSB Protein
Data Bank under the ID code 2Q8W.
 | Table IData collection and refinement
statistics. |
Table I
Data collection and refinement
statistics.
| X-ray data | Value |
|---|
| Wavelength (Å) | 0.8033 |
| Resolution (Å) | 29.73–1.70
(1.74–1.70) |
| Space group | I222 |
| Unit-cell
dimensions |
| a (Å) | 78.63 |
| b (Å) | 84.19 |
| c (Å) | 90.88 |
| Overall
reflections | 417,357 |
| Unique
reflections | 33,596 |
| Multiplicity | 12.4 |
| Completeness
(%) | 99.9 (100.0) |
|
Rmergea (%) | 4.3 (38.9) |
| I/σ (I) | 55.7 (7.1) |
|
| Model refinement
statistics | Value |
|
| Resolution (Å) | 29.73–1.70
(1.78–1.70) |
| No. of
reflections | 32,752 (1,625) |
| Overall
completeness | 97.7 (93.4) |
|
Rcrystb | 0.169 (0.230) |
|
Rfreeb | 0.211 (0.277) |
| R.m.s.d. from ideal
geometry |
| Bonds (Å) | 0.012 |
| Angles (Å) | 1.7 |
| Protein atoms | 2,103 |
| GlcNAc atoms | 14 |
| Solvent atoms | 464 |
| Ramachandran
plotc |
| Favored (%) | 96.2 |
| Allowed (%) | 99.6 |
| Disallowed
(%) | 0.4 |
Results
Preliminary protein sequencing of
PAP-S1aci
Initial purification of an approximate equimolar
mixture of seed PAP isoenzymes, PAP-S1aci and
PAP-S2aci, was reported (18). Since the two isoenzymes were of
similar size as determined by SDS-PAGE, they were separated by
reversed-phase HPLC. Different gradients were used in an attempt to
optimize separation, however, it was difficult to identify optimal
conditions; hence the PAP-S1aci sample used for
primary structure analysis had an estimated final purity of ~95%.
In order to define the N-terminus of PAP-S1aci,
samples from reversed-phase HPLC were separated by SDS-PAGE,
electroblotted onto PVDF and used for PVDF sequencing, producing
the initial sequence INTITFDAGXATIN, where X was an empty cycle
(see below). Since the sequence length obtainable from a PVDF blot
is limited, Protein Support sequencing was performed on the bulk
PAP-S1aci sample obtained from HPLC. This yielded
the more extensive N-terminal sequence INTITFDAGXATINKYATEMESLR
NEAKDPSLKXYGXPXXP (X=unknown amino acid). A protein-protein BLAST
search (29) on the N-terminal
sequence revealed 85% sequence identity to the seed isoenzyme
PAP-S1 from P. americana (30). The full sequence of this protein
was used for subsequent planning of the sequencing strategy.
As the PAP-S1 homolog from P. americana has
261 amino-acid residues and five internal Met residues, CNBr
cleavage was performed followed by fragment separation and
analysis. Of the eight peaks that appeared during HPLC separation
following CNBr cleavage, M1, M2 and M8 represented nonpeptide
contaminants. Fragment M3 covered the sequence Glu21-Gly36 (residue
numbering reflects the final sequence of
PAP-S1aci), fragment M4 (Mr ~10
kDa by MALDI-MS) encompassed the entire C-terminal region of the
protein (Val173-Thr261) and fragment M5 (Mr ~10
kDa by MALDI-MS) had the extended internal sequence Gly75-Gln171.
The fragment MU6 (Ile1-Met20) was already determined by N-terminal
sequencing and fragment MU7 comprised Leu40-Met74. Thus, the five
CNBr fragments covered the entire PAP-S1aci
protein sequence. The sixth Met peptide anticipated from the
homology to PAP-S1 does not exist in PAP-S1aci
due to the presence of an Ile residue (Ile65) in this position.
Arg-C-generated peptides were used to complete the amino acid
sequence in areas not available for MS or MS/MS analysis. Partial
sequencing of two of these peptides covered the regions Asn25-Asn44
and Asn135-Lys164. Peptide resequencing following Cys modification
confirmed the positions of four Cys residues within the sequence.
Finally, the C-terminal sequence of the protein was derived from
peptide sequencing following Cys modification and immobilization on
arylamino-PVDF. Altogether, 241 of 261 amino-acid residues were
covered by Edman degradation, yielding a sequence coverage of
~92%.
Protein sequence verification and
completion by mass spectrometry
Certain sequences within PAP-S1aci
would be difficult to cover by Edman methods due to the requirement
for multiple rounds of cleavage and purification. To circumvent
this complication, peptide MS was used to identify the small
percentage of missing sequences and to verify sequences derived
from automated Edman degradation. By employing μLC-ESI-MS/MS
tandem mass spectrometry on different protease digests (trypsin,
Glu-C and Asp-N) additional sequence data were generated for
Asn70-Met74, Lys128-Arg134, Phe165-Met172 and Asp232-Arg240. In all
instances, an overlap with the Edman data was evident. Altogether
the MS/MS data covered ~90% of the PAP-S1aci
sequence. Subsequent MALDI-MS was used to verify the pre-determined
amino acid sequence by measuring the mass of peptide fragments.
This analysis covered the PAP-S1aci sequence by
another 93%.
Analysis of N-glycosylation sites
It was difficult to identify amino-acid residues at
three positions within the PAP-S1aci sequence
(residues 10, 44 and 255). These positions were characterized by
low yields of phenylthiohydantoin (PTH)-amino acids with a similar
elution profile to PTH-Asp. Although this may be indicative of any
post-translational modification, they could be identified as
Asn-GlcNAc linkages - an extremely rare type of
N-glycosylation. Large carbohydrates attached to Asn
residues may prevent extraction of the corresponding
PTH-Asn-oligosaccharide from the cartridge (genuine 'empty' cycle),
however, PTH-Asn-GlcNAc is extractable (albeit at a lower yield),
and would possibly elute earlier than PTH-Asn in reversed-phase
HPLC. Definitive evidence for this assignment at each position in
the sequence was provided by tandem MS. An example of the MS/MS
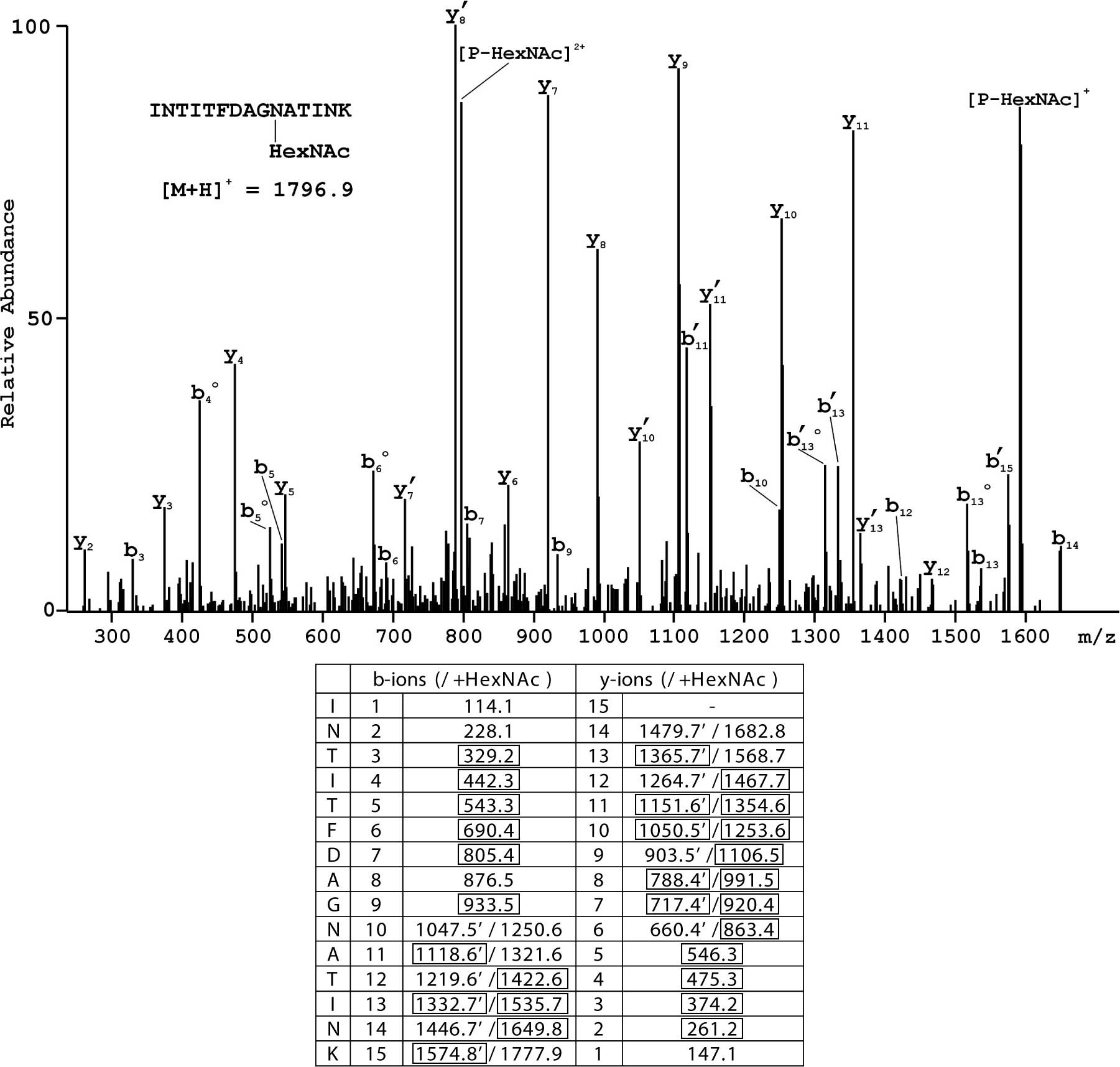
spectrum for the N-terminal glycosylated peptide having the
structure INTITFDAG [N-HexNAc
(L-asparagine-N-acetyl-hexosamine)]ATINK is shown in Fig. 1, where extensive sequence coverage
by fragments generated following the initial loss of HexNAc as well
as those containing the modification is evident.
Correction of Edman/MS sequencing errors
by X-ray sequencing
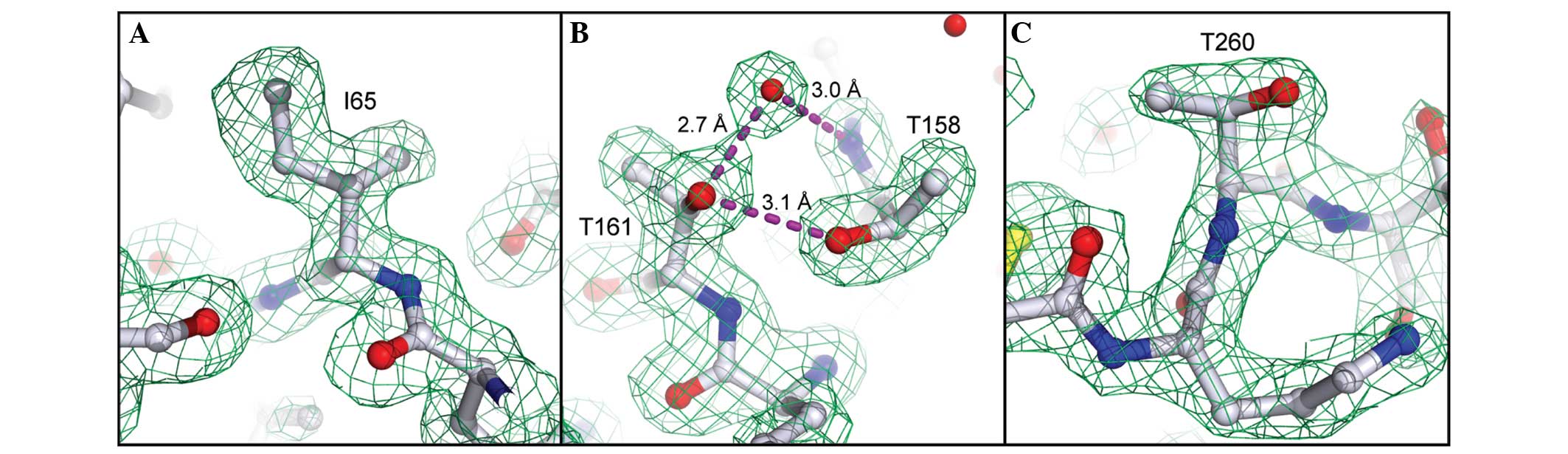
The high-quality X-ray data allowed us to identify
three positions in the sequence where the Edman/MS data were
evidently erroneous (residues 65, 161 and 260). On the basis of
clear electron density and/or H-bonding features, the original
assignments of Leu65 (derived by Edman and MS/MS), Ile161 (Edman)
and Ala260 (Edman) were corrected to Ile65, Thr161 and Thr260.
Electron density at these positions is presented in Fig. 2, illustrating the utility of
high-quality X-ray data as a complementary sequencing method,
particularly for direct visualization of isomeric (Ile/Leu) or
isobaric (Gln/Lys) residues, which are frequently misannotated by
other protein sequencing methods.
Sequence and structure of
PAP-S1aci
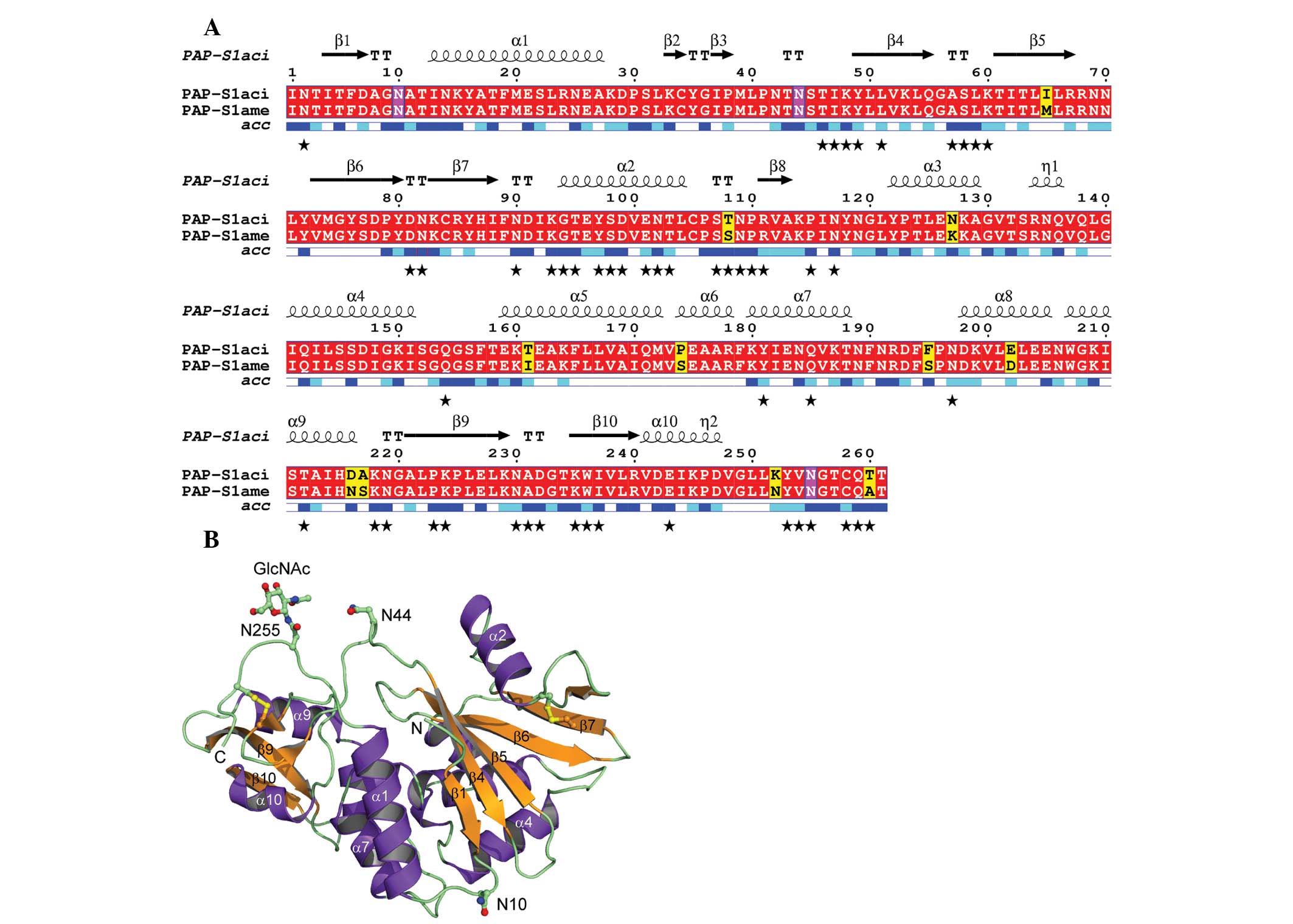
The sequence and X-ray structure of
PAP-S1aci are presented in Fig. 3. The figure was generated with DSSP
(25), Multalin (31) and ESPript 2.2 (32). PAP-S1aci contains
invariant active-site residues of the RIP superfamily
(corresponding to Tyr72, Tyr122, Glu175 and Arg178 in
PAP-S1aci), which are critical for catalysis
(15,33). Four conserved Cys residues forming
two intramolecular disulfide bonds (Cys34-Cys258 and Cys84-Cys105)
are also present. A protein-protein BLAST search (34) was performed to determine the
closest known sequence relative, identifying P. americana
PAP-S1 (30) with 96% sequence
identity (250 of 261 residues) to PAP-S1aci
(Fig. 3A). The 1.8 Å crystal
structure of PAP-S1 from P. americana has been elucidated
and includes a fully resolved carbohydrate structure consisting of
three GlcNAc monosaccharide linkages at Asn10, Asn44 and Asn255
(17). The current results
demonstrated that the same three N-glycosylation sequons are
also found in PAP-S1aci and that each site is
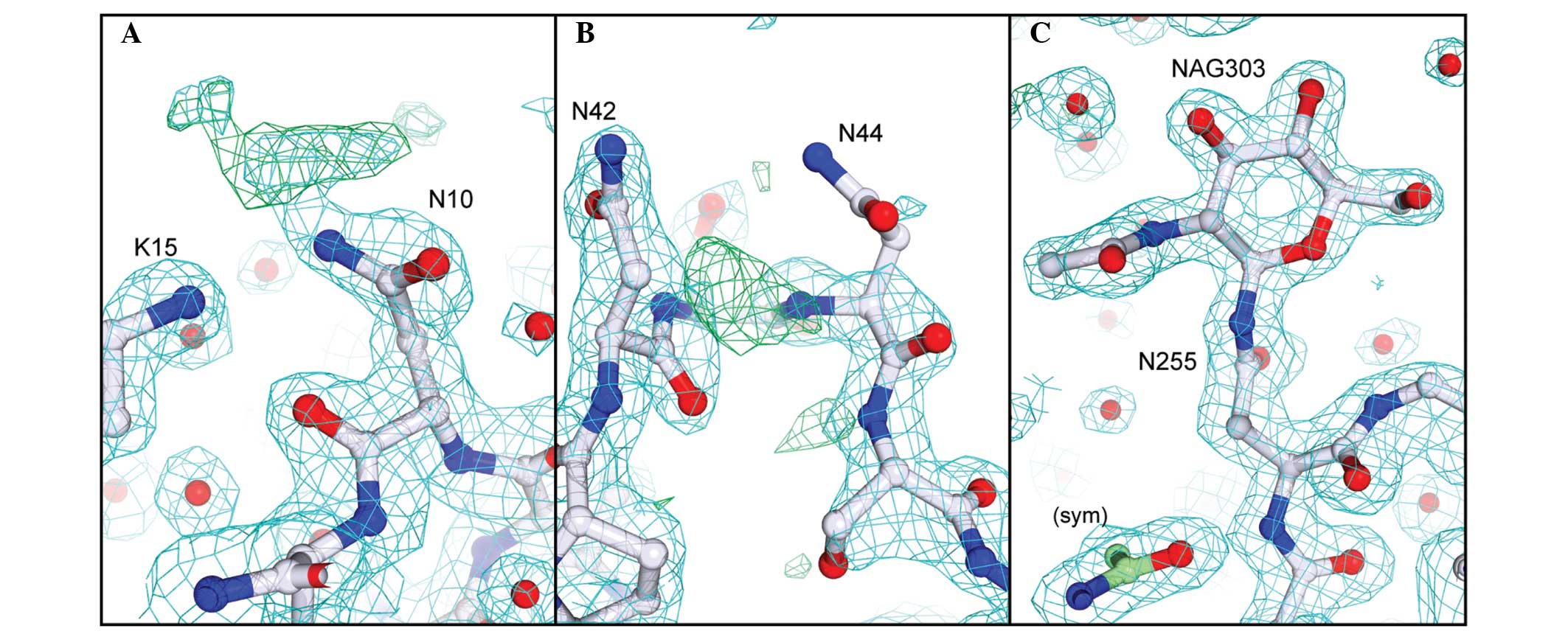
similarly modified with a single GlcNAc residue. In our crystal
form there is clear electron density for the sugar at Asn255 due to
its involvement in crystal packing (18,35),
however, Asn10 and Asn45 are freely exposed to solvent and the
corresponding electron density for the linked GlcNAc residues is
weak (Fig. 4).
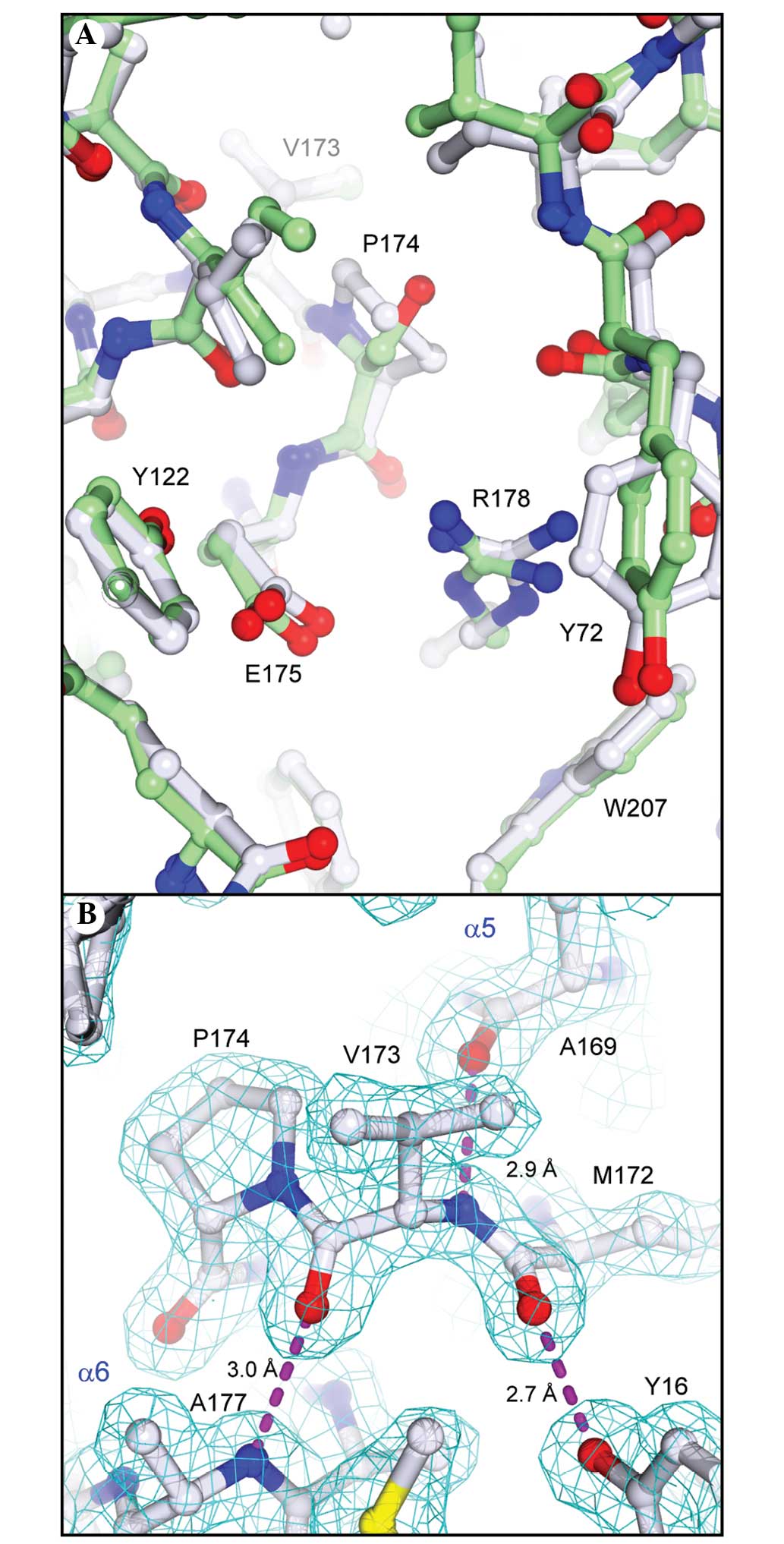
As expected by the high sequence identity between
PAP-S1aci and PAP-S1, the overall folds of the
two proteins are identical within the limits of experimental
detection. An overall Cα least-squares superposition between the
two provided a Cα root-mean-square deviation (r.m.s.d.) of 0.48 Å,
comparable with the summed cross-validated Luzatti estimates of the
coordinate errors in the two structures (0.52 Å). A few small
regional Cα displacements on the order of 0.8–1.3 Å can be
attributed to localized areas of poor density or differences in
crystal packing between the two crystal forms. A Cα geometrical
validation of our refined model with MolProbity (24) indicated a significant outlier,
Val173 (φ,Ψ= −114°, −83°), which lies outside the small α region of
the pre-proline Ramachandran plot. Given that the nearby Glu175 is
a conserved active-site residue believed to stabilize the
developing oxocarbonium ion during substrate cleavage (15), the present study aimed to determine
whether the intervening Pro174 was commonly found in RIP sequences.
An extensive protein-protein BLAST search and alignment of
non-redundant RIP sequences indicated the residue preceding the
catalytic Glu175 was almost invariably Ser or Ala (corresponding to
Ser174 in PAP-S1; Fig. 3A). Only
three sequences out of all known bacterial and plant RIPs contain a
Pro residue in this position: Ribosome inactivating protein 2 from
Phytolacca insularis (Korean pokeweed), curcin from
Jatropha curcas (spurge family) and ebulin from Sambucus
ebulus (Dwarf Elder). Since the geometrically strained Val173
is situated in a break between helices α5 and α6 (Fig. 3A), the present study aimed to
determine whether the Pro174 substitution in
PAP-S1aci led to any notable structural
deviations in the α5/α6 region or if the refined geometry of Val173
is fully supported by the X-ray data. A superposition analysis
between the structures of PAP-S1aci and PAP-S1
revealed the Ser→Pro substitution in PAP-S1aci is
accommodated without significant alterations in the positioning of
Glu175 or in the overall topology of the active site (Fig. 5A). In addition, the lower than
average temperature factors in this region (B 2 Val173=15.4 Å ;
BProtein=22.3 Å2), coupled with excellent electron
density, strongly supports the refined backbone conformation at
Val173-Pro174 (Fig. 5B). Although
the main-chain conformation does appear to be rather unusual, a
more recent statistical analysis of protein pre-Pro geometry
indicated that the φ,Ψ values observed for Val173 are within a
broad sterically allowed area extending from the α region towards
the lower left corner of the Ramachandran plot (36).
Discussion
Using complementary methods, the present study
detected the presence of N-GlcNAc residues in mature
PAP-S1aci. This rare type of
N-glycosylation has been previously identified in fungi
(37), animals (38,39)
and plants (40), including RIPs
from the seeds of Phytolacca americana and Luffa
cylindrica (sponge gourd) (41). The metabolic origin of this
modification remains to be elucidated, although certain
investigators have hypothesized that it originates from endogenous
endo-β-N-acetylglucosaminidase (ENGase) activity on proteins
bearing N-linked oligosaccharide chains (39,41).
However, this theory has been recently challenged by a study
demonstrating that proteins with N-GlcNAc modifications are
produced in ENGase knockout strains of Arabidopsis,
indicating the presence of a separate and as yet unresolved pathway
(40). The homologous
O-linked GlcNAc found in certain eukaryotic glycoproteins
has gained recognition as a regulatory post-translational
modification akin to reversible phosphorylation (42). O-GlcNAc linkages are
produced by a single conserved nucleocytoplasmic enzyme,
O-GlcNAc transferase, which catalyzes GlcNAc transfer from
UDP-GlcNAc to the hydroxyl oxygen of Ser/Thr (42). Previous biochemical studies also
described the isolation of a eukaryotic N-GlcNAc transferase
(NGT) that transfers GlcNAc from UDP-GlcNAc to the carboxamide
nitrogen of Asn in Asn-Xxx-Ser/Thr sequons (EC 2.4.1.94) (43–45).
According to these studies, NGT activity was detected in various
organisms and subcellular locations. Therefore, it remains
plausible that the N-GlcNAc modifications identified in the
present study, and in previous studies (37–41),
may be associated with the activity of an uncharacterized NGT
homolog.
Further studies are required to determine whether
N-GlcNAc modifications affect glycoprotein function by
regulating enzyme activity, protein localization or protein-protein
interactions. In the present study, a molecular modeling approach
was undertaken to examine whether the N-GlcNAc modifications
of PAP-S1aci or PAP-S1 could affect interactions
with the ribosome. Previous studies have revealed the involvement
of regions Asn42-Thr43 and Asn253-Val255 of PAP-I in the binding of
the SRL of ribosomal RNA (33,46).
The structural similarity between PAP-I and
PAP-S1aci allowed us to map these rRNA-binding
regions in PAP-I to the corresponding Asn44-GlcNAc and
Asn255-GlcNAc residues in PAP-S1aci. Since PAP-I
lacks glycosylation sequons and does not contain N-GlcNAc
modifications, the present study aimed to determine whether larger
oligosaccharide linkages at these positions in
PAP-S1aci and PAP-S1 are able to impair ribosomal
interactions. Our modeling studies suggested that the ribosome
depurination activity of seed PAPs would be adversely affected by
the N-glycosylation of Asn44 and Asn255 with larger and more
typical oligosaccharide chains, as they would shield the
rRNA-binding sites on the protein. Given our current structural
data, the earlier in vitro evidence that PAP-I and
PAP-S1aci exhibit similar ribosomal depurination
activities (47,48) supports the hypothesis that smaller
N-linked monosaccharides do not substantially affect
ribosome binding. Taken together, this evidence suggests that
N-GlcNAc modifications observed in certain type I seed RIPs,
including PAP-S1aci, may serve a different
biological function while also preserving ribotoxin activity.
Suggestions at a more plausible role for
N-GlcNAc modifications stem from the observation that PAP-S1
exhibits far greater cytotoxicity in vivo than PAP-I or the
A-chain of ricin (14). RIPs are
hypothesized to be internalized by cells either through
receptor-mediated or fluid-phase endocytosis. Double-chain RIPs,
including ricin, are primarily taken up by hepatocytes via two main
routes: i) The additional lectin B-chain interacts with galactosyl
residues of the cell membrane and ii) mannose-containing
oligosaccharides on the two chains exploit the
mannose/GlcNAc-receptor-mediated uptake pathway (49). Non-glycosylated single-chain RIPs,
including PAP-I, are hypothesized to enter cells by the less
efficient route of fluid-phase endocytosis. The absence of
carbohydrate chains on PAP-I, a cell-wall protein, may in fact
reflect a specialized antiviral role involving a local suicide
mechanism for compromised cells. Evidence demonstrating that PAP
can efficiently inactivate pokeweed ribosomes supports this
hypothesis (47). The superior
cytotoxicity and biodistribution of PAP-S1 in animal models,
however, favors an active receptor-mediated uptake mechanism
characteristic of type-2 RIPs or fully glycosylated type-1 RIPs.
Nevertheless, the question remains: Can N-linked GlcNAc
residues effectively facilitate receptor-mediated uptake of seed
RIPs, including PAP-S1, PAP-S1aci, α-luffin and
β-luffin? A previous study on unrelated glycoproteins have
demonstrated that ENGase-treated glycoproteins, which are trimmed
to contain only Asn-GlcNAc monosaccharide linkages, are
internalized via mannose/GlcNAc receptor-mediated pinocytosis with
an even greater efficiency than their oligosaccharide-bearing
counterparts (50). Collectively,
this body of evidence supports our hypothesis that N-GlcNAc
modifications may have evolved to enhance the cytotoxicity of type
I seed RIPs by exploiting receptor-mediated uptake pathways of seed
predators without compromising ribosome affinity. A deeper
understanding of the different structural characteristics and
uptake mechanisms that confer cytotoxic variability may prove
beneficial in the development of modified RIPs tailored for
specific therapeutic applications.
Abbreviations:
|
PAP
|
pokeweed antiviral protein
|
|
GlcNAc
|
N-acetyl-D-glucosamine
|
|
HexNAc
|
N-acetylhexosamine
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
r.m.s.d
|
root-mean square deviation
|
Acknowledgments
The authors would like to thank Dr. R. Hilgenfeld
for support and early interest in the present study, Dr. K.
Bezouska for protein sequencing and mass-spectrometry analysis and
Dr. I.K. Smatanova for the preliminary crystallization
experiments.
References
|
1
|
Puri M, Kaur I, Perugini MA and Gupta RC:
Ribosome-inactivating proteins: current status and biomedical
applications. Drug Discov Today. 17:774–783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stirpe F: Ribosome-inactivating proteins:
from toxins to useful proteins. Toxicon. 67:12–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Endo Y, Tsurugi K and Lambert JM: The site
of action of six different ribosome-inactivating proteins from
plants on eukaryotic ribosomes: the RNA N-glycosidase activity of
the proteins. Biochem Biophys Res Commun. 150:1032–1036. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gessner SL and Irvin JD: Inhibition of
elongation factor 2-dependent translocation by the pokeweed
antiviral protein and ricin. J Biol Chem. 255:3251–3253.
1980.PubMed/NCBI
|
|
5
|
Stirpe F and Battelli MG:
Ribosome-inactivating proteins: progress and problems. Cell Mol
Life Sci. 63:1850–1866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rajamohan F, Kurinov IV, Venkatachalam TK
and Uckun FM: Deguanylation of human immunodeficiency virus (HIV-1)
RNA by recombinant pokeweed antiviral protein. Biochem Biophys Res
Commun. 263:419–424. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hudak KA, Bauman JD and Tumer NE: Pokeweed
antiviral protein binds to the cap structure of eukaryotic mRNA and
depurinates the mRNA downstream of the cap. RNA. 8:1148–1159. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parikh BA, Coetzer C and Tumer NE:
Pokeweed antiviral protein regulates the stability of its own mRNA
by a mechanism that requires depurination but can be separated from
depurination of the alpha-sarcin/ricin loop of rRNA. J Biol Chem.
277:41428–41437. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parikh BA and Tumer NE: Antiviral activity
of ribosome inactivating proteins in medicine. Mini Rev Med Chem.
4:523–543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mansouri S, Choudhary G, Sarzala PM,
Ratner L and Hudak KA: Suppression of human T-cell leukemia virus I
gene expression by pokeweed antiviral protein. J Biol Chem.
284:31453–31462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
D'Cruz OJ, Waurzyniakt B and Uckun FM: A
13-week subchronic intravaginal toxicity study of pokeweed
antiviral protein in mice. Phytomedicine. 11:342–351. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uckun FM, Rustamova L, Vassilev AO,
Tibbles HE and Petkevich AS: CNS activity of pokeweed anti-viral
protein (PAP) in mice infected with lymphocytic choriomeningitis
virus (LCMV). BMC Infect Dis. 5:92005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shapira A and Benhar I: Toxin-based
therapeutic approaches. Toxins (Basel). 2:2519–2583. 2010.
View Article : Google Scholar
|
|
14
|
Benigni F, Canevari S, Gadina M, Adobati
E, Ferreri AJ, Di Celle EF, Comolli R and Colnaghi MI: Preclinical
evaluation of the ribosome-inactivating proteins PAP-1, PAP-S and
RTA in mice. Int J Immunopharmacol. 17:829–839. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Monzingo AF, Collins EJ, Ernst SR, Irvin
JD and Robertus JD: The 2.5 A structure of pokeweed antiviral
protein. J Mol Biol. 233:705–715. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kurinov IV, Myers DE, Irvin JD and Uckun
FM: X-ray crystallographic analysis of the structural basis for the
interactions of pokeweed antiviral protein with its active site
inhibitor and ribosomal RNA substrate analogs. Protein Sci.
8:1765–1772. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng ZH, He XL, Li HM, Hu Z and Wang DC:
Crystal structure of pokeweed antiviral protein with well-defined
sugars from seeds at 1.8 A resolution. J Struct Biol. 141:171–178.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hogg T, Kuta Smatanova I, Bezouska K,
Ulbrich N and Hilgenfeld R: Sugar-mediated lattice contacts in
crystals of a plant glycoprotein. Acta Crystallogr D Biol
Crystallogr. 58:1734–1739. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brune DC: Alkylation of cysteine with
acrylamide for protein sequence analysis. Anal Biochem.
207:285–290. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perkins DN, Pappin DJ, Creasy DM and
Cottrell JS: Probability-based protein identification by searching
sequence databases using mass spectrometry data. Electrophoresis.
20:3551–3567. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Read RJ: Improved Fourier coefficients for
maps using phases from partial structures with errors. Acta Cryst
A. 42:140–149. 1986. View Article : Google Scholar
|
|
22
|
Emsley P and Cowtan K: Coot:
model-building tools for molecular graphics. Acta Crystallogr D
Biol Crystallogr. 60:2126–2132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brünger AT, Adams PD, Clore GM, DeLano WL,
Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu
NS, et al: Crystallography & NMR system: A new software suite
for macromolecular structure determination. Acta Crystallogr D Biol
Crystallogr. 54:905–921. 1998. View Article : Google Scholar
|
|
24
|
Lovell SC, Davis IW, Arendall WB 3rd, de
Bakker PI, Word JM, Prisant MG, Richardson JS and Richardson DC:
Structure validation by Calpha geometry: phi, psi and Cbeta
deviation. Proteins. 50:437–450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kabsch W and Sander C: Dictionary of
protein secondary structure: pattern recognition of hydrogen-bonded
and geometrical features. Biopolymers. 22:2577–2637. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kabsch W: A solution for the best rotation
to relate two sets of vectors. Acta Cryst A. 32:922–923. 1976.
View Article : Google Scholar
|
|
27
|
Collaborative Computational Project,
Number 4: The CCP4 suite: programs for protein crystallography.
Acta Crystallogr D Biol Crystallogr. 50:760–763. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
DeLano WL: The PyMOL Molecular Graphics
System. DeLano Scientific; Palo Alto, USA: 2002, http://www.pymol.org.
|
|
29
|
McGinnis S and Madden TL: BLAST: at the
core of a powerful and diverse set of sequence analysis tools.
Nucleic Acids Res. 32:W20–W25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Honjo E, Dong D, Motoshima H and Watanabe
K: Genomic clones encoding two isoforms of pokeweed antiviral
protein in seeds (PAP-S1 and S2) and the N-glycosidase activities
of their recombinant proteins on ribosomes and DNA in comparison
with other isoforms. J Biochem. 131:225–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Corpet F: Multiple sequence alignment with
hierarchical clustering. Nucl Acids Res. 16:10881–10890. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gouet P, Courcelle E, Stuart DI and Métoz
F: ESPript: analysis of multiple sequence alignments in PostScript.
Bioinformatics. 15:305–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rajamohan F, Pugmire MJ, Kurinov IV and
Uckun FM: Modeling and alanine scanning mutagenesis studies of
recombinant pokeweed antiviral protein. J Biol Chem. 275:3382–3390.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Altschul SF, Gish W, Miller W, Myers EW
and Lipman DJ: Basic local alignment search tool. J Mol Biol.
215:403–410. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hogg T and Hilgenfeld R: Protein
crystallography in drug discovery. Comprehensive Medicinal
Chemistry II. Taylor JB, Triggle DJ and Kubinyi H: 3. 2nd edition.
Elsevier; Amsterdam: pp. 875–900. 2007, View Article : Google Scholar
|
|
36
|
Ho BK and Brasseur R: The Ramachandran
plots of glycine and pre-proline. BMC Struct Biol. 5:142005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hase S, Fujimura K, Kanoh M and Ikenaka T:
Studies on heterogeneity of Taka-amylase A: isolation of an amylase
having one N-acetylglucosamine residue as the sugar chain. J
Biochem. 92:265–270. 1982.PubMed/NCBI
|
|
38
|
Chalkley RJ, Thalhammer A, Schoepfer R and
Burlingame AL: Identification of protein O-GlcNAcylation sites
using electron transfer dissociation mass spectrometry on native
peptides. Proc Natl Acad Sci USA. 106:8894–8899. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Z, Udeshi ND, Slawson C, Compton PD,
Sakabe K, Cheung WD, Shabanowitz J, Hunt DF and Hart GW: Extensive
crosstalk between O-GlcNAcylation and phosphorylation regulates
cytokinesis. Sci Signal. 3:ra22010.PubMed/NCBI
|
|
40
|
Kim YC, Jahren N, Stone MD, Udeshi ND,
Markowski TW, Witthuhn BA, Shabanowitz J, Hunt DF and Olszewski NE:
Identification and origin of N-linked β-D-N-acetylglucosamine
monosaccharide modifications on Arabidopsis proteins. Plant
Physiol. 161:455–464. 2013. View Article : Google Scholar :
|
|
41
|
Islam MR, Kung SS, Kimura Y and Funatsu G:
N-acetyl-D-glu cosamine-asparagine structure in
ribosome-inactivating proteins from the seeds of Luffa cylindrica
and Phytolacca americana. Agric Biol Chem. 55:1375–1381. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wells L, Whelan SA and Hart GW: O-GlcNAc:
a regulatory post-translational modification. Biochem Biophys Res
Commun. 302:435–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khalkhall Z and Marshall RD: Glycosylation
of ribonuclease A catalysed by rabbit liver extracts. Biochem J.
146:299–307. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Khalkhali Z and Marshall RD:
UDP-N-acetyl-D-glucosamine-as paragine sequon
N-acetyl-b-D-glucosaminyl-transferase-activity in human serum.
Carbohydr Res. 49:455–473. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Khalkhali Z, Marshall RD, Reuvers F,
Habets-Willems C and Boer P: Glycosylation in vitro of an
asparagine sequon catalysed by preparations of yeast cell
membranes. Biochem J. 160:37–41. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rajamohan F, Mao C and Uckun FM: Binding
interactions between the active center cleft of recombinant
pokeweed antiviral protein and the alpha-sarcin/ricin stem loop of
ribosomal RNA. J Biol Chem. 276:24075–24081. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bonness MS, Ready MP, Irvin JD and Mabry
TJ: Pokeweed antiviral protein inactivates pokeweed ribosomes;
implications for the antiviral mechanism. Plant J. 5:173–183. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Poyet JL and Hoeveler A: cDNA cloning and
expression of pokeweed antiviral protein from seeds in Escherichia
coli and its inhibition of protein synthesis in vitro. FEBS Lett.
406:97–100. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Barbieri L, Battelli MG and Stirpe F:
Ribosome-inactivating proteins from plants. Biochem Biophys Acta.
1154:237–282. 1993.PubMed/NCBI
|
|
50
|
Gross V, Heinrich PC, vom Berq D, Steube
K, Andus T, Tran-Thi TA, Decker K and Gerok W: Involvement of
various organs in the initial plasma clearance of differently
glycosylated rat liver secretory proteins. Eur J Biochem.
173:653–659. 1988. View Article : Google Scholar : PubMed/NCBI
|