Introduction
Aldosterone is the principal mineralocorticoid
hormone in humans, which is synthesized predominantly in the
adrenal zona glomerulosa. It regulates sodium excretion and
intravas-cular volume via distal convoluted tubules and renal
cortex collecting ducts (1).
Aldosterone synthase (CYP11B2), a cytochrome P450 enzyme encoded by
the CYP11B2 gene, is a key enzyme that controls the final
three steps of aldosterone biosynthesis (2). The first step is the
11-β-hydroxylation of 11-deoxycorticosterone to corticosterone; the
second step is the 18-hydroxylation of corticosterone to
18-hydroxycorticosterone (18OHB), and the third step is the
18-oxidation of 18-hydroxy-corticosterone to aldosterone (2). CYP11B2 (OMIM124080) is located
at 8q24.3 and contains nine exons encoding steroid
11-β-hydroxylase, 18-hydroxylase and 18-oxidase. CYP11B2
exhibits a high level of homology to the CYP11B1 gene, which
encodes the steroid, 11-β-hydroxylase (2–4).
Mutations in the CYP11B2 gene cause
aldosterone synthase deficiency (ASD); the clinical manifestations
of which vary with age (2).
Affected infants may develop symptoms of miner-alocorticoid
deficiency, and present clinically with frequent vomiting, a
variable degree of hyponatremia, hyperkalemia, and metabolic
acidosis combined with poor growth. Older children and adults
usually have normal serum electrolytes, even if untreated (1–3). The
clinical features of ASD are easily confused with several other
endocrine disorders, particularly prior to obtaining definitive
biochemical results. These endocrine disorders include
pseudohypoaldosteronism type 1 (PHA1), which is characterized by
resistance to the actions of aldosterone, and congenital adrenal
hyperplasia (CAH), associated with 21-hydroxylase deficiency,
11-β-hydroxylase deficiency, or 3-β-hydroxysteroid dehydrogenase
deficiency. For the clinical diagnosis of ASD, it may difficult to
determine the differential diagnosis from the above-mentioned
confusion. In addition, patients may succumb to mortality as a
result of salt wasting and high blood potassium (5–8).
Therefore, the establishment of a prompt and accurate diagnostic
method is required.
The present study describes the clinical features of
a 4 month-old boy with the clinical symptoms described above. Using
whole exome sequencing (WES) technology, two novel variants of the
CYP11B2 gene were identified, and the functional effects of
the mutation were assessed. The pathogenicity of the c.240–1G>A
mutation was further investigated by analyzing its effect on
splicing in a minigene construct.
Subjects and methods
Clinical description of the patient
A 4-month-old male infant was recruited to the
Gastroenterology department of the Shanghai Children's Medical
Center, Shanghai Jiaotong University School of Medicine (Shanghai,
China) due to frequent vomiting (6–8 times/day) without
inducements, shortly following birth, and had not improved with
age. The patient was a full-term baby and had an uncomplicated
delivery with a birth weight of 2,960 g. His parents were physical
healthy, with non-consanguineous marriage. Physical examination
showed his weight was 3,300 g, height was 54 cm, heart rate was 168
bpm, respiratory rate was 42 bpm, blood pressure was 82/55 mmHg,
and body temperature was 37.5°C. He had severe malnutrition and
moderate dehydration. The patient had male-appearing genitalia,
with marginally darkened color scrotal skin, and ~1 ml bilateral
testicles. Neither heart murmur, nor hepatosplenomegaly were found.
Congenital anomaly of the digestive tract and inborn metabolic
diseases were excluded. The patient had initially been treated for
gastroesophageal reflux and allergy to formula in another hospital,
without a satisfactory improvement. The present study was approved
by the ethics committee of the Shanghai Children's Medical Center,
Shanghai Jiaotong University School of Medicine.
The results of the laboratory examinations performed
on the first visit to hospital are shown in Table I. Based on the examination results,
the baby was treated intravenously for rehydration, supplementation
with sodium salt, correction of acidosis and nutritional support.
However, it was difficult to correct hyponatremia and hyperkalemia
following shifting to oral therapy, even with sodium
supplementation to 18 mEq/kg/day. Problems in adrenal hormone
production were considered, although the laboratory data did not
provide evidence to confirm a diagnosis, and hormone detection can
be affected by several factors (9). In order to obtain a clinical
diagnosis, mutant genes were screened using WES, and CYP11B2
gene mutations were identified, however, due to the high levels of
aldosterone at the first visit, aldosterone levels were evaluated
again. Unlike the first visit, a low aldosterone level (38.12
pg/ml) was found. Subsequently, the patient was simultaneously
treated with 9α-fluorohydrocortisone, and electrolyte levels
rapidly reached equilibrium (data not shown). The effectiveness of
the treatment supported the genetic diagnosis and the use of
molecular genetic assessments, the patient was finally diagnosed
with ASD.
 | Table ILaboratory results during initial
hospital visit. |
Table I
Laboratory results during initial
hospital visit.
| Factor | Value | Normal range |
|---|
| Na (mmol/l) | 113.00 | 137.00–145.00 |
| K (mmol/l) | 6.30 | 3.50–5.10 |
| CL (mmol/l) | 77.00 | 98.00–107.00 |
| Lactic acid
(mmol/l) | 2.70 | 0.70–2.10 |
| Aldosterone
(pg/ml) | 361.00 | 50.00–313.00 |
| PRA (ng/ml/h) | 0.03 | 0.13–1.94 |
| Cortisol
(µg/dl) | 26.90 | 5.70–16.60 |
| ACTH (pg/ml) | 33.00 | 8.00–80.00 |
|
17-hydroxyprogesterone (nmol/l) | 1.10 | <30.00 |
WES and data analysis
The steps of the WES experiment were based on the
report by Wang J et al (10). The genomic DNA of the patient and
parents were isolated from 2 ml peripheral blood samples collected
from the cubital veins using a QIAamp Blood DNA Mini
kit® (Qiagen GmbH, Hilden, Germany). A total of 3
µg DNA from the patient was processed through shearing using
a Covarias® M220 Ultrasonicator system (Covaris, Inc.
Woburn, MA, USA) to result in sizes of 150–200 bp. An
adapter-ligated library was prepared, and enrichment of the coding
exons and flanking intronic regions was performed. Clusters were
then generated by isothermal bridge amplification using an Illumina
cBot station, and sequencing was performed on an Illumina HiSeq
2000 system (Illumina, Inc., San Diego, CA, USA).
Base calling and sequence read quality assessment
were performed using Illumina HCS 2.2.58 software (Illumina, Inc.)
for the Illumina HiSeq 2000 system which included new versions of
HiSeq control software and Real Time Analysis. Alignment of the
sequence reads to a reference human genome (Human 37.3; SNP135) was
performed using NextGENe® (SoftGenetics LLC, State
College, PA, USA). All single nucleotide variants (SNVs) and indels
were saved in a VCF format file, and uploaded for
Ingenuity® Variant Analysis™ (Ingenuity Systems,
Mountain View, CA, USA) for biological analysis and
interpretation.
Sanger sequencing verification of the
CYP11B2 gene
The primers for amplification of the CYP11B2
gene (GenBank accession no. NM_000498.3) were designed using UCSC
ExonPrimer online software (http://genome.ucsc.edu/index.html) and synthesized by
Map Biotechnology, Co., Ltd., Shanghai, China. The primers designed
for exon 2 were as follows: Forward 5′-CAG AGA AAA CCC CAG CTC
AC-3′ and reverse 5′-CAA CCC ACA GTG CAG ACG-3′; and the primers
designed to amplify exon 6 were as follows: Forward 5′-CCC AAG TGG
TCA TCA AGGTT-3′ and reverse 5′-GGT GTT GAA GAG GGA TTC CA-3. The
exons and the exonintron boundaries were amplified using polymerase
chain reaction (PCR; Takara Biotechnology, Co., Ltd., Dalian,
China). The reaction mixture for each amplification contained 1X
Premix Taq (Ex Taq Version 2.0; cat. no. RR003; Takara
Biotechnology, Co., Ltd.), 100 ng genomic DNA, and 1 pmol forward
and reverse primer in a final volume of 25 µl. The reaction
was carried out with the following PCR conditions: Initial
denaturation at 95°C for 5 min, then 19 cycles of 95°C for 30 sec,
65°C for 30 sec, 72°C for 45 sec, 14 cycles of 95°C for 30 sec,
55°C for 30 sec, 72°C for 45 sec, and a final elongation step at
72°C for 5 min using a C1000™ Thermal Cycler PCR instrument
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PCR products
(5 µl) were examined on a 1% agarose gel and purified using
a QIAquick Gel Extraction kit (Qiagen GmbH). The resulting DNA was
sequenced using the ABI3730XL sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) with the forward and
reverse primers. The sequence data were analyzed using Mutation
Surveyor® software version 4.0.4 (SoftGenetics,
LLC).
Minigene construction
The splice region (c.240-1G>A; intron 1) of the
CYP11B2 gene was amplified from the patient's genomic DNA
using the following specific primer pair: Forward 5′-GCgaattcAGA
GTC TCA GGC AGG TCCA-3′ and reverse 5′-GCggatccGAT GTG CTT TTG GGT
CCT AC-3′. This was performed using PrimeSTAR HS DNA Polymerase
(Takara Biotechnology, Co., Ltd.). The forward and reverse primer
sequences were located in the upstream of the promoter region and
in intron 2, respectively. The length of the PCR product was 986
bp. The restriction enzyme sites EcoRI and BamHI were
inserted into the primer sequences to enable directional cloning.
DNA from the patient's father who did not carry the genetic variant
was used as a control. Target fragments were ligated into the
pcDNA3.1/Myc-His B vector (Invitrogen; Thermo Fisher Scientific,
Inc.) using solution I ligase (Takara Biotechnology, Co., Ltd.),
according to the manufacturer's protocol.
Cell culture and cell transfection
HEK 293 cells were grown in Dulbecco's modified
Eagle's medium supplemented with 10% (v/v) fetal bovine serum
(Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) in a 5% CO2
incubator at 37°C. Cell transfection was performed using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol.
Total RNA isolation, cDNA synthesis and
splice site analysis
At 48 h post- plasmid transfection, the total RNA
from the HEK 293 cells was isolated using an RNeasy Mini kit
(Qiagen GmbH), and the RNA samples were treated with 5 U/µl
DNase I (Takara Biotechnology, Co., Ltd.). cDNA chains were
obtained from 1 µg RNA using C1000™ Thermal Cycler PCR
instrument (Bio-Rad Laboratories, Inc.) with a Primer Script™ RT
Reagent kit (cat. no. RR037A; Takara Biotechnology, Co., Ltd.). The
following primers were used to amplify a 330 bp PCR product for the
target cDNA fragment of the CYP11B2 gene: Forward 5′-GCA GAG
GTG TGC GTG GCAG-3′ and reverse 5′-CCA GGG CTC CAG GAT CATC-3′. The
forward and reverse primer sequences were located in exon 1 and
exon 2, respectively. Subsequently, the PCR products were
visualized by electrophoresis on a 2% agarose gel (Sangon Biotech,
Co., Ltd., Shanghai, China) following staining with 0.5
µg/ml ethidium bromide (Sigma-Aldrich, St. Louis, MO, USA).
In addition, the PCR products were sequenced for further
identification. GAPDH was used as a control for RNA quality,
and the following primers were used to amplify a 245 bp PCR product
for GAPDH: Forward 5′-GTC AGT GGT GGA CCT GAC CT-3′ and
reverse 5′-TGC TGT AGC CAA ATT CGT TG-3′.
Results
Identification and confirmation of the
mutated gene
To obtain a rapid and accurate clinical diagnosis,
the patient was screened for causal variants using WES. Exome
sequencing yielded a total of 101,306,626 reads, and the mean
target coverage was 136 reads with 95.48% having 20x coverage, and
99.71% having 1x coverage. The candidate variants were first
screened with criterion of a minor allele frequency under 3% in the
1000 Genomes Project, the NHLBI exome variant server, or in 50
HapMap control exomes, the area of analysis area included each exon
and ~20 bp of exonintron boundaries. Subsequently, clinical
symptoms of frequent vomiting, hyponatremia, and hyperkalemia were
chosen as the filtering indexes to analyze the candidate variants.
Ultimately, the results demonstrated that the patient had compound
heterozygous mutations in the CYP11B2 gene, which were
considered to contribute to the patient's condition. Of the two
mutations, one was a heterozygous point transition (c.1009C>T)
in exon 6. Sequence analysis results showed that the c.1009C>T
mutation at codon 337 of exon 6 was a nonsense mutation, which led
to early termination of the protein translation process (p.Q337X).
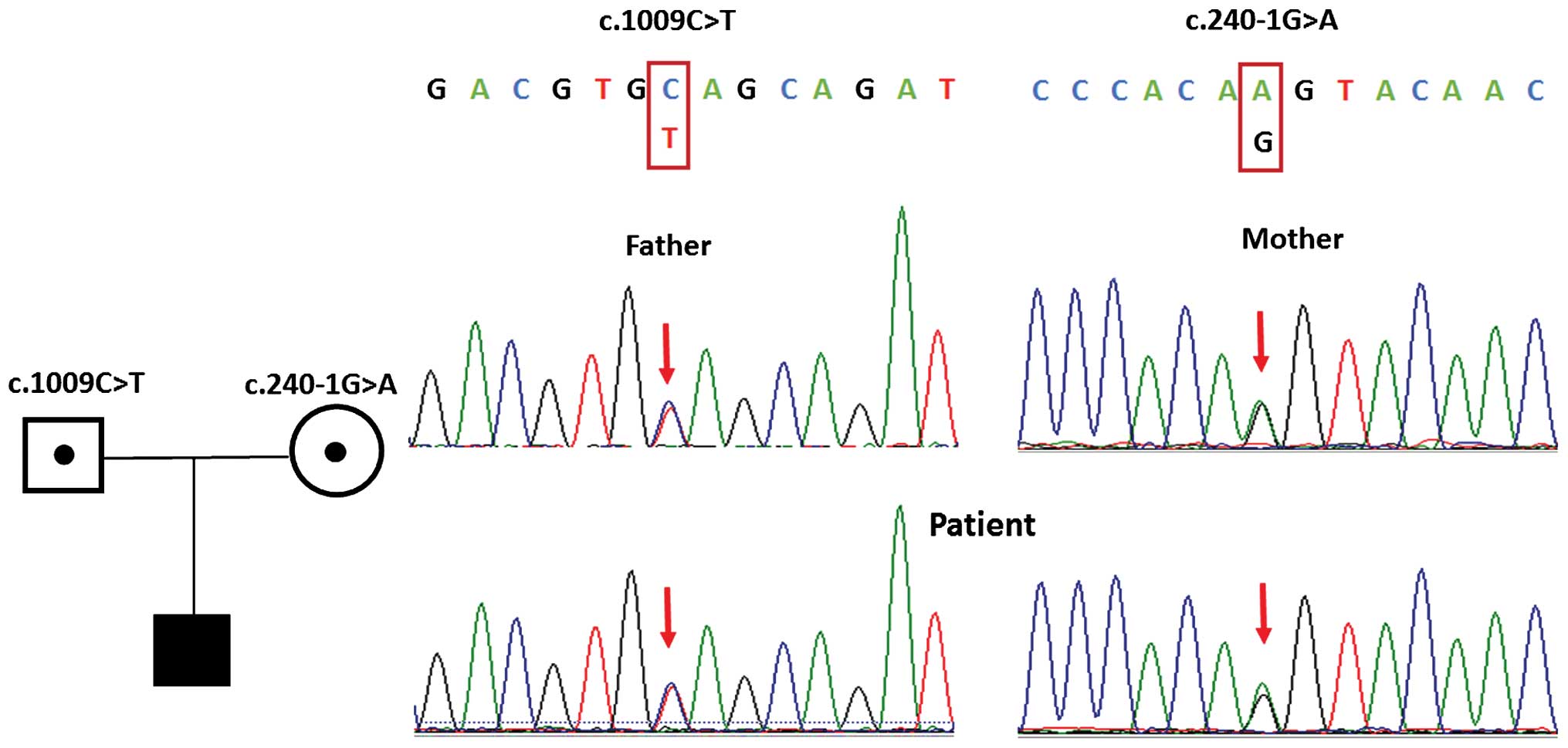
The other was a transversion of guanine to adenine (c.240-1G>A)
in the first base of the 3′-splice site of intron 1 (Fig. 1). Further analysis revealed that
c.240-1G>A was an intronic variation, which was involved in the
agGT-aaGT splicing site, which occurs at a highly conserved 'ag-GT'
site of the intron-exon junction region. The two mutations were
identified as novel mutations, according to the Human Gene Mutation
Database (HGMD; http://www.hgmd.cf.ac.uk). In addition, several genes
associated with the clinical symptoms mentioned above are
wild-type, including CYP11B1, CYP17A1, CYP21A2, 3βHSD, SCNN1A,
SCNN1B, SCNN1 G, NR3C2, DAX1.
To assess the identified variations in the patient's
parents, and further confirm the results of the WES, the proband
and parents were examined using PCR Sanger sequencing. Direct
sequencing results revealed that the patient's father was
heterozygous for the c.1009C>T and p.Q337X mutation, and his
mother was heterozygous for the c.240-1G>A mutation, which
indicated that the gene mutations in the pediatric patient were
inherited from her parents. These results led to the conclusion
that these two novel compound heterozygous mutations in the
CYP11B2 gene were the genetic cause for the patient's
condition.
Splicing assays of the c.240-1G>A
mutation
To evaluate the effect of the c.240-1G>A mutation
on splicing, a minigene construct was generated, which was
transiently transfected into cultured HEK 293 cells (Fig. 2A and B). The HEK 293 cells were
transfected with either a mutant-type CYP11B2, wild-type
CYP11B2 or empty pcDNA3.1 vector (negative control). As
shown in Fig. 2C, a difference in
the size of the cDNA fragment was observed between the wild-type
and mutant-type CYP11B2, as expected. The wild-type
CYP11B2 produced a PCR product of ~330 bp, whereas the
mutant-type construct produced a longer band of 717 bp.
GAPDH was used as a control for RNA quality and quantity.
Clone sequencing verified that the mutant CYP11B2 construct
contained the 387 nucleotides, which constituted intron 1.
Therefore, it was concluded that the c.240-1G>A mutation of the
3′-splice site at the intronexon boundary resulted in the retention
of intron 1 in the RNA.
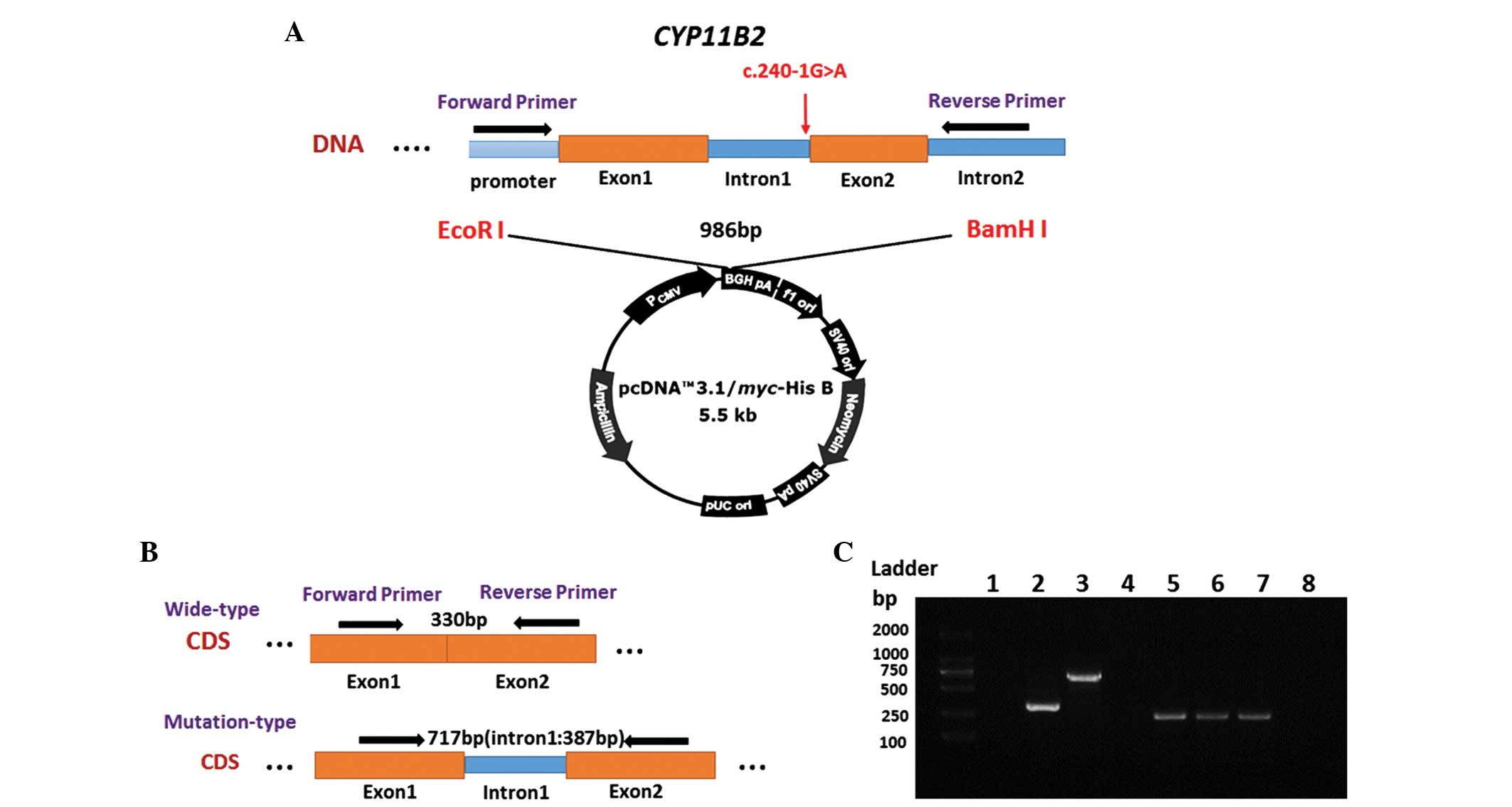 | Figure 2Splicing assays of the c.240-1G>A
mutation. (A) Schematic representation of the mutant-type plasmid
construct. The splice region (c.240-1G>A, intron1) of the
CYP11B2 gene was amplified, and the products were ligated
into the pcDNA3.1/Myc-His B vector. (B) Schematic representation of
expected RT-PCR results in HEK 293 cells transfected with different
plasmids. The c.240-1G>A mutant type produced a longer band. (C)
Electrophoresis was used to analyze the RT-PCR results. Lane 1,
empty pcDNA3.1 vector; lane 2, wild-type CYP11B2 (330 bp);
lane 3, c.240-1G>A mutant (717 bp); lanes 4 and 8,
ddH2O (negative control, lanes 5, 6, and 7, GAPDH
(control; 245 bp). RT-PCR, reverse transcription polymerase chain
reaction. |
Discussion
ASD has been classified into two phenotypes. Type 1
ASD is defined as a total loss of enzyme activity caused by a
CYP11B2 mutation. Patients with Type 1 ASD have low to
normal levels of 18OHB, undetectable to low levels of plasma
aldosterone and an increased B/18OHB ratio, whereas type 2 ASD is
reported to involve only defects in 18-hydroxylase and 18-oxidase
activities, but not in 11-β-hydroxylase activity, and serum levels
of aldosterone are usually within normal limits, particularly in
older children and adults. Affected individuals are often
characterized by an increased 18OHB/aldosterone ratio (2,11–14).
The predominant method of treatment for patients with ASD is
mineralocorticoid and sodium supplementation (15). During the course of treatment,
clinicians gradually adjust the dose according to levels of serum
electrolytes and hormones, particularly potassium (15). When treatment is effective,
patients with ASD are able to maintain a satisfactory clinical and
metabolic state, without salt-wasting crisis, and exhibit normal
growth and development (16).
To date, ~49 mutations have been identified in the
CYP11B2 gene, including missense/nonsense mutations,
splicing mutations, regulatory mutations, small
insertions/deletions, gross deletions and complex rearrangements
(data from HGMD). The proportion of missense/nonsense mutations
accounts for ~70% (33/49). ASD is a rare, autosomal recessive
disease, and cases have been identified in the Iranian Jewish,
European and North American populations. It has also been reported
in Asian populations, including Thai, Japanese, and Indian
individuals (8,14,15).
However, to the best of our knowledge, only one Chinese patient
with ASD has been reported with a heterozygous missense mutation
(c.977C>A) and a heterozygous small deletion mutation
(c.523_525delAAG) of the CYP11B2 gene (17). In the present study, two mutations
(c.1009C>T and c.240-1G>A) in the CYP11B2 gene were
successfully identified and, using familial analysis, it was
confirmed that the patient was a compound heterozygote, with the
two mutations confirmed as novel mutations. Sequence analysis
indicated that the c.1009C>T (p.Q337X) mutation on exon 6
resulted in a truncate protein, which may be due to premature stop
codon formation or nonsense-mediated RNA decay. The c.240-1G>A
mutation occurred at the conserved ag-GT site of the intronexon
boundary region, and minigene construction analysis revealed that
the mutation resulted in aberrant splicing, in which intron 1 was
preserved and the 3′-splice site of intron 1 disappeared.
Following a review of the patient's hormone levels a
second time, the first set of results may have been a result of the
stress response resulting from the child's poor health condition,
which introduced complexity in clinical judgment. The second set of
examination results in the outpatient department showed that the
electrolyte balance was well-controlled (Table II). The identified nonsense
mutation and intron retention mutation were expected to result in
two nonfunctional alleles, however, the serum levels of aldosterone
indicated that aldosterone synthase remained, indicating that the
mutations may have affected only the somatic cells of the adrenal
gland, and the aldosterone synthesis catalyzed by the
CYP11B2 gene in other systems, including the subcapsular
aldosterone-producing cell clusters are involved in the
compensatory effect (18,19).
| Table IIResults of the second set of
laboratory examinations, obtained in the outpatient department. |
Table II
Results of the second set of
laboratory examinations, obtained in the outpatient department.
| Factor | Value | Normal range |
|---|
| Aldosterone
(pg/ml) | 29.00 | 50.00–313.00 |
| Cortisol
(µg/dl) | 6.60 | 5.70–16.60 |
| ACTH (pg/ml) | 10.00 | 8.00–80.00 |
| Na (mmol/l) | 137.40 | 137.00–145.00 |
| K (mmol/l) | 4.70 | 3.50–5.10 |
| CL (mmol/l) | 105.00 | 98.00–107.00 |
It is known that the precondition of effective
treatment is a definitive clinical diagnosis (20). The high efficiency of conventional
Sanger sequencing is based on defined clinical manifestations in
line with expectations, and it may not apply to the diagnosis of
polygenic diseases or complicated rare diseases (21). It is difficult to distinguish ASD
from several other endocrine genopathies, including PHA1 and CAH,
particularly following hormone therapy in the affected individuals.
In addition, it is difficult to survey the actual hormone levels in
a 4-month-old infant, which is critical for clinical diagnosis and
treatment. The present study demonstrated that WES, which is a type
of next generation sequencing, may be an effective and rapid
detection technology for certain complicated rare diseases.
In the present study, two novel mutations were
identified in the CYP11B2 gene in a Chinese pediatric
patient with ASD. The c.1009C>T mutation was a nonsense
mutation, which led to a premature stop codon (p.Q337X). The
pathogenicity of the c.240-1G>A mutation was further
demonstrated by analyzing its effect on splicing in a minigene
construct, which resulted in the preservation of intron 1, and the
disappearance of the 3′-splice site during post-transcriptional
processing of the mRNA. The present study not only enhances our
understanding of the CYP11B2 mutation, but also demonstrated
that WES may serve as a powerful tool for the clinical diagnosis of
complex diseases.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81201353 and 81472051), and
the Project of Shanghai Municipal Science and Technology Commission
(grant no. 12411950402). The authors would like to thank LetPub
(www.letpub.com) for its linguistic assistance during
manuscript preparation.
References
|
1
|
Curnow KM, Tusie-Luna MT, Pascoe L,
Natarajan R, Gu JL, Nadler JL and White PC: The product of the
CYP11B2 gene is required for aldosterone biosynthesis in the human
adrenal cortex. Mol Endocrinol. 5:1513–1522. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
White PC: Aldosterone synthase deficiency
and related disorders. Mol Cell Endocrinol. 217:81–87. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peter M: Congenital adrenal hyperplasia:
11beta-hydroxylase deficiency. Semin Reprod Med. 20:249–254. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kondo E, Nakamura A, Homma K, Hasegawa T,
Yamaguchi T, Narugami M, Hattori T, Aoyagi H, Ishizu K and Tajima
T: Two novel mutations of the CYP11B2 gene in a Japanese patient
with aldosterone deficiency type 1. Endocr J. 60:51–55. 2013.
View Article : Google Scholar
|
|
5
|
Bin-Abbas B, Al-Humaida D, Al-Sagheir A,
Qasem E, Almohanna M and Alzahrani AS: Divergent gender identity in
three siblings with 46XX karyotype and severely virilizing
congenital adrenal hyperplasia caused by a novel CYP11B1 mutation.
Endocr Pract. 20:e191–e197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wasniewska M and De Luca F, Valenzise M,
Lombardo F and De Luca F: Aldosterone synthase deficiency type I
with no documented homozygous mutations in the CYP11B2 gene. Eur J
Endocrinol. 144:59–62. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Yu T, Yin L, Li J, Yu L, Shen Y,
Yu Y, Shen Y and Fu Q: Novel mutations in the SCNN1A gene causing
Pseudohypoaldosteronism type 1. PLoS One. 8:e656762013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Portrat S, Mulatero P, Curnow KM,
Chaussain JL, Morel Y and Pascoe L: Deletion hybrid genes, due to
unequal crossing over between CYP11B1 (11beta-hydroxylase) and
CYP11B2 (aldosterone synthase) cause steroid 11 beta-hydroxylase
deficiency and congenital adrenal hyperplasia. J Clin Endocrinol
Metab. 86:3197–3201. 2001.PubMed/NCBI
|
|
9
|
Rodrigues SM, LeDoux JE and Sapolsky RM:
The influence of stress hormones on fear circuitry. Annu Rev
Neurosci. 32:289–313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang J, Zhang W, Jiang H, Wu BL, Primary
Ovarian and Insufficiency Collaboration: Mutations in HFM1 in
recessive primary ovarian insufficiency. N Engl J Med. 370:972–974.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown NJ: Contribution of aldosterone to
cardiovascular and renal inflammation and fibrosis. Nat Rev
Nephrol. 9:459–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peter M, Dubuis JM and Sippell WG:
Disorders of the aldo-sterone synthase and steroid
11beta-hydroxylase deficiencies. Horm Res. 51:211–222. 1999.
View Article : Google Scholar
|
|
13
|
Nguyen HH, Hannemann F, Hartmann MF,
Malunowicz EM, Wudy SA and Bernhardt R: Five novel mutations in
CYP11B2 gene detected in patients with aldosterone synthase
deficiency type I: Functional characterization and structural
analyses. Mol Genet Metab. 100:357–364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ezquieta B and Luzuriaga C: Neonatal
salt-wasting and 11 beta-hydroxylase deficiency in a child carrying
a homozygous deletion hybrid CYP11B2 (aldosterone synthase)-CYP11B1
(11 beta-hydroxylase). Clin Genet. 66:229–235. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kondo E, Nakamura A, Homma K, Hasegawa T,
Yamaguchi T, Narugami M, Hattori T, Aoyagi H, Ishizu K and Tajima
T: Two novel mutations of the CYP11B2 gene in a Japanese patient
with aldosterone deficiency type 1. Endocr J. 60:51–55. 2013.
View Article : Google Scholar
|
|
16
|
Stapenhorst L: 9alpha-Fluorohydrocortisone
therapy in aldosterone synthase deficiency. Pediatr Nephrol.
20:8392005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hui E, Yeung MC, Cheung PT, Kwan E, Low L,
Tan KC, Lam KS and Chan AO: The clinical significance of
aldosterone synthase deficiency: Report of a novel mutation in the
CYP11B2 gene. BMC Endocr Disord. 14:292014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schiffer L, Anderko S, Hannemann F,
Eiden-Plach A and Bernhardt R: The CYP11B subfamily. J Steroid
Biochem Mol Biol. 151:38–51. 2015. View Article : Google Scholar
|
|
19
|
Nishimoto K, Nakagawa K, Li D, Kosaka T,
Oya M, Mikami S, Shibata H, Itoh H, Mitani F, Yamazaki T, et al:
Adrenocortical zonation in humans under normal and pathological
conditions. J Clin Endocrinol Metab. 95:2296–2305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang J and Shen Y: When a “disease-causing
mutation” is not a pathogenic variant. Clin Chem. 5:711–713. 2014.
View Article : Google Scholar
|
|
21
|
Mamanova L, Coffey AJ, Scott CE, Kozarewa
I, Turner EH, Kumar A, Howard E, Shendure J and Turner DJ:
Target-enrichment strategies for next-generation sequencing. Nat
Methods. 7:111–118. 2010. View Article : Google Scholar : PubMed/NCBI
|