Introduction
Sepsis is defined as a serious, uncontrolled,
systemic inflammatory response syndrome (SIRS), which is caused by
infection. Infection itself is not usually the cause of damage to
the body; rather, it is the abnormally high levels of
infection-induced inflammation that result in injury. Furthermore,
late sepsis results in systemic damage, which is often complicated
by multiple organ dysfunction and septic shock (1), resulting in further deterioration of
the functional status and internal environment of the patient, the
mechanism of which may be associated with endothelial cell (EC)
damage (2–4). The microvascular EC environment
provides an important interface for the role of inflammatory
cytokines (5). In addition,
endothelial injury can amplify inflammatory processes, enhancing
its effects (6).
It has been demonstrated that high mobility group
protein B1 (HMGB1), which is released in late endotoxemia (7), is a lethal inflammatory mediator
(8) that is sustained in the
blood, and maintains and extends the pathological process of
sepsis. High serum levels of HMGB1 are associated with mortality
rates of patients with sepsis (9).
Advanced glycation end product receptor (RAGE) is a receptor for
HMGB1, which is expressed on the EC membrane (10). HMGB1 interacts with ECs via RAGE,
and is involved in endothelial activation and injury (11). Early EC injury leads to endothelial
apoptosis, and in the late stage results in endothelial death in
vivo, which can cause increased EC permeability (12,13).
ECs possess a high degree of biological activity and
are involved in various physiological processes within the body
(14). Endothelial apoptosis not
only destroys the barrier function of ECs, but also further
aggravates the inflammatory response (14). Therefore, endothelial apoptosis
significantly affects the pathological process of sepsis. When
endotoxin is injected into the blood, it has been shown to cause
microvascular EC damage, resulting in the shedding of ECs in animal
experiments (15). Shedding of ECs
can also be detected in the peripheral blood of patients with
sepsis, and the degree of shedding is often correlated with patient
mortality rates (16).
As a feature of sepsis, capillary leakage represents
an endothelial barrier dysfunction, which is induced by elevated
levels of inflammatory cytokines that, in turn, increase
endothelial permeability. Proinflammatory cytokines, including
tumor necrosis factor (TNF)-α and inflammatory mediators, including
thrombin, can lead to increased EC permeability (17,18).
HMGB1 has been shown to increase the permeability of ECs cultured
in vitro (19). However,
whether HMGB1 also induces endothelial apoptosis remains to be
elucidated. In the present study, the expression levels of HMGB1
were detected in the serum of patients with sepsis at various time
points using western blotting. EC apoptosis and permeability were
also assessed following stimulation with septic serum.
Patients and methods
Subjects
The subjects involved in the present study were
included healthy volunteers and patients with a diagnosis of
sepsis, who were admitted to the intensive care unit (ICU) of
Chongming Branch, Xin Hua Hospital, Shanghai Jiao Tong University
School of Medicine (Shanghai, China) within 12 h of onset. Sepsis
was defined as SIRS caused by infection, which was confirmed by the
presence of bacteria or clinically suspicious foci. Disease
severity was classified as sepsis, severe sepsis or septic shock,
according to the Surviving Sepsis Campaign (SSC) guidelines
(20). The investigation period
ran between August 2012 and July 2013. The inclusion criteria were
also defined according to SSC guidelines (20): Patients with the following
characteristics were excluded from the present study: (1) A history of coronary heart disease,
chronic obstructive pulmonary disease, diabetes, hypertension,
blood diseases, hyperlipidemia, autoimmune diseases, cancer,
rheumatism/connective tissue disease, chronic renal insufficiency,
liver cirrhosis; (2) unable to
participate to the end of the clinical trial when serum samples
were collected; (3) patients
<18 years or >75 years of age; (4) smoking or (and) alcohol consumption;
(5) patients with a medication
history of >1 month. According to the above standards, 38
patients with sepsis (21 men and 17 women; age, 45±3.4 years) and
32 healthy volunteers (17 men and 15 women; age, 48±4.2 years) were
enrolled in the present study, which was approved by the ethics
committee of the ChongMing branch of the Xin Hua hospital, Shanghai
Jiao Tong University School of Medicine (Shanghai, China). All the
subjects provided written informed consent prior to commencement of
the investigation.
Human serum
Peripheral venous blood (5 ml) was collected from
the subjects and healthy volunteers at 6, 12, 18, 24, 36, 48 and 96
h following hospital admission. All blood samples were subjected to
natural clotting at room temperature, followed by centrifugal serum
separation (2,000 × g for 8 min) and complement inactivation at
56°C. The aliquots were stored at −70°C until further use.
Chemicals and reagents
A human umbilical vein EC (HUVEC) line was purchased
from Shanghai Institute of Biochemistry and Cell Biology, Chinese
Academy of Sciences (Shanghai, China). The cell line was originally
isolated from human embryonic umbilical vein ECs. Ethyl pyruvate
(EP), fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse
and anti-rabbit secondary antibodies, and tetramethylrhodamine
(TRITC)-phalloidin were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Anti-vascular endothelial (VE)-cadherin antibodies
(polyclonal goat anti-human; cat. no. sc-6458), anti-B-cell
lymphoma 2 (BCL-2; monoclonal mouse anti-human; cat. no. sc-7382)
and anti-BCL-2-associated X protein (BAX; monoclonal mouse
anti-human; cat. no. sc-70407) antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Polyclonal rabbit
anti-human HMGB1 antibody (cat. no. LS-C31535-100) was purchased
from LifeSpan Biosciences, Inc. (Seattle, WA, USA). Invitrogen
Annexin V-FITC/propidium iodide (PI) was purchased from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA).
Cell culture
The HUVECs (1×106 cells) were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS; Biochrom AG, Berlin, Germany), 150 U/ml
penicillin (Beyotime Institute of Biotechnology, Hangzhou, China)
and 150 U/ml streptomycin at 37°C in an atmosphere containing 5%
CO2. Cell morphology was observed under a microscope
(IX70; Olympus Corporation, Tokyo, Japan) and identified using
factor VIII immunofluorescence staining (2%) with anti-factor VIII
rabbit polyclonal primary antibody (Hua Yi Biotechology, Shanghai,
China; cat. no. hy032974). Briefly, ECs were seeded onto slides and
cultured for 24 h, and the slides were subsequently fixed with 4%
paraformaldehyde at room temperature for 15 min. The slides were
then washed with phosphate-buffered saline (PBS) and treated with
0.2% Triton X-100 (Liansuo Biological Technology Co., Ltd.,
Shanghai, China), prior to being blocked with 1% bovine serum
albumin (BSA; LiRui Biological Technology Co., Ltd., Shanghai,
China) at room temperature for 30 min, and incubated with rabbit
anti-human factor VIII polyclonal primary antibody (100 µl;
1:50) at 4°C for 12–14 h. Following washing with PBS, the slides
were incubated with PE-labeled mouse anti-rabbit IgG 100 µl
(1:50) in the dark at room temperature for 1 h. Images of the
slides were captured using a IX71 fluorescence microscope (Olympus,
Tokyo, Japan). Once grown to 80% confluence, the cells were
pretreated with EP (5 µM) in DMEM with 10% FBS for 24 h at
37°C. The culture medium was subsequently removed by washing twice
with PBS (pH 7.4), and the cells were exposed to 20% septic serum
obtained from patient blood samples, diluted in culture medium for
6 h at 37°C. The present study included the following experimental
groups: A 20% septic serum-treated group and 20% septic serum + EP
(5 µM) pre-treated group; and the following control groups:
A 20% control serum-treated (healthy volunteers) group and a 20%
control serum + EP (5 µM)-treated group. Cell viability was
determined using trypan blue staining (40%; Beyotime Institute of
Biotechnology).
Endothelial monolayer permeability
The ECs (1×105 cells) were grown on 3
µm pore Transwell filters (Costar®, Corning
Incorporated, Corning, NY, USA) until confluent, and were then
transferred to starvation medium (Biochrom AG) containing 1% FBS
for 2 h. FITC-dextran (Mr, 42,000; Sigma-Aldrich) was applied
apically at 1 µg/ml, and allowed to equilibrate for 30 min
at 25°C prior to 200 µl sample of the medium being removed
from the lower chamber, to measure basal permeability. The
monolayers were pretreated with EP (5 µM) for 24 h at 37°C,
and were then either untreated (control) or were stimulated in
triplicate with 20% septic serum for 6 h at 37°C. Samples (200
µl) were removed from the lower chamber for fluorescence
measurements, which were compared with those from the control
monolayers. The fluorescence intensity of FITC was detected using a
fluorescence spectrometer (LS-50B; PerkinElmer, Waltham, MA, USA),
operating at an excitation wavelength of 492 nm and a detection
wavelength of 520 nm.
VE-cadherin and filamentous (F)-actin
immunofluorescence staining
The ECs (1×104 cells) were serum-starved
for 2 h, pretreated with EP for 24 h, and then either remained
untreated or were stimulated with 20% septic serum for 6 h. For
F-actin staining, the ECs were fixed in 3.7% formaldehyde (Liansuo
Biological Technology Co., Ltd.) at 4°C for 10 min. The cells were
permeabilized in 0.2% Triton-X-100 for 5 min and then blocked in 1%
BSA in PBS, prior to being incubated with TRITC-phalloidin (2
kµ/l) at room temperature in the dark for 1 h. For
VE-cadherin staining, following dewaxing, samples were heated to
92–98°C for 15–20 min in 0.01 M sodium citrate buffer solution (pH
6.0), and then cooled to room temperature for 20–30 minutes.
Finally, the slice was rinsed with distilled water and PBS,
respectively. The ECs were fixed and blocked, followed by
incubation with mouse anti-VE-cadherin antibody (1:100 dilution)
overnight at room temperature. The culture medium was removed by
washing twice with PBS (pH 7.4) and the ECs were subsequently
exposed to FITC-labeled goat anti-mouse IgG secondary antibody
(1:200 dilution; cat. no. sc-2010; Santa Cruz Biotechnology, Inc.)
at room temperature in the dark for 1 h. Images were captured using
a confocal laser scanning microscope (LSM410; Carl Zeiss AG,
Oberkochen, Germany).
Western blotting
The ECs (3×106 cells) were serum-starved
for 2 h, pretreated with EP for 24 h, and either remained untreated
or were stimulated with 20% septic serum for 6 h. The ECs were
subsequently lysed with SDS sample buffer (Beijing Taize Technology
Development Co., Ltd., Beijing, China). Protein concentration was
determined using a Bicinchoninic acid assay method, and the
supernatants (100 µl protein) were separated by 10%
SDS-PAGE. The proteins were then transferred to nitrocellulose
membranes (Mai Bio Co., Ltd., Shanghai, China), which were blocked
with 10% non-fat dry milk in Tris-buffered saline and Tween-20
(Double Helix Biotechnology Co., Ltd., Shanghai, China), containing
20 mmol/l Tris (pH 8.0); 137 mmol/l NaCl and 10% Tween-20, and
incubated for 12 h at 4°C with monoclonal mouse anti-human BAX,
monoclonal mouse anti-human BCL-2 antibody, and polyclonal rabbit
anti-human HMGB1 primary antibodies, and then incubated for 2 h at
25°C with horseradish peroxidase (HRP)-conjugated goat anti-mouse
IgG (cat. no. B1706516; ShangHaiyelibio) and HRP-conjugated goat
anti-rabbit IgG (cat. no. 96692; Baijibio Beijing). Bound proteins
were detected using enhanced chemiluminescence (Beyotime Institute
of Biotechnology), according to the manufacturer's instructions.
The chemiluminescence signal was detected using a Quantitative Gel
and Western Blot Imaging system, and the results were analyzed
using FluorChem Q software (ProteinSimple, San Jose, CA, USA).
Flow cytometry
The ECs were serum-starved for 2 h, pretreated with
EP (5 µM) for 24 h, and then either remained untreated or
were stimulated with 20% septic serum for 6 h. The ECs were then
diluted to a suspension of 1×106 cells/ml in 1X binding
buffer (Ebioeasy Co., Ltd., Shanghai, China) and were lysed twice
with cold PBS buffer. Purified recombinant Annexin V (5–15
µg) was mixed gently with the cell suspension (100
µl) at room temperature for 15 min. Subsequently, Annexin
V-FITC (5 µl) and PI (10 µl) were added to the cell
suspension and mixed gently for 15 min at room temperature in the
dark, prior to adding 1X binding buffer (400 µl). The rate
of EC apoptosis was measured using flow cytometry (Epics XL;
Beckman Coulter, Inc., Brea, CA, USA) within 1 h.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean ±
standard deviation. Statistically significance differences between
groups were identified using one-way analysis of variance, and a
least significant difference test was used for inter-group
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Serum expression levels of HMGB1 in
patients with sepsis
The serum expression levels of HMGB1 in patients
with sepsis were detected using western blot analysis. As shown in
Fig. 1, HMGB1 was detected in the
sera of the patients with sepsis; however, it was not expressed
until 24 h post-onset (Fig. 1),
following which its expression gradually increased, peaking at 48 h
(Fig. 1). The expression levels of
HMGB1 remained elevated for a long period of time, prior to gradual
weakening. The elevated expression levels were ultimately sustained
for ~96 h. These results suggested that HMGB1 was not released
until sepsis is fairly well established (24 h), and continued to be
expressed from that point, maintaining and extending the
pathological process of sepsis.
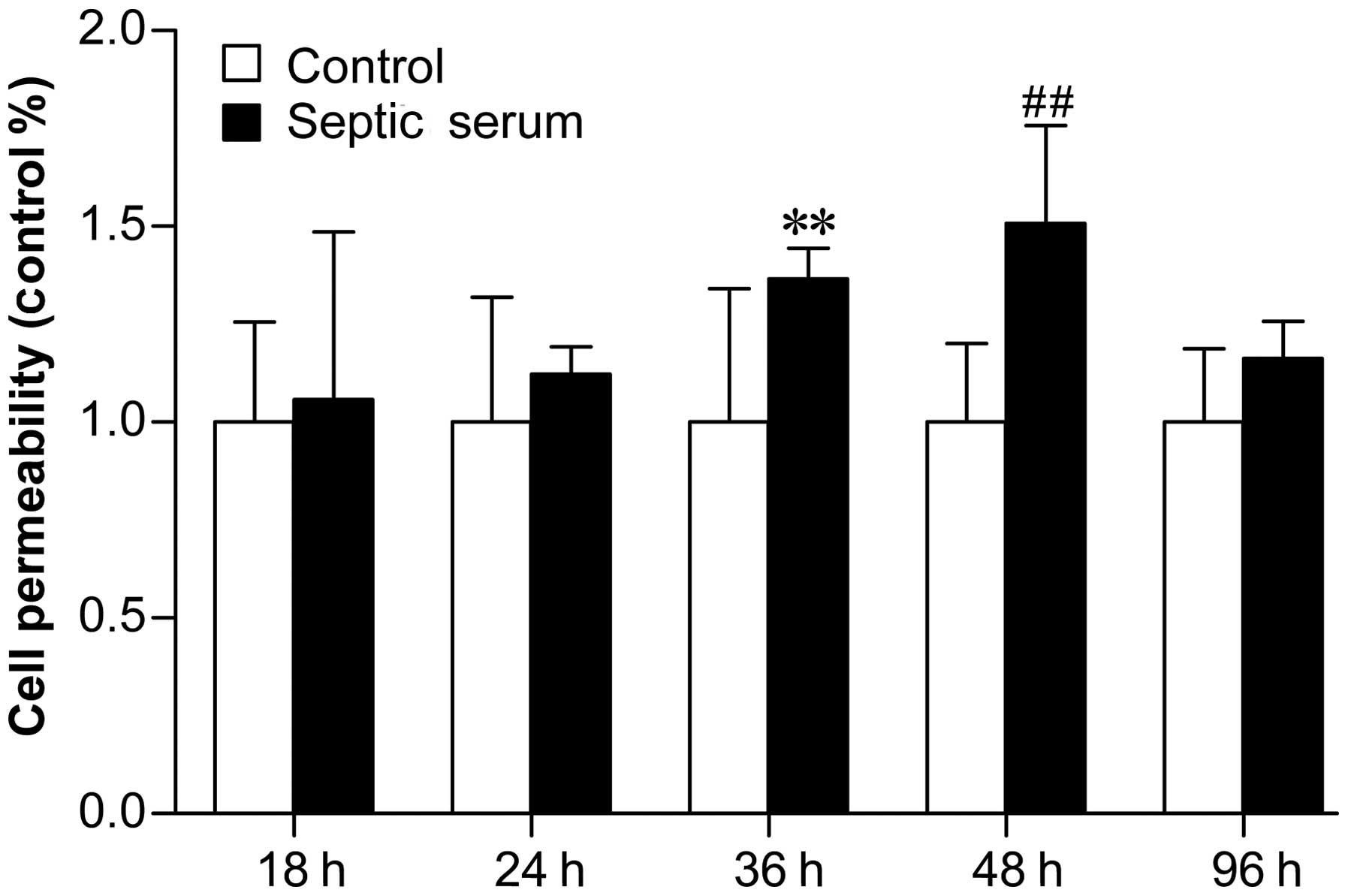
Effects of septic serum on the
permeability of HUVECs in vitro
Treatment of ECs with septic patient sera increased
cell permeability, as determined using immunofluorescence. As shown
in Fig. 2, septic serum induced EC
permeability, compared with control serum, after 18 and 24 h;
however, the difference was only significant after 36 h (Fig. 2; P<0.05), and peaked at 48 h
(Fig. 2; P<0.01). These results
suggested that HMGB1 was released in late sepsis and was involved
in the activation and injury of ECs, predominantly by inducing
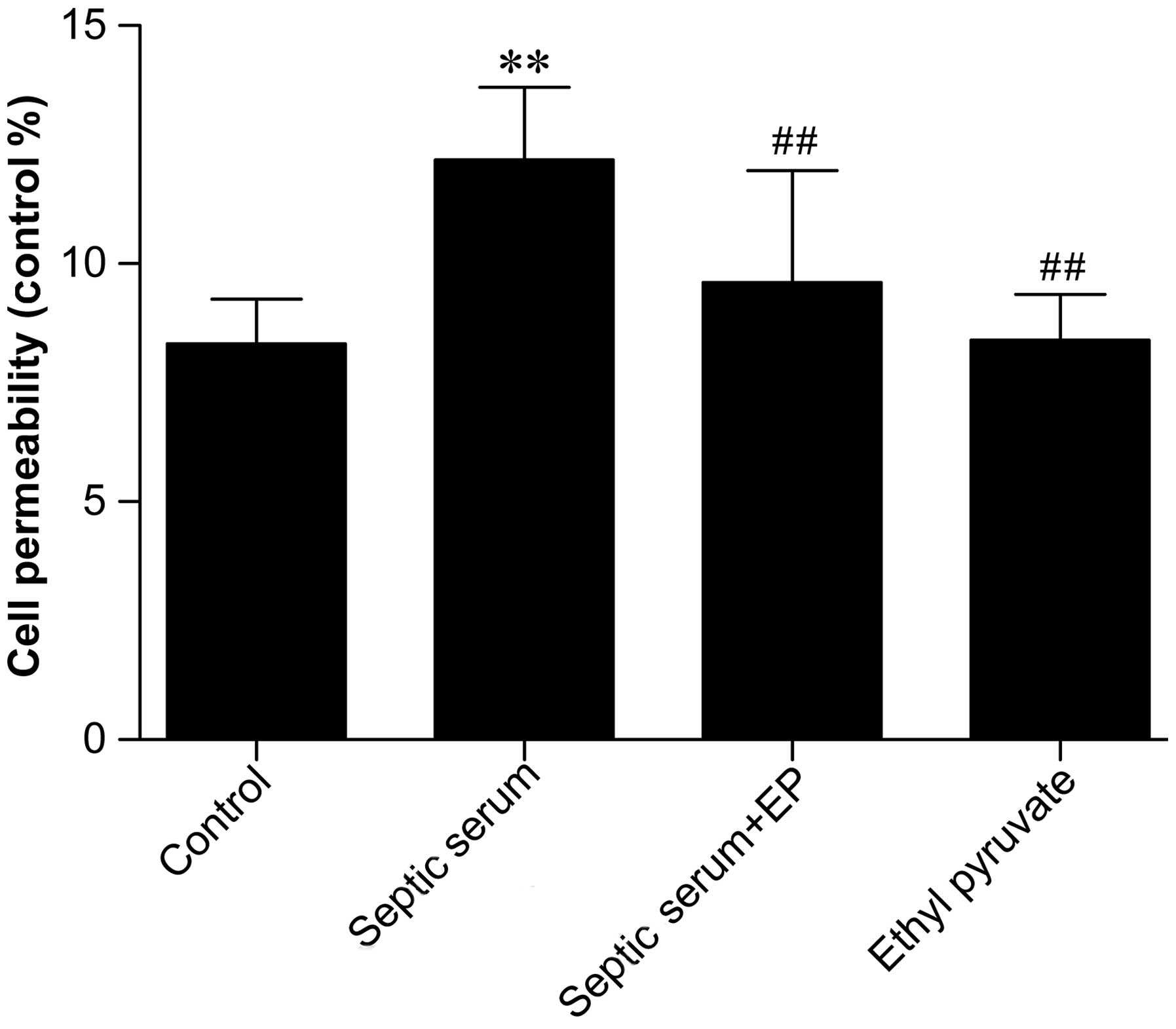
endothelial permeability. In addition, as shown in Fig. 3, treatment with septic serum
significantly increased endothelial monolayer permeability after 6
h, compared with cells treated with control serum (P<0.01).
However, pretreatment with 5 µM EP (an inhibitor of HMGB1)
for 24 h prior to treatment with serum significantly reduced
serum-induced permeability. HMGB1 was detected readily in the sera
from the patients with sepsis, and its inhibition decreased
permeability. These results indicated that the functions of HMGB1
were likely responsible for the increased EC permeability
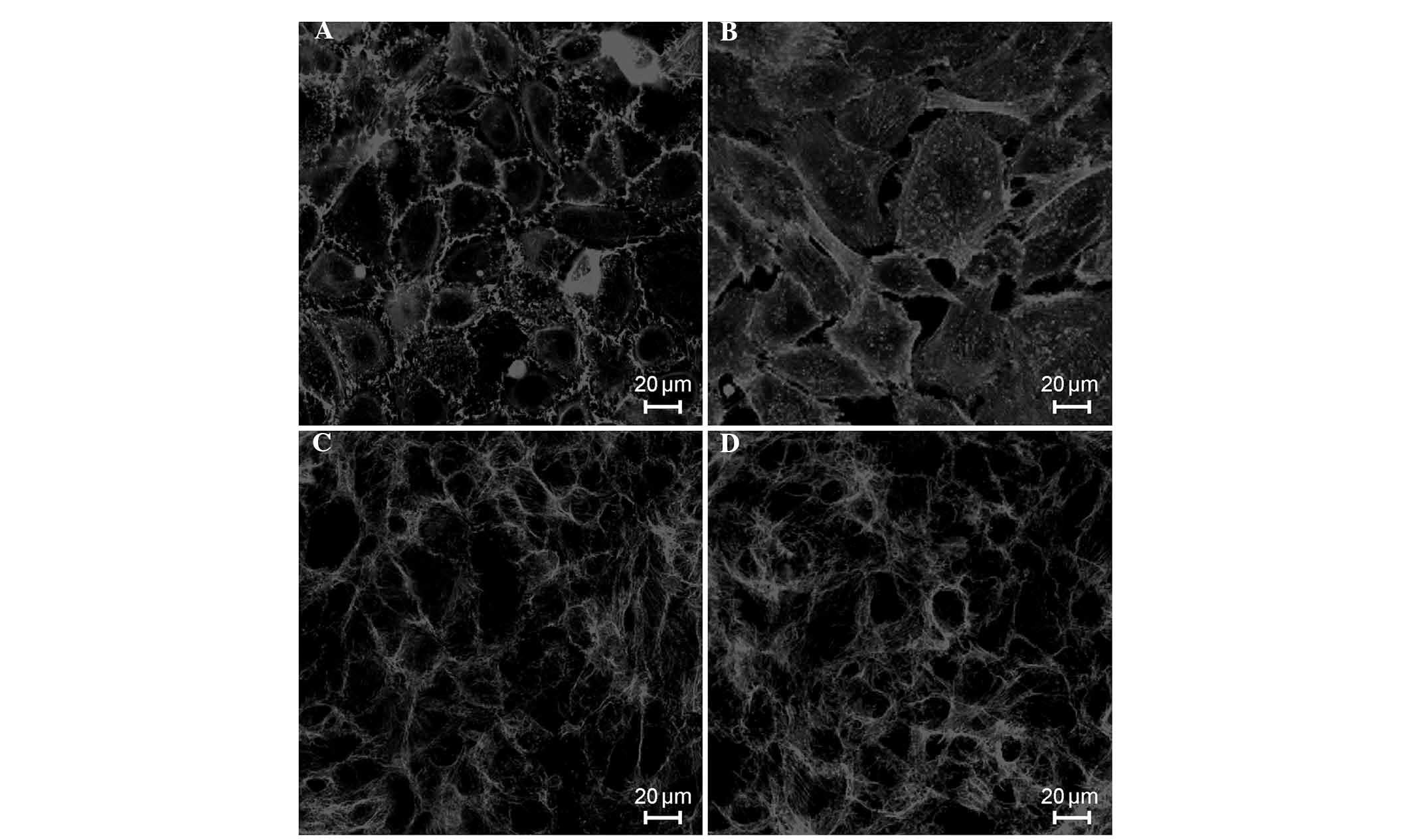
Effects of septic serum on the
morphologies of endothelial cytoskeletal actin and VE-cadherin
In the control cells, the majority of F-actin was
localized to the cell periphery, parallel to cell-cell junctions,
with no clear gaps between the cells (Fig. 4A). When the ECs were exposed to
septic serum for 6 h, almost all of cells became elongated, with
thick actin stress fibers that traversed the cells in the direction
of cell elongation (F-actin reorganization), increasing the
intercellular gap distances (Fig.
4B). However, pretreatment of the cells with EP (5 µM)
for 24 h inhibited F-actin reorganization, as shown in Fig. 4C. Treatment with EP alone did not
cause F-actin reorganization, and the morphology of endothelial
F-actin in these cells was similar to the normal morphology
(Fig. 4D). Similarly, in the
control cells VE-cadherin was located at the cell periphery and
predominantly formed a continuous line at the cell-cell junctions
with occasional gaps (Fig. 5A).
However, upon exposure to septic serum for 6 h, the junctions
became segmented and discontinuous, despite the fact that
VE-cadherin remained localized to areas of cell-cell contact,
indicating that the adhesive connection integrity was disrupted
(Fig. 5B). However, pretreatment
with EP partially inhibited the diffuse redistribution of
VE-cadherin (Fig. 5C), whereas EP
treatment alone had no effect on the morphology of VE-cadherin
(Fig. 5D).
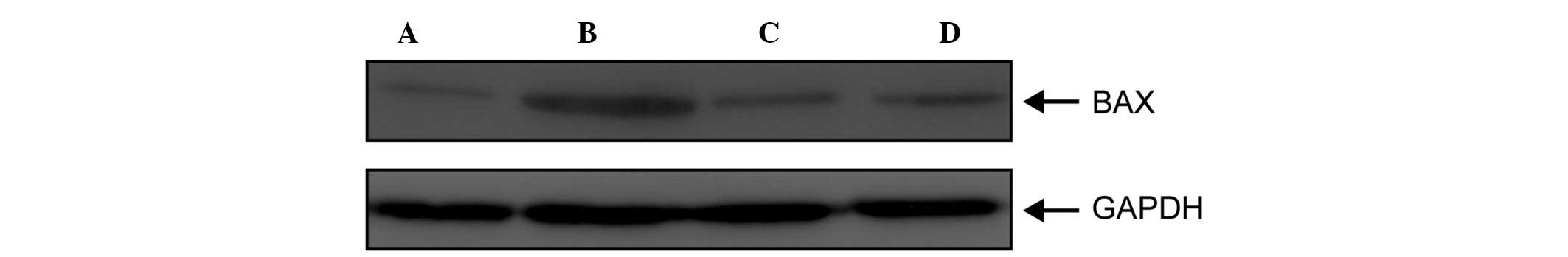
Effects of septic serum on the protein
expression levels of endothelial BAX and BCL-2
As shown in Fig.
6A, BAX, a pro-apoptotic protein, was not expressed in the ECs
treated with control serum; however, its expression was markedly
upregulated following treatment with septic serum (Fig. 6B). In addition, the upregulation in
the expression of BAX was inhibited by pretreatment with 5
µM EP (Fig. 6C), whereas
treatment with EP alone had no effect on the expression levels of
BAX (Fig. 6D). As shown in
Fig. 7, the protein expression
levels of BCL-2, an inhibitor of apoptosis, were markedly reduced
when the ECs were stimulated with septic serum, compared with those
treated with control serum (Fig. 7A
and B). However, the protein expression levels of BCL-2 were
restored following EP pretreatment (Fig. 7C). These results suggested that
HMGB1-rich serum induced EC apoptosis by downregulating the
expression of BCL-2 and upregulating the expression of BAX.
Effects of septic serum on endothelial
apoptosis
The effects of septic serum on endothelial apoptosis
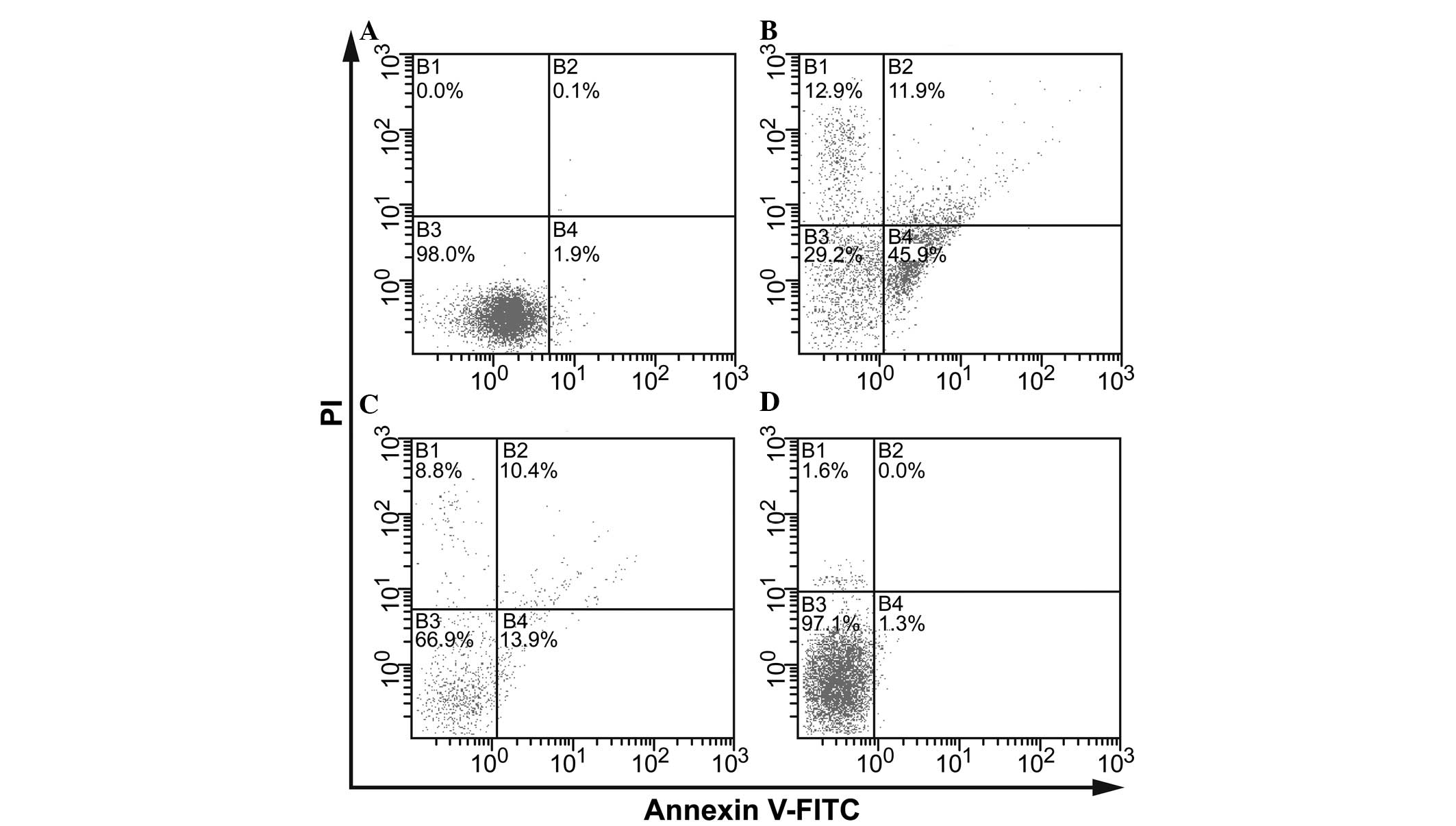
were evaluated using flow cytometry. In Fig. 8, the lower left quadrant represents
normal cells, whereas the upper left quadrant represents dead
cells, and the lower right quadrant represents early apoptotic
cells, whereas the upper right quadrant represents late apoptotic
cells. Treatment with septic serum markedly increased the rate of
endothelial apoptosis; with early apoptotic cells accounting for
45.9% of the total cells and late apoptotic cells accounted for
11.9%. In addition, compared with the control group (Fig. 8A), necrotic cells accounted for
12.9% of the cells (Fig. 8B).
However, endothelial apoptosis was markedly reduced following EP
pretreatment, particularly the early apoptotic cells, which
accounted for only 13.9% of the cells (Fig. 8C). However, treatment with EP alone
had no effect on endothelial apoptosis (Fig. 8D). These results indicated that
high levels of HMGB1 in the septic serum may be involved in
inducing endothelial apoptosis and EC permeability, particularly
early apoptosis. However, with prolonged exposure, EC death
occurs.
Discussion
Among critical illnesses, sepsis accounts for the
highest mortality rate of non-cardiac diseases in the ICU (21), which is often complicated by acute
respiratory distress syndrome, septic shock, multiple organ
dysfunction syndrome (MODS) and/or multiple organ failure (MOF)
(1). The reason for this is that
the uncontrolled release of inflammatory cytokines leads to SIRS
and compensatory anti-inflammatory response syndrome, which
ultimately results in the patient succumbing to mortality.
Proinflammatory cytokines, including early stage TNF-α and
interleukin-1β, and late stage HMGB1, interact with other
inflammatory effector cells, including polymorphonuclear
neutrophils and ECs, to form the inflammatory cascade effect, which
causes damage to body organs and affects their functions (22). It has been demonstrated that EC
dysfunction is an important factor that contributes to the
mortality rates of patients with sepsis (3).
As a late release proinflammatory cytokine, compared
with early release inflammatory cytokines, HMGB1 exhibits distinct
kinetic effects due to its relatively late release and sustained
duration in the blood (8,23). The expression of HMGB1 is often
associated with the mortality rates of patients with sepsis; and
high levels of plasma HMGB1 are positively correlated with
mortality rate (9,24). HMGB1 interacts with ECs via RAGE,
which is expressed on ECs (10,25).
The present study demonstrated that the expression of HMGB1 was not
detected until 24 h following the onset of sepsis; however, serum
expression levels were sustained until 96 h. Furthermore, ECs
treated with septic serum, containing highly expression levels of
HMGB1, exhibited increased endothelial permeability. When the
function of HMGB1 was inhibited by EP pretreatment, the effects of
septic serum were significantly attenuated. These results indicated
that HMGB1 is a pro-inflammatory cytokine secreted in late sepsis,
which may be involved in activating and injuring ECs, and
maintaining and extending the pathological process of sepsis.
As a feature of sepsis, capillary leakage represents
an endothelial barrier dysfunction, which can be induced by the
secretion of inflammatory cytokines that increase endothelial
permeability (26). The dangers of
capillary leakage include increased tissue edema, cell hypoxia and
the excessive local accumulation of inflammatory cytokines, which
directly lead to tissue and cell damage and can induce MODS or MOF
(27). The pathological basis of
increased endothelial permeability is the loss of or damage to EC
barrier function integrity (28).
The key factor in maintaining integrity is the state of the actin
cytoskeleton and connexin (29,30).
Under normal conditions, F-actin appears in the form of dense bands
around the periphery, and ECs appear relaxed enabling their barrier
functions to maintain integrity (31). When ECs are subjected to
inflammatory stimuli, F-actin can undergo rearrangement and
aggregate into thick stress fibers across the cell, resulting in
increased EC contraction and the formation of gaps between the ECs
(17). In turn, the paracellular
permeability of the ECs increase, and macromolecules, which usually
cannot pass through, will leak outside the capillary through gaps
between the ECs, resulting in capillary leakage. Cadherin is a
calcium-dependent endothelial cell-specific transmembrane adhesion
protein, which connects to the actin cytoskeleton via catenin
(32). VE-cadherin undergoes
morphological changes when the tension generated by F-actin
rearrangement passes to VE-cadherin through catenin (33). As a result, VE-cadherin is
endocytosed or degraded, causing ECs to lose integrity.
The present study demonstrated that EC permeability
increased significantly (P<0.01) when the ECs cultured in
vitro were stimulated with septic serum, compared with control
serum. In addition, endothelial F-actin rearrangement occurred; in
which the dense filaments disappeared at the periphery and thick
stress fibers formed across the ECs, creating gaps between the ECs
and altering VE-cadherin morphology. Green fluorescence intensity
was significantly reduced, suggesting that VE-cadherin underwent
degradation or endocytosis to account for the loss of EC integrity,
however, these effects were largely inhibited by EP pretreatment.
These results suggested that HMGB1 affected cell permeability, and
may be considered a therapeutic target to reduce the detrimental
effects of sepsis and subsequent inflammation.
Previous studies have demonstrated that apoptosis is
an important pathological change, which occurs during sepsis
(34,35). ECs are important in the host
response to sepsis, and endothelial system dysfunction is an
important feature of sepsis-associated organ failure and mortality
(4). At the cellular level, the
loss of endothelial function in the present study was predominantly
due to the increased apoptosis of ECs. EC apoptosis is an important
part of the pathological process of sepsis. Vascular ECs (VECs)
encounter various inflammatory cytokines and toxins, the exposure
to which may result in VEC activation and damage. If the stimulus
cannot be removed, the VECs will inevitably move toward apoptosis
and, once EC apoptosis begins, apoptotic cells enter the
bloodstream and form circulating endothelial cells (36). Animal experiments have previously
demonstrated that the injection of endotoxin into the blood can
damage microvascular ECs and lead to EC shedding (16). In addition, EC shedding can be
detected in the peripheral blood of patients with sepsis, and the
extent of shedding is associated with mortality rates (16). VEC apoptosis can lead to the loss
of endothelial barrier function, resulting in vascular leakage and
edema in patients with sepsis. The present study demonstrated that
the number of apoptotic ECs in the peripheral blood of patients
with late sepsis was significantly increased, indicating that the
apoptosis of ECs in late sepsis may be an important characteristic
contributing to multiple organ dysfunction and sepsis-associated
conditions. However, EP pretreatment reduced apoptosis, suggesting
that HMGB1 is also likely to be associated with apoptosis in
patients with sepsis.
The relative expression levels of BCL-2 and BAX,
which are encoded by genes upstream of apoptotic signaling
pathways, are important factors, which determine whether apoptosis
takes place in a cell (37). For
this reason, these proteins have increased in interest. The results
of the present study revealed that when ECs were stimulated with
septic serum, the expression levels of BCL-2 were significantly
decreased, whereas the expression levels of BAX were significantly
increased. However, these expression patterns were restored to
normal levels following EP pretreatment, suggesting that HMGB1 is
involved in inducing apoptosis by affecting the expression levels
of BCL-2 and BAX. Additional investigations are required to
elucidate the precise molecular mechanism underlying the expression
of apoptotic genes/proteins.
The results of the present require caution in
interpretation, as sera from patients with sepsis, which occurs at
different time points, may have different compositions of
inflammatory mediators affecting EC permeability (38). In addition, HMGB1, which is a major
proinflammatory cytokine in late sepsis, can induce the secretion
of other inflammatory cytokines (39). Therefore, it is difficult to
distinguish the direct effect of HMGB1 alone, compared with the
effects of other cytokines/mediators present in whole serum.
In conclusion, the present study demonstrated that
HMGB1 was released in the sera of patients with late sepsis, which
increased the permeability of ECs cultured in vitro. The
underlying mechanism of action may be associated with the induction
of EC apoptosis by altering the expression levels of BAX and
BCL-2.
Acknowledgments
The authors of the present study would like to thank
the Duoease Scientific Service Center for their language editing
and suggestions for figure revision. The study was supported, in
part, by funds from the Shanghai Municipal Commission of Health and
Family Planning Research Fund (grant. no. 20124318) and the
Chongming County Science and Technology Development Fund (grant.
no. CKY 2012-02), and predominantly by the Shanghai Jiao Tong
University School of Medicine Foundation (grant no. 12XJ22016).
References
|
1
|
Yao YM and Sheng ZY: Progress of basis
research on traumatic sepsis in China. Chin J Trauma. 19:9–12.
2003.
|
|
2
|
Koh IH, Menchaca-Diaz JL, Koh TH, Souza
RL, Shu CM, Rogerio VE and Liberatore AM: Microcirculatory
evaluation in sepsis: A difficult task. Shock. 34(Suppl 1): 27–33.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aird WC: The role of the endothelium in
severe sepsis and multiple organ dysfunction syndrome. Blood.
101:3765–3777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee WL and Liles WC: Endothelial
activation, dysfunction and permeability during severe infections.
Curr Opin Hematol. 18:191–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee WL and Slutsky AS: Sepsis and
endothelial permeability. N Engl J Med. 363:689–691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schouten M, Wiersinga WJ, Levi M and van
der Poll T: Inflammation, endothelium, and coagulation in sepsis. J
Leukoc Biol. 83:536–545. 2008. View Article : Google Scholar
|
|
7
|
Wang H, Yang H and Tracey KJ:
Extracellular role of HMGB1 in inflammation and sepsis. J Intern
Med. 255:320–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang H, Yang H, Czura CJ, Sama AE and
Tracey KJ: HMGB1 as a late mediator of lethal systemic
inflammation. Am J Respir Crit Care Med. 164:1768–1773. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Karlsson S, Pettilä V, Tenhunen J,
Laru-Sompa R, Hynninen M and Ruokonen E: HMGB1 as a predictor of
organ dysfunction and outcome in patients with severe sepsis.
Intensive Care Med. 34:1046–1053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kokkola R, Andersson A, Mullins G, Ostberg
T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA and
Harris HE: RAGE is the major receptor for the proinflammatory
activity of HMGB1 in rodent macrophages. Scand J Immunol. 61:1–9.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mullins GE, Sunden-Cullberg J, Johansson
AS, Rouhiainen A, Erlandsson-Harris H, Yang H, Tracey KJ, Rauvala
H, Palmblad J, Andersson J and Treutiger CJ: Activation of human
umbilical vein endothelial cells leads to relocation and release of
high-mobility group box chromosomal protein 1. Scand J Immunol.
60:566–573. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gill SE, Taneja R, Rohan M, Wang L and
Mehta S: Pulmonary microvascular albumin leak is associated with
endothelial cell death in murine sepsis-induced lung injury in
vivo. PloS One. 9:e885012014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hornburger MC, Mayer BA, Leonhardt S,
Willer EA, Zahler S, Beyerle A, Rajalingam K, Vollmar AM and Fürst
R: A novel role for inhibitor of apoptosis (IAP) proteins as
regulators of endothelial barrier function by mediating RhoA
activation. FASEB J. 28:1938–1946. 2014. View Article : Google Scholar
|
|
14
|
Aird WC: Endothelium as an organ system.
Crit Care Med. 32(5 Suppl): S271–S279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leclerc J, Pu Q, Corseaux D, Haddad E,
Decoene C, Bordet R, Six I, Jude B and Vallet B: A single endotoxin
injection in the rabbit causes prolonged blood vessel dysfunction
and a procoagulant state. Crit Care Med. 28:3672–3678. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mutunga M, Fulton B, Bullock R, Batchelor
A, Gascoigne A, Gillespie JI and Baudouin SV: Circulating
endothelial cells in patients with septic shock. Am J Respir Crit
Care Med. 163:195–200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McKenzie JA and Ridley AJ: Roles of
Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial
morphology and permeability. J Cell Physiol. 213:221–228. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Nieuw Amerongen GP, van Delft S,
Vermeer MA, Collard JG and van Hinsbergh VW: Activation of RhoA by
thrombin in endothelial hyperpermeability: Role of Rho kinase and
protein tyrosine kinases. Circ Res. 87:335–340. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng YJ, Zhou B, Song ZF, Li L, Wu J,
Zhang RY and Tang YQ: Study of Astragalus mongholicus
polysaccharides on endothelial cells permeability induced by HMGB1.
Carbohydr Polym. 92:934–941. 2013. View Article : Google Scholar
|
|
20
|
Dellinger RP, Levy MM, Rhodes A, Annane D,
Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke
R, et al: Surviving Sepsis Campaign: International guidelines for
management of severe sepsis and septic shock, 2012. Intensive Care
Med. 39:165–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lagu T, Rothberg MB, Shieh MS, Pekow PS,
Steingrub JS and Lindenauer PK: Hospitalizations, costs, and
outcomes of severe sepsis in the United States 2003 to 2007. Crit
Care Med. 40:754–761. 2012. View Article : Google Scholar
|
|
22
|
Bilgin YM, van de Watering LM, Versteegh
MI, van Oers MH and Brand A: Effects of allogeneic leukocytes in
blood transfusions during cardiac surgery on inflammatory mediators
and postoperative complications. Crit Care Med. 38:546–552. 2010.
View Article : Google Scholar
|
|
23
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bitto A, Barone M, David A, Polito F,
Familiari D, Monaco F, Giardina M, David T, Messina R, Noto A, et
al: High mobility group box-1 expression correlates with poor
outcome in lung injury patients. Pharmacol Res. 61:116–120. 2010.
View Article : Google Scholar
|
|
25
|
Fiuza C, Bustin M, Talwar S, Tropea M,
Gerstenberger E, Shelhamer JH and Suffredini AF:
Inflammation-promoting activity of HMGB1 on human microvascular
endothelial cells. Blood. 101:2652–2660. 2003. View Article : Google Scholar
|
|
26
|
Liu MW, Wang YH, Qian CY and Li H:
Xuebijing exerts protective effects on lung permeability leakage
and lung injury by upregulating Toll-interacting protein expression
in rats with sepsis. Int J Mol Med. 34:1492–1504. 2014.PubMed/NCBI
|
|
27
|
Bannerman DD and Goldblum SE: Direct
effects of endotoxin on the endothelium: Barrier function and
injury. Lab Invest. 79:1181–1199. 1999.PubMed/NCBI
|
|
28
|
Goldenberg NM, Steinberg BE, Slutsky AS
and Lee WL: Broken barriers: A new take on sepsis pathogenesis. Sci
Transl Med. 3:88ps252011.PubMed/NCBI
|
|
29
|
Jones TJ, Adapala RK, Geldenhuys WJ,
Bursley C, AbouAlaiwi WA, Nauli SM and Thodeti CK: Primary cilia
regulates the directional migration and barrier integrity of
endothelial cells through the modulation of hsp27 dependent actin
cytoskeletal organization. J Cell Physiol. 227:70–76. 2012.
View Article : Google Scholar
|
|
30
|
Corada M, Mariotti M, Thurston G, Smith K,
Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro
A, Ruco L, et al: Vascular endothelial-cadherin is an important
determinant of microvascular integrity in vivo. Proc Natl Acad Sci
USA. 96:9815–9820. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ng CT, Fong LY, Sulaiman MR, Moklas MA,
Yong YK, Hakim MN and Ahmad Z: Interferon-gamma increases
endothelial permeability by causing activation of p38 MAP kinase
and actin cytoskeleton alteration. J Interferon Cytokine Res.
35:513–522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Muradashvili N, Tyagi N, Tyagi R, Munjal C
and Lominadze D: Fibrinogen alters mouse brain endothelial cell
layer integrity affecting vascular endothelial cadherin. Biochem
Biophys Res Commun. 413:509–514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wesche-Soldato DE, Swan RZ, Chung CS and
Ayala A: The apoptotic pathway as a therapeutic target in sepsis.
Curr Drug Targets. 8:493–500. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmid FX, Vudattu N, Floerchinger B,
Hilker M, Eissner G, Hoenicka M, Holler E and Birnbaum DE:
Endothelial apoptosis and circulating endothelial cells after
bypass grafting with and without cardiopulmonary bypass. Eur J
Cardiothorac Surg. 29:496–500. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu H, Wang QS, Luo Q, Tan S, Su H, Tang
SL, Zhao ZL and Huang LP: Simvastatin inhibits apoptosis of
endothelial cells induced by sepsis through upregulating the
expression of Bcl-2 and downregulating Bax. World J Emerg Med.
4:291–297. 2014. View Article : Google Scholar
|
|
36
|
Kluz J, Kopeć W, Jakobsche-Policht U and
Adamiec R: Circulating endothelial cells, endothelial apoptosis and
soluble markers of endothelial dysfunction in patients with
systemic lupus erythematosus-related vasculitis. Int Angiol.
28:192–201. 2009.PubMed/NCBI
|
|
37
|
Xiao D and Zhang L: Upregulation of Bax
and Bcl-2 following prenatal cocaine exposure induces apoptosis in
fetal rat brain. Int J Med Sci. 5:295–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang Y, Li X, Zhang X, Li Z, Wang L, Sun
Y, Liu Z and Ma X: Elevated levels of plasma TNF-α are associated
with microvascular endothelial dysfunction in patients with sepsis
through activating the NF-κB and p38 mitogen-activated protein
kinase in endothelial cells. Shock. 41:275–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Andersson U, Wang H, Palmblad K, Aveberger
AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M,
Yang H and Tracey KJ: High mobility group 1 protein (HMG-1)
stimulates proinflammatory cytokine synthesis in human monocytes. J
Exp Med. 192:565–570. 2000. View Article : Google Scholar : PubMed/NCBI
|