Introduction
Parkinson's disease (PD), the second most common
neurodegenerative disease in the world, is characterized by the
loss of dopaminergic neurons and muscular rigidity (1). A major product of the oxidation of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is
1-methyl-4-phenylpyridinium (MPP+), which has been
extensively used in a variety of in vitro systems to model
PD (2). MPP+ has been
reported to be actively transported into dopaminergic neurons via
the plasma membrane in a similar manner to dopamine transporters
(3). Neurotoxicity of
MPTP/MPP+ is complex, and the overproduction of nitric
oxide (NO), hydroxyl radical generation and apoptosis have all been
associated with the neurotoxicity of MPP+ (4). Furthermore, MPP+ was
reported to induce mitochondrial dysfunction by inhibiting the
activity of complex I (5).
However, the intracellular mechanisms of MPP+-induced
neurotoxicity underlying the degenerative process require further
elucidation.
Propofol (2,6-diisopropylphenol) is an intravenous
anesthetic agent that has been widely administered as a
short-acting intravenous anesthetic since the late 1980s (6). In addition to its application for
maintenance of sedative effects as an anesthetic, various
characteristics of propofol have been investigated in recent years.
Notably, propofol demonstrates anti-oxidative (7) and anti-inflammatory properties
(8). Propofol is chemically
similar to the endogenous antioxidant, α-tocopherol (vitamin E),
therefore should theoretically demonstrate similar properties
(9). Increasing evidence has
indicated that propofol scavenges oxygen free radicals, and
inhibits oxidative damage and the release of inflammatory factors
(10). Notably, a previous study
demonstrated the neuroprotective effects of propofol against
amyloid β (Aβ) toxicity in Alzheimer's disease (AD). In addition,
propofol has been shown to protect the brain from
ischemia-reperfusion injury (11).
However, to the best of our knowledge, it is unknown whether
propofol has similar protective effects on the pathophysiological
changes in PD. Therefore, in the present study, the effect of
propofol in reducing MPP+-induced toxicity was
investigated in human SH-SY5Y cells.
Materials and methods
Cell culture, treatment and
transfection
SH-SY5Y human neuroblastoma cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Eagle's
minimum essential medium (American Type Culture Collection) and
Ham's F12 medium (American Type Culture Collection) containing 10%
fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA) and 1%
penicillin (100 U/ml) and streptomycin (100 µg/ml; Lonza,
Walkersville, MD, USA). Cells were exposed to 50 µM
MPP+ (Sigma-Aldrich) in the presence or absence of 25 or
50 µM propofol (Sigma-Aldrich) for 24 h.
Western blot analysis
Proteins were extracted from cultured cells using
cell lysis buffer (Cell Signaling Technology, Inc., Danvers, MA,
USA). Protein concentration was determined using a bicinchoninic
acid protein assay kit (Sigma-Aldrich). The soluble protein
solutions were mixed with 4X sample buffer (0.25 M Tris-HCl, 20%
mercaptoethanol, 8% SDS, 20% sucrose, 0.008% bromophenol blue; pH
6.8) and boiled for 5 min. Equal quantities of protein (20
µg) were loaded onto SDS-PAGE (10% separation gel and 5%
spacer gel). The separation gel was then run at 80 V for 15 min and
the spacer gel was run at 120 V for 1 h with 1X running buffer (25
mM Tris-HCl, 200 mM glycine, 0.1% (w/v) SDS; Bio-Rad, Hercules, CA,
USA). The gel was then transferred onto a polyvinylidene fluoride
membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using a
transfer buffer (25 mM Tris, 192 mM glycine and 20% methanol) at 80
V for 3 h in an ice-cold environment. Following blocking in 5%
skimmed milk in Tris-buffered saline with Tween-20 (TBST; 20 mM
Tris-HCl, 150 mM sodium chloride, 0.1% Tween-20), the membranes
were incubated with the following primary antibodies at 4°C
overnight: Mouse monoclonal anti-human B cell lymphoma 2 (Bcl-2;
1:2,000; cat. no. sc-7382; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA); mouse monoclonal anti-human Bcl-2-associated X protein
(Bax; 1:3,000; cat. no. sc-20067; Santa Cruz Biotechnology, Inc.);
mouse monoclonal anti-human cytochrome c (1:2,000; cat. no.
sc-514435; Santa Cruz Biotechnology, Inc.); mouse monoclonal
anti-human cytochrome c oxidase subunit IV isoform 1 (COX4;
1:5,000; cat. no. sc-376731; Santa Cruz Biotechnology, Inc.);
rabbit polyclonal anti-human cleaved caspase-3 (1:1,000; cat. no.
sc-22171-R; Santa Cruz Biotechnology, Inc.); mouse monoclonal
anti-human β-actin (1:10,000; cat. no. sc-130065; Santa Cruz
Biotechnology, Inc.). The membranes were subsequently incubated
with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG
(1:5,000; cat. no. sc-2005; Santa Cruz Biotechnology, Inc.) or
HRP-conjugated goat anti-rabbit IgG (1:5,000; cat. no. sc-2004;
Santa Cruz Biotechnology, Inc.) secondary antibodies for 1 h at
room temperature. The membranes were washed three times with TBST
and the proteins were detected with the enhanced chemiluminescence
system (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
relative densities of protein bands were analyzed using Image J 2.1
software (National Institutes of Health, Bethesda, MA, USA).
Protein carbonyl assay
Cells were lysed with cell lysis buffer (Cell
Signaling Technology, Inc.) and protein samples from cells were
adsorbed to wells of an ELISA plate (Thermo Fisher Scientific,
Inc.) and reacted with 2,4-dinitrophenylhydrazine (DNPH;
Sigma-Aldrich). The hydrazone adducts were incubated with an
anti-DNPH antibody (1:1,000; Sigma-Aldrich; cat. no. HPA029675) for
1 h at 37°C, and a secondary rat anti-mouse monoclonal antibody
conjugated with HRP (1:3,000; Thermo Fisher Scientific, Inc.; cat.
no. 04-6120) for 1 h at 37°C. The method was calibrated using
oxidized bovine serum albumin (BSA; Sigma-Aldrich), and prepared as
previously described (12).
Reactive oxygen species (ROS)
determination
Fluorescent dye, 2′,7′-dichlorofluorescein-diacetate
(DCFH-DA; Sigma-Aldrich) was used to measure the intracellular ROS
level (13). Following the cell
treatment with 50 µM MPP+ in the presence or
absence of 25 or 50 µM propofol for 24 h, SH-SY5Y cells were
loaded with 10 µM DCFH-DA and incubated in a CO2
incubator for 30 min at 37°C. The cells were washed with phosphate
buffered saline (PBS) three times, and fluorescence signals were
recorded using a fluorescence microscope (IX71SIF-2; Olympus
Corporation, Tokyo, Japan).
4-Hydroxy-2-nonenal (4-HNE)
immunofluorescence staining
Following the indicated treatment, the cells were
fixed in 4% paraformaldehyde (Sigma-Aldrich) for 10 min at room
temperature (RT) followed by permeabilization with 0.4% Triton
X-100 (Sigma-Aldrich) on ice for 15 min. Cells were blocked with 5%
BSA and 2.5% FBS in PBS with Tween-20 (Sigma-Aldrich).
Subsequently, cells were incubated with anti-4-HNE (Cell Signaling
Technology, Inc.) for 2 h at RT followed by incubation with
Invitrogen Alexa-594-conjugated secondary antibodies (Thermo Fisher
Scientific) for 1 h at RT. Staining signals were recorded using a
fluorescence microscope.
Statistical analysis
Results are presented as the mean ± standard error
of the mean of at least three experiments. Results obtained from
different experiments were analyzed using one-way analysis of
variance and P<0.05 was considered to indicate a statistically
significant difference.
Cell viability determination
An MTT (Sigma-Aldrich) reduction assay was used to
determine cell viability incubated at 37°C at 5,000 per well. The
cells were cultured in 96-well plates and exposed to 50 µM
MPP+ in the presence or absence of 25 or 50 µM
propofol for 24 h. MTT (at a final concentration of 1.0 mg/ml) was
then added to each well and incubated for 2 h at 37°C in the dark.
The formed formazane crystal was dissolved in dimethyl sulfoxide
(Sigma-Aldrich). The solution was agitated at room temperature for
10 min, and absor-bance was determined at 570 nm using a iMark
Microplate Absorbance Reader (Bio-Rad Laboratories) to index cell
viability.
Determination of lactate dehydrogenase
(LDH) release
Cells were cultured in 96-well plates and exposed to
50 µM MPP+ in the presence or absence of 25 or 50
µM propofol for 24 h. A Cytotoxicity Detection kit
(Sigma-Aldrich) was used to determine the levels of LDH released
from the damaged cells, according to the manufacturer's
instructions. Absorbance was determined at 490 nm using a
microplate reader to quantify the levels of LDH.
Intracellular NO determination
The cells were exposed to 50 µM
MPP+ in the presence or absence of 25 or 50 µM
propofol for 24 h. The levels of intracellular NO was then
determined suing a diaminofluorescein-FM diacetate (DAF-FM DA)
cell-permeable fluorescent probe (Sigma-Aldrich). Briefly, 10
µM DAF-FM DA was loaded and incubated at 37°C for 30 min in
the dark. Fluorescence signals were captured using a a IX71SIF-2
fluorescence microscope.
Mitochondrial membrane potential (MMP)
determination
Cells were exposed to 50 µM MPP+
in the presence or absence of 25 or 50 µM propofol for 24 h.
The MMP was then determined using a tetramethylrhodamine methyl
ester (TMRM) fluorescence dye (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Briefly, the cells were
treated with 20 nmol/l TMRM and incubated for 60 min at room
temperature. Following three washes in PBS, fluorescence signals
were captured using a IX71SIF-2 fluorescence microscope.
Determination of ATP levels using a
bioluminescence assay
The cells were treated with 50 µM
MPP+ in the presence or absence of 25 or 50 µM
propofol for 24 h. The levels of ATP were determined using an ATP
bioluminescence assay kit (Roche Diagnositics, Basel, Switzerland)
according to the manufacturer's instructions. Briefly, the cells
were lysed and centrifuged at 10,000 × g for 10 min at 4°C. The
supernatants were then collected and mixed with equal amount of
luciferase reagent (Promega Corporation, Madison, WI, USA), which
catalyzed light production from ATP and luciferin. The signals were
recorded using a microplate luminometer (GloMax 96 Microplate
Luminometer; Promega Corporation) and used to index ATP
concentration.
Results
Propofol attenuates
MPP+-induced oxidative stress
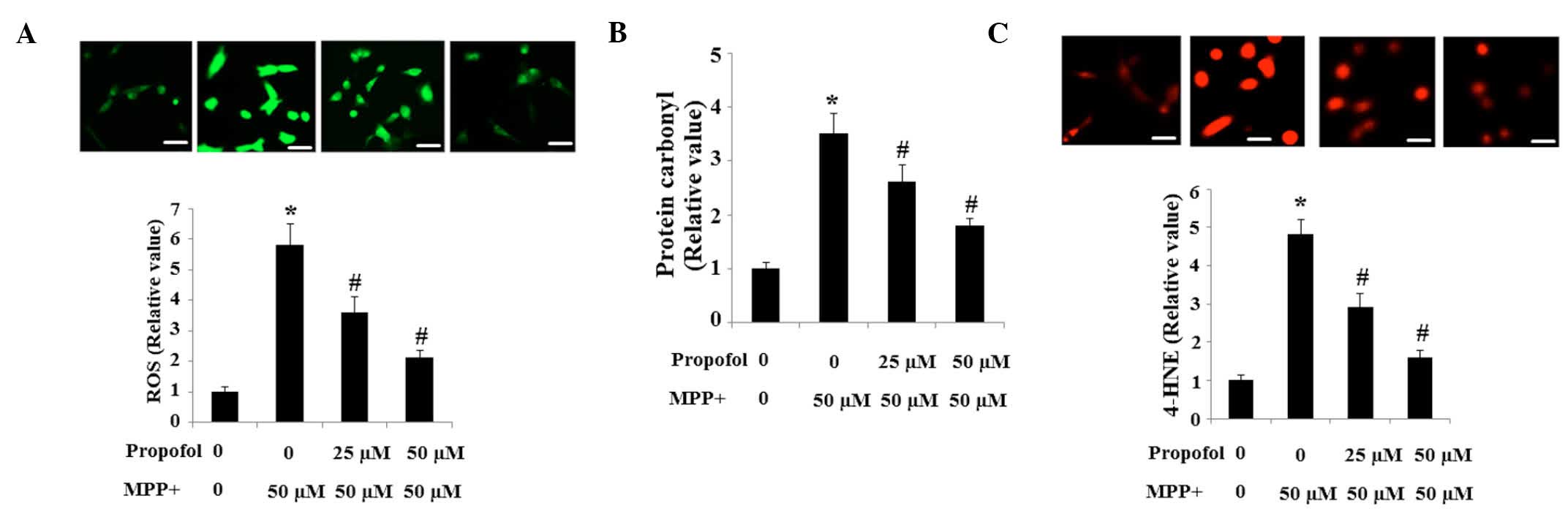
The production of intracellular ROS in SH-SY5Y cells
was assessed using the fluorescence probe, DCFH-DA. The results in
the present study indicate that the intracellular ROS level in
cells exposed to MPP+ is significantly higher when
compared with the controls, which was reduced by propofol treatment
in a dose-dependent manner (Fig.
1A). Consistent with these results, the basal level of protein
carbonyl (Fig. 1B) and 4-HNE
(Fig. 1C) was increased by
exposure to MPP+, and these increases were prevented by
pretreatment with propofol.
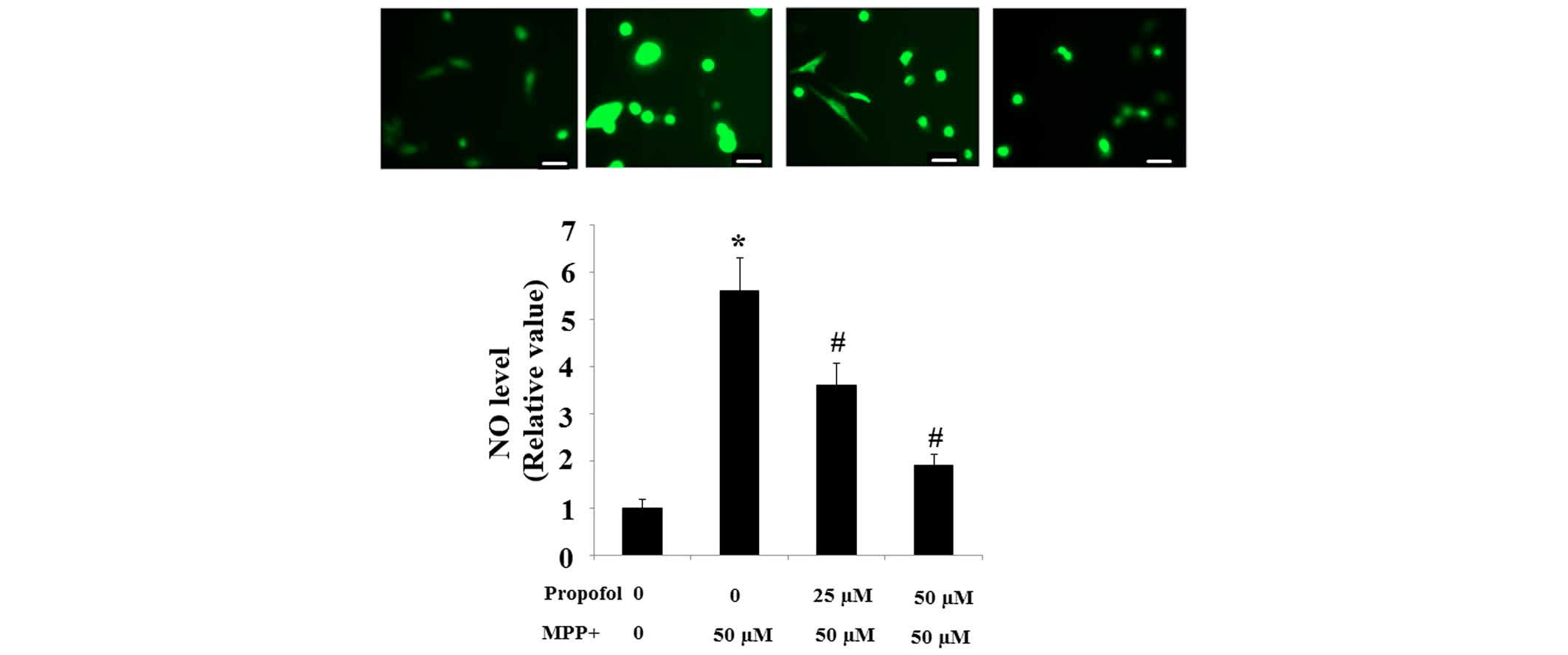
Propofol ameliorates the generation of NO
induced by MPP+
Intracellular production of NO is the main component
of reactive nitrogen species (RNS). The intracellular NO level in
SH-SY5Y cells exposed to MPP+ cells was observed to be
significantly higher than in control cells; however, increases in
the NO level were suppressed in a dose-dependent manner by
pretreatment with propofol (Fig.
2).
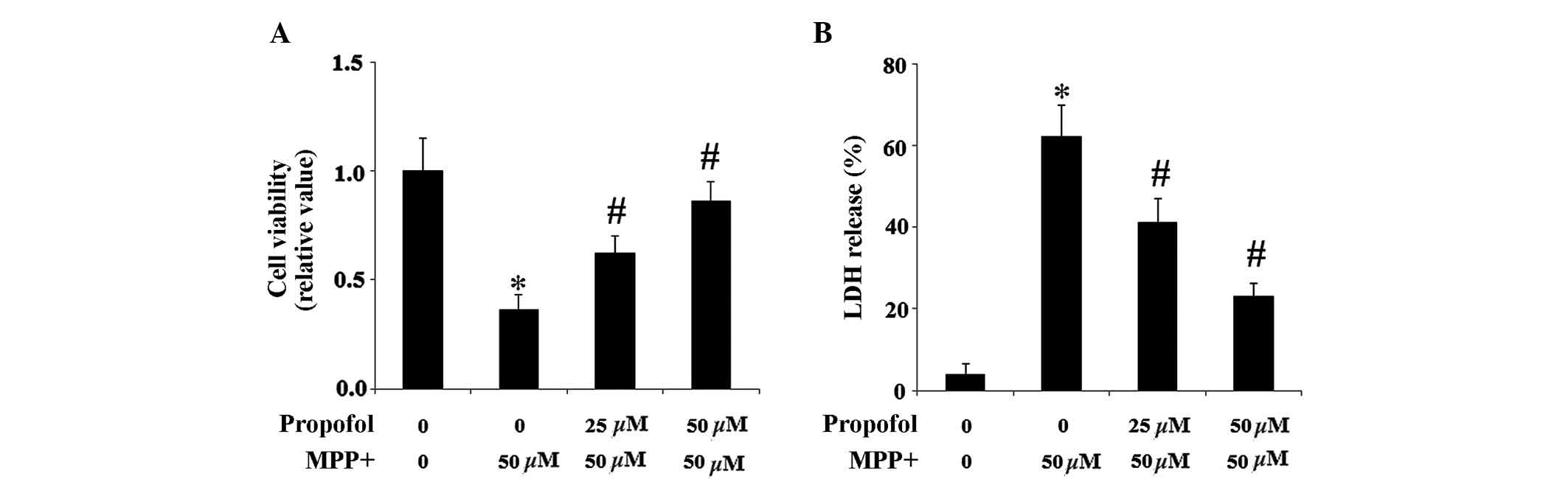
Propofol inhibits MPP+-induced
cell death
The MTT reduction assay was used to determine cell
viability. As presented in Fig.
3A, propofol treatment ameliorated the impaired cell viability
induced by 50 µM MPP+ exposure. To further
demonstrate that propofol increased cell resistance to
MPP+, the levels of cellular toxicity were determined
using a LDH assay. Pretreatment with propofol in SH-SY5Y cells
mitigated MPP+-induced LDH release following a 24-h
incubation (Fig. 3B).
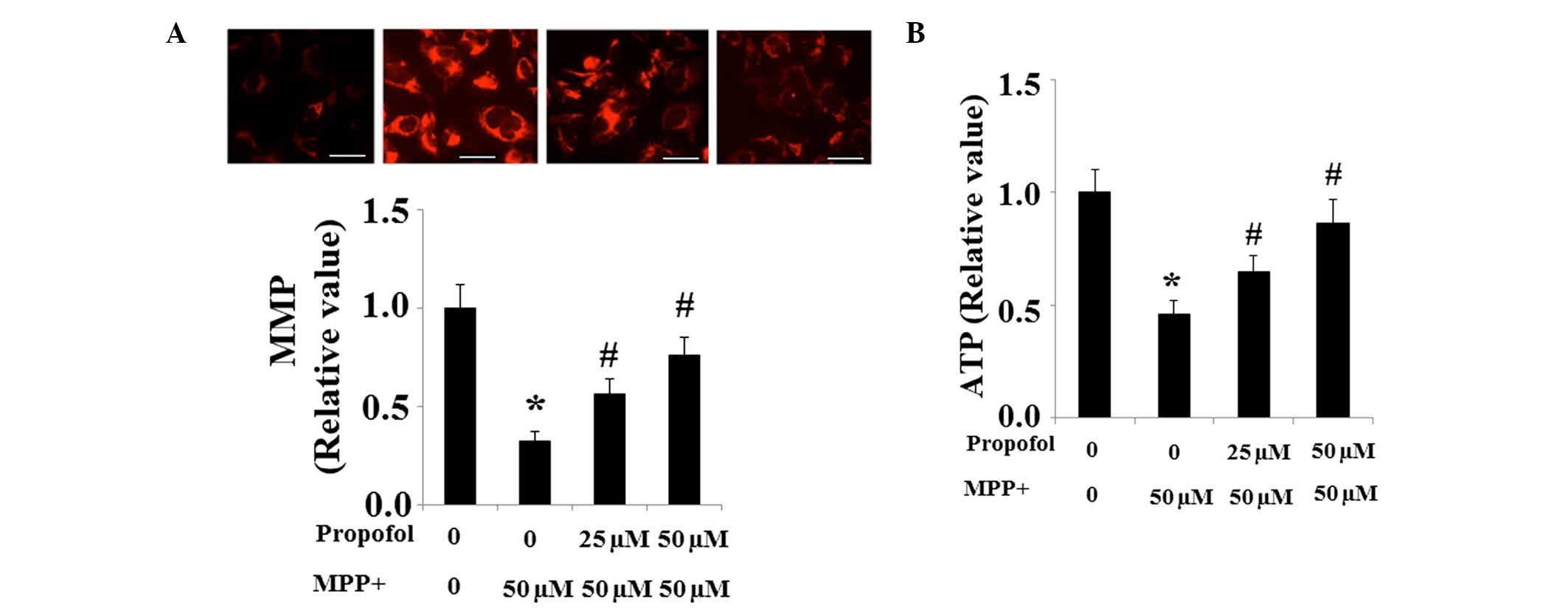
Propofol rescues MPP+-induced
mitochondrial dysfunction
The levels of MMP were investigated to determine
mitochondrial function. Significantly reduced MMP in SH-SY5Y cells
was observed following MPP+ exposure, which was
partially restored by pretreatment with propofol (Fig. 4A). Decreased levels of ATP
demonstrate mitochondrial dysfunction and results from the present
study indicate that the level of ATP was significantly reduced in
SH-SY5Y cells exposed to MPP+ when compared with the
non-treated controls. However, the decreased production of ATP was
partially ameliorated by propofol treatment (Fig. 4B).
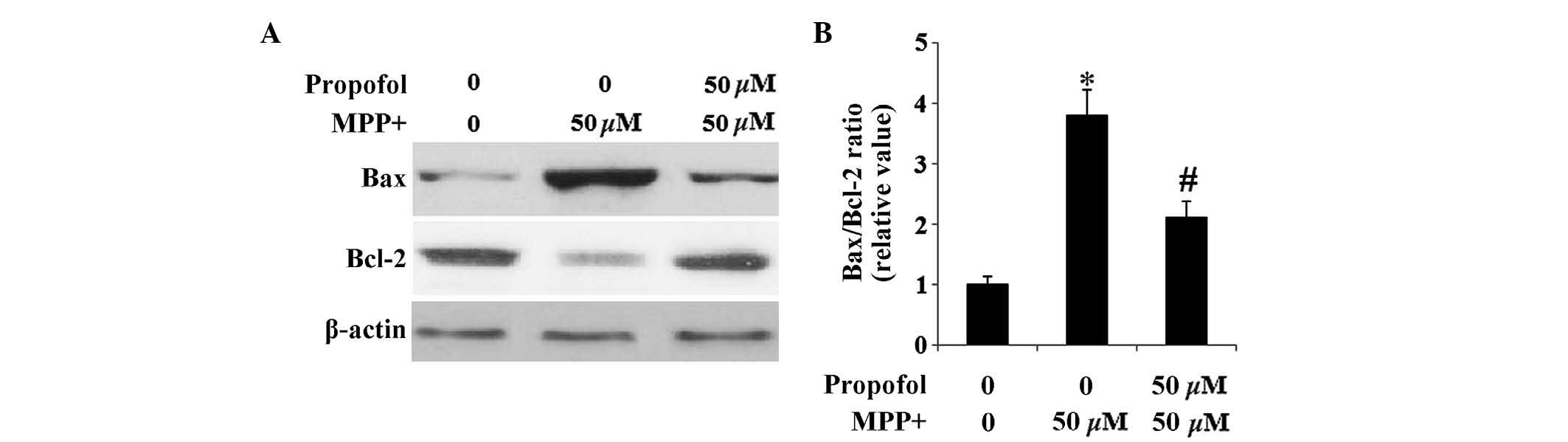
Propofol reverses the alteration of Bax
and Bcl-2 induced by MPP+
The difference between the effect of propofol on the
pro- and anti-apoptotic pathways in SH-SY5Y cells was examined. The
Bcl-2 family members are essential in the mitochondrial pathway of
apoptosis (14). It has been
previously reported that Bax is significantly upregulated and Bcl-2
is downregulated in MPP+-induced neurotoxicity (15). Results from the current study
indicate that SH-SY5Y cells exhibit an increase in the expression
levels of Bax and a marked decrease in the expression levels of
Bcl-2 following MPP+ exposure. Furthermore, these
expression levels were restored by administration of propofol
(Fig. 5).
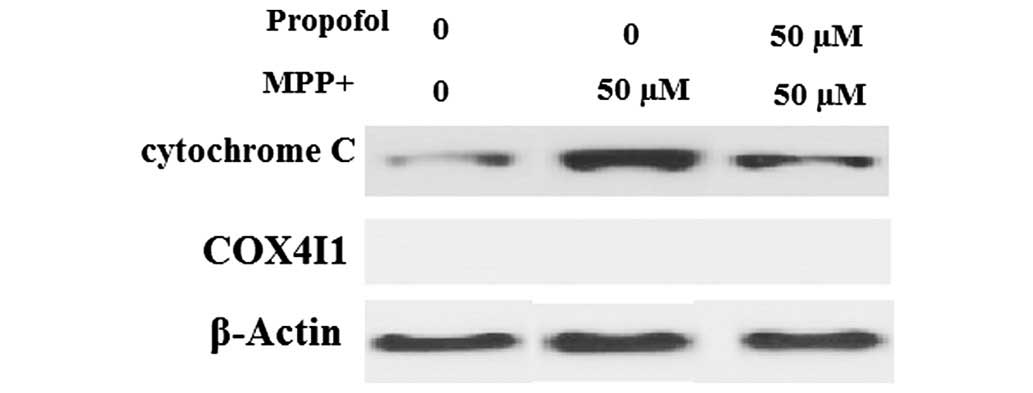
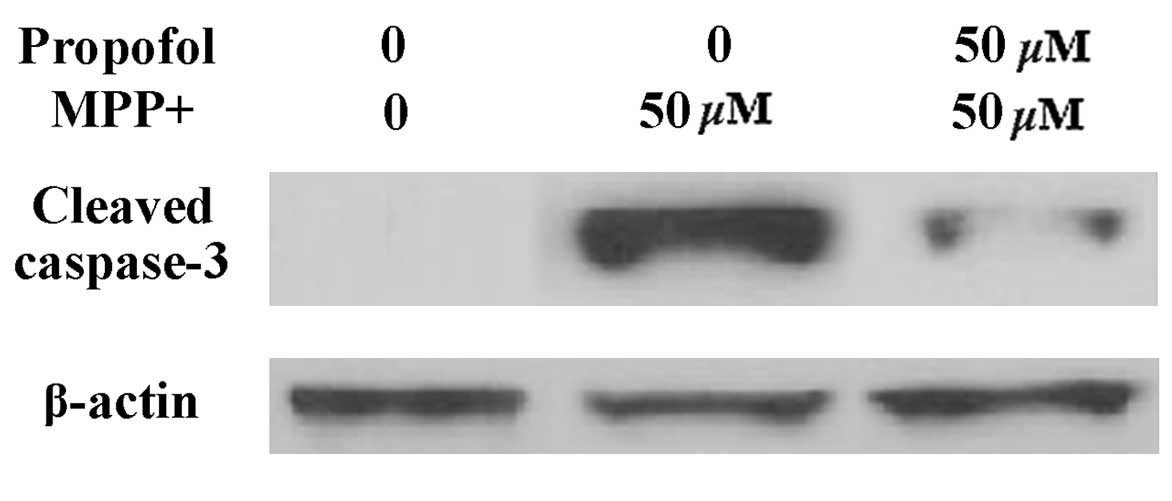
Propofol suppresses the release of
cytochrome c and caspase-3 cleavage
Impaired MMP results in the release of cytochrome
c into the cytoplasm, followed by activation of caspase-3.
Therefore, the effects of propofol on cytochrome c release
were investigated. As presented in Fig. 6, cytochrome c levels in the
cytosol were significantly increased in cells following
MPP+ exposure, which was markedly prevented by propofol
treatment. COX4 served as the internal control for cytosolic
fractions. Notably, it was observed that caspase-3 expression
levels were increased in SH-SY5Y cells following MPP+
exposure; however, the increased expression was significantly
attenuated by treatment with propofol (Fig. 7).
Discussion
PD has the second highest incidence of the
progressive degenerative central nervous system disorders. PD is
characterized by the loss of dopaminergic neurons and muscular
rigidity (16). The causes and
underlying mechanisms of PD are complex and remain to be
elucidated, however, free radicals produced during oxidative stress
contribute to the mechanism of cell death in PD. Antioxidant and
anti-apoptosis therapeutic strategies have become attractive
adjunct methods for PD treatment (17). MPP+ has been widely
demonstrated to result in hydroxyl radical generation (4), mitochondrial dysfunction (18), and apoptosis (19) in in vitro PD studies. In the
present study, the effects of propofol on the neurotoxicity of
PD-associated MPP+ were investigated. Patterns of
oxidative stress, mitochondrial dysfunction and cell susceptibility
were analyzed. Data from the present study demonstrates that
MPP+-induced oxidative stress, mitochondrial dysfunction
and apoptosis may be rescued by treatment with propofol.
Propofol is widely adopted for clinical studies as
an intravenous general anesthetic (20). It is administered in anesthesia and
sedation, as it is fast acting and patients rapidly regain full
consciousness, as the agent does not accumulate over time with
continuous infusion (21).
Notably, increasing evidence has demonstrated the anti-inflammatory
properties of propofol (8).
Corcoran et al (22)
reported that propofol may reduce neutrophil adhesion to vascular
endothelial cells by inhibiting the expression of adhesion
molecule, P-selectin. A previous study demonstrated that propofol
may inhibit oxidative damage by scavenging oxygen free radicals,
which is consistent with the findings of the present study that
propofol may possess an anti-oxidative stress property against
MPP+.
Notably, as an important neurotoxin, MPP+
induces apoptotic activity in dopaminergic neurons. In the present
study, apoptosis induced by MPP+ is partially restored
by propofol treatment. Propofol was also demonstrated to decrease
the expression of Bax and increase the expression of Bcl-2 in
SH-SY5Y cells exposed to MPP+. This may explain the
results in the present study that propofol treatment also inhibited
cytochrome c release and caspase-3 activation. The results
indicate that propofol protected the SH-SY5Y cells against
MPP+-induced apoptosis via the inhibition of the
mitochondrial apoptosis pathway. In addition, propofol has been
demonstrated to inhibit cardiomyocyte apoptosis and the release of
inflammatory factors (10).
Previous studies have reported the neuroprotective
effects of propofol in neurological disorders. For example, a
previous study demonstrated that propofol treatment may inhibit AD
pathogenesis. The apoptosis rate was significantly decreased when
cells were administered with Aβ25-35 and propofol, and
an increase in Bcl-2 expression and a decrease in tau
phosphorylation were also observed, when compared with
Aβ25-35 treatment alone (23). In addition, previous studies have
suggested that propofol may exert neuroprotective effects,
particularly in ischemia-reperfusion injury conditions, including
hypoxemia and hypothermia (11).
In conclusion, the results of the present study demonstrate that
propofol is a potential therapeutic agent for the treatment of PD.
Further investigation may elucidate the underlying mechanisms of
PD, as well as those of additional neurode-generative diseases.
References
|
1
|
Dauer W and Przedborski S: Parkinson's
disease: Mechanisms and models. Neuron. 39:889–909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chiba K, Trevor AJ and Castagnoli N Jr:
Active uptake of MPP+, a metabolite of MPTP, by brain
synaptosomes. Biochem Biophys Res Commun. 128:1228–1232. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kopin IJ and Markey SP: MPTP toxicity:
Implications for research in Parkinson's disease. Annu Rev
Neurosci. 11:81–96. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Obata T: Nitric oxide and MPP+-induced
hydroxyl radical generation. J Neural Transm. 113:1131–1144. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dawson TM and Dawson VL: Molecular
pathways of neurodegeneration in Parkinson's disease. Science.
302:819–822. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saraghi M, Badner VM, Golden LR and Hersh
EV: Propofol: An overview of its risks and benefits. Compend Contin
Educ Dent. 34:252–258. 2013.PubMed/NCBI
|
|
7
|
Yamaguchi S, Hamaguchi S, Mishio M, Okuda
Y and Kitajima T: Propofol prevents lipid peroxidation following
transient forebrain ischemia in gerbils. Can J Anaesth.
47:1025–1030. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inada T, Kubo K and Shingu K: Possible
link between cyclooxygenase-inhibiting and antitumor properties of
propofol. J Anesth. 25:569–575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aarts L, van der Hee R, Dekker I, de Jong
J, Langemeijer H and Bast A: The widely used anesthetic agent
propofol can replace alpha-tocopherol as an antioxidant. FEBS Lett.
357:83–85. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Tan J, Zou Z, Huang CG and Shi XY:
Propofol post-conditioning protects against cardiomyocyte apoptosis
in hypoxia/reoxygenation injury by suppressing nuclear factor-kappa
B translocation via extracellular signal-regulated kinase
mitogen-activated protein kinase pathway. Eur J Anaesthesiol.
28:525–534. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harman F, Hasturk AE, Yaman M, Arca T,
Kilinc K, Sargon MF and Kaptanoglu E: Neuroprotective effects of
propofol, thiopental, etomidate, and midazolam in fetal rat brain
in ischemia-reperfusion model. Childs Nerv Syst. 28:1055–1062.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sheng B, Gong K, Niu Y, Liu L, Yan Y, Lu
G, Zhang L, Hu M, Zhao N, Zhang X, et al: Inhibition of γ-secretase
activity reduces Abeta production, reduces oxidative stress,
increases mitochondrial activity and leads to reduced vulnerability
to apoptosis: implications for the treatment of Alzheimer's
disease. Free Radic Biol Med. 46:1362–1375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sheng BY, Niu Y, Zhou H, Yan JX, Zhao NM,
Zhang XF and Gong YD: The mitochondrial function was impaired in
APP knockout mouse embryo fibroblast cells. Chin Sci Bull.
54:1725–1731. 2009. View Article : Google Scholar
|
|
14
|
Park JR and Hockenbery DM: BCL-2, a novel
regulator of apoptosis. J Cell Biochem. 60:12–17. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou J, Sun Y, Zhao X, Deng Z and P u X:
3-O-demethylswertipunicoside inhibits MPP+-induced
oxidative stress and apoptosis in PC12 cells. Brain Res.
1508:53–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crosiers D, Theuns J, Cras P and Van
Broeckhoven C: Parkinson disease: Insights in clinical, genetic and
pathological features of monogenic disease subtypes. J Chem
Neuroanat. 42:131–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moore DJ, West AB, Dawson VL and Dawson
TM: Molecular pathophysiology of Parkinson's disease. Annu Rev
Neurosci. 28:57–87. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boada J, Cutillas B, Roig T, Bermúdez J
and Ambrosio S: MPP(+)-induced mitochondrial dysfunction is
potentiated by dopamine. Biochem Biophys Res Commun. 268:916–920.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheehan JP, Palmer PE, Helm GA and Tuttle
JB: MPP+ induced apoptotic cell death in SH-SY5Y
neuroblastoma cells: An electron microscope study. J Neurosci Res.
48:226–237. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanto JH: Propofol, the newest induction
agent of anesthesia. Int J Clin Pharmacol Ther Toxicol. 26:41–57.
1988.PubMed/NCBI
|
|
21
|
McNeir DA, Mainous EG and Trieger N:
Propofol as an intravenous agent in general anesthesia and
conscious sedation. Anesth Prog. 35:147–151. 1988.PubMed/NCBI
|
|
22
|
Corcoran TB, O'Shea A, Engel A and Shorten
GD: The influence of propofol on P-selectin expression and nitric
oxide production in re-oxygenated human umbilical vein endothelial
cells. Acta Anaesthesiol Scand. 50:348–354. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang R, Xu J, Liu YY, Zuo PP, Yang N, Ji
C, Wang Y, Wang H, Wu AS and Yue Y: Propofol may protect PC12 cells
from β-amyloid25–35 induced apoptosis through the GSK-3β
signaling pathway. Chin Med J (Engl). 126:1884–1889. 2013.
|