Introduction
Osteoarthritis (OA) is a painful joint disease that
develops due to insufficient and aberrant repair of damaged
synovial joint tissue (1). The
articular cartilage is altered to a certain extent in response to
aging, inappropriate mechanical stress and low-grade inflammation
associated with obesity (2).
Obesity has long been recognized as a risk factor for the
development of OA. Excess body weight may lead to cartilage
degeneration and dysfunction of chondrocytes, due to increased
mechanical forces. However, a strong association between obesity
and OA in non-weight-bearing joints, including those in the fingers
and wrists, has also been demonstrated (3). A previous study indicated that
adipokines released by white adipose tissue (WAT) may be associated
with the high prevalence of OA among obese people (4).
Leptin centrally regulates body weight and eating
behavior, and is the most researched adipokine secreted by WAT
(5). In addition, the role of
leptin as a proinflammatory regulator in OA and the expression of
leptin receptors in chondrocytes have previously been reported
(6). During the development of OA,
chondrocytes undergo gradual dedifferentiation, apoptosis is
triggered by low-grade inflammation (7), and reactive oxygen species (ROS) are
generated (8). Leptin has been
demonstrated to enhance the expression of inducible nitric oxide
synthase (iNOS) (9) and
cartilage-degrading matrix metalloproteinases (MMP), including
MMP-9 and MMP-13, which are involved in OA cartilage damage
(10). This requires the
activation of the Janus kinase 2 (JAK2)-signal transducer and
activator of transcription 3 (STAT3),
phosphatidylinositol-4,5-bisphos-phate 3-kinase (PI3K) and
mitogen-activated protein kinase (MAPK) signaling pathways
(9,11).
The underlying mechanism by which leptin induces
chondrocyte apoptosis remains to be elucidated. In the present
study, the role of the JAK2-STAT3 signaling pathway in
leptin-induced chondrocyte apoptosis was investigated. To further
understand the mechanism of action, characteristic autophagy
markers were also investigated.
Materials and methods
Primary culture of chondrocytes from a
model of OA
The present study was approved by the ethics
committee of Xin Hua Hospital Affiliated to Shanghai Jiaotong
University. A total of 12 male Sprague-Dawley rats (SD) rats (age,
10 weeks) were obtained from Shanghai Experimental Animal Center
(Shanghai, China), and were housed at ambient temperature (23°C)
under a 12 h/12 h light-dark cycle, with free access to chow and
water. SD rats were anesthetized with 4% halothane in oxygen, and
were subsequently shaved and disinfected with 70% ethanol. The
right knee joint was exposed by the medial parapatellar approach.
The patella was dislocated laterally and the knee placed in full
flexion, followed by anterior cruciate ligament transection (ACLT)
using micro-scissors, in order to induce ACLT-mediated knee OA, as
previously described (12). In the
control group, the right knee joint was exposed and incisions were
sutured following subluxation of the patella and washing of the
joint surface with saline. Following full post-operative recovery,
rats were sacrificed by anesthesia (4% halothane in oxygen) and
cartilage samples from the knee joints of the OA model and control
group rats were removed and washed several times with
phosphate-buffered saline (PBS). Type II collagenase
(Sigma-Aldrich, St. Louis, MO, USA) was used to digest the
cartilage at 37°C for 5 h, and the solution was centrifuged for 5
min at 200 × g, 4°C. Primary chondrocytes were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 1%
fetal bovine serum (Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA). Cells were seeded in a monolayer at 50–60% density for
use in further studies.
Identification of chondrocytes
Freshly isolated chondrocytes from the control group
were cultured to 50–60% density and fixed with 4% paraformaldehyde
at room temperature for 30 min. Following several washes with PBS,
the cells were treated with 3% H2O2 and
blocked with non-specific normal anti-goat serum (Beyotime
Institute of Biotechnology, Shanghai, China). A previous study
indicated that expression of collagen II was a marker for
functional chondrocytes (13). The
rabbit anti-rat polyclonal collagen II antibody (1:200; cat. no.
ab34712; Abcam, Cambridge, MA, USA) was applied overnight at 4°C,
followed by incubation with horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (1:1,000; cat. no. ab6721;
Abcam) at 37°C for 20 min. Cells incubated with immunoglobulin
(Ig)G (1:200; cat. no. A7016; Beyotime Institute of Biotechnology)
were used as negative controls for the immunocytochemistry.
3,3′-Diaminobenzidine staining was used to indicate the presence of
collagen II and images were captured using an Eclipse Ti-S
microscope (Nikon Corporation, Tokyo, Japan) at a magnification of
×200. ImageJ version 1.49 software (imagej.nih.gov/ij) was used for quantitative analysis
of positive areas.
Cell viability assay
Chondrocytes (3 × 103 per well) were
plated in 96-well plates. A series of leptin concentrations (2.5,
5, 10, 20 or 50 ng/ml; Sigma-Aldrich) or 20 ng/l leptin in
combination with AG490 (10 nM; Selleck Chemicals, Houston, TX, USA)
were added. At various time points following this (12, 24, 48 or 72
h), 10% Cell Counting kit (CCK)-8 medium (Beyotime Institute of
Biotechnology) diluted in serum-free DMEM was added to each well
for 1 h. The absorbance of each sample was measured at a wavelength
of 450 nm using a Labsystems MK3 microplate reader (Thermo Fisher
Scientific Inc., Waltham, MA, USA) to detect cell viability,
according to the manufacturer's protocols. Cells not treated with
leptin or AG490 served as the control group, and all experiments
were performed in triplicate.
Apoptosis assay
Cells were washed with pre-warmed PBS, trypsinized
and suspended in binding buffer (BD Biosciences, Franklin Lakes,
NJ, USA) and the density was adjusted to 5–10×105/ml.
Apoptosis was measured by staining with Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI). The cell
suspension (195 µl) was added to 5 µl Annexin V-FITC
and 10 µl PI (BD Biosciences). Stained cells were analyzed
by fluorescence-activated cell-sorting (FACS) (FACSCalibur, BD
Biosciences). The apoptotic cells were reported as the percentage
of cells that were Annexin-positive and PI-negative.
Measurement of intracellular ROS
levels
The levels of intracellular ROS were measured using
a ROS assay kit (Beyotime Institute of Biotechnology), performed
according to the manufacturer's protocol. Treated chondrocytes were
washed three times with pre-warmed PBS and harvested for staining.
Dihydroethidium (50 µM) was added to the cells and incubated
at room temperature for 30 min prior to analysis using the
FACSCalibur flow cytometer. The fluorescence intensity was
monitored at an excitation wavelength of 488 nm and an emission
wavelength of 605 nm.
Western blot analysis
Chondrocytes were washed with PBS several times then
added to lysis buffer (Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) with proteinase and phosphatase
inhibitors (Sigma-Aldrich). The concentration of protein was
quantified using the bicinchoninic acid method (Thermo Fisher
Scientific, Inc.). A 35 µg protein sample from each group
was separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, prior to transfer to polyvinylidene fluoride
membranes (Merck Millipore, Darmstadt, Germany). The blots were
blocked with 5% non-fat milk and incubated overnight with primary
antibodies at 4°C, as follows: Rabbit polyclonal anti-rat MMP-13
(1:2,500; cat. no. ab39012; Abcam); rabbit polyclonal anti-rat
Beclin-1 (1:800; cat. no. ab62557; Abcam), rabbit polyclonal
anti-rat B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax;
1:100; cat. no. sc-493; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA); rabbit polyclonal anti-rat Bcl-2 (1:150; cat. no. sc-492;
Santa Cruz Biotechnology, Inc.); rabbit polyclonal
anti-microtubule-associated protein 1A/1B-light chain 3 (LC3)
(1:200; cat. no. ab58610; Abcam); rabbit monoclonal anti-rat
phosphorylated (p)-JAK2 (1:1,000; cat. no. 3776; Cell Signaling
Technology, Inc., Danvers, MA, USA); rabbit monoclonal anti-rat
JAK2 (1:1,000; cat. no. 3230; Cell Signaling Technology, Inc.);
rabbit monoclonal anti-rat p-STAT3 (1:1,000; cat. no. ab76315;
Abcam); mouse monoclonal anti-rat STAT3 (1:1,000; ab119352; Abcam);
and rabbit monoclonal anti-rat glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; 1:1,500; cat. no. 5174; Cell Signaling
Technology, Inc.). GAPDH served to normalize protein loading.
Following three washes with Tris-buffered saline with 0.1% Tween
20, the blots were incubated for 1 h at room temperature with
horseradish peroxidase-conjugated goat anti-mouse (1:1,000; cat.
no. A0216; Beyotime Institute of Biotechnology) or goat anti-rabbit
(1:1,000; cat. no. A0208; Beyotime Institute of Biotechnology)
secondary antibodies. The blots were washed again and the signals
were analyzed using enhanced chemiluminescence (EMD Millipore,
Billerica, MA, USA). Intensities were reported as the relative
pixels normalized to those of GAPDH and measured using ImageJ
software.
Statistical analysis
Statistical analysis was performed with one-way
analysis of variance using GraphPad Prism version 6.0 software
(GraphPad Software, Inc., La Jolla, CA, USA). Data were expressed
as the mean ± standard deviation of triplicate samples. All results
were observed in at least three repeated experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
Leptin induces chondrocyte apoptosis and
decreases cell viability
In order to investigate the function of leptin in
OA, an in vitro chondrocyte model was established from rat
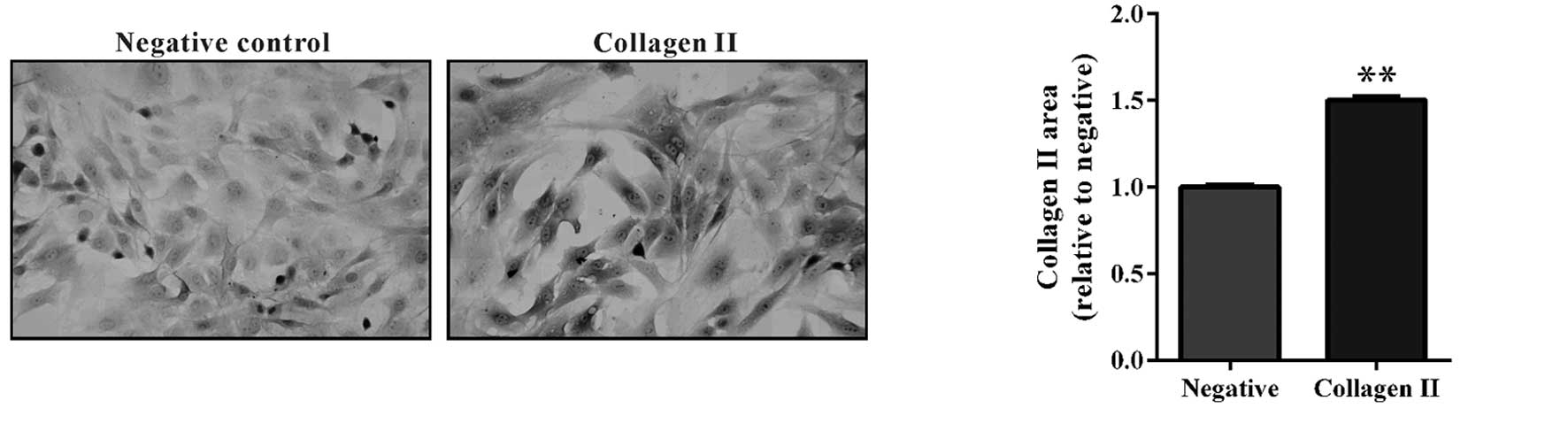
knee joints following ACLT surgery. Collagen II is abundantly
expressed in functional chondrocytes, and in joint diseases
characterized by low-grade inflammation and severe mechanical
stress, including OA. Immunocytochemistry demonstrated that
collagen II was expressed in primary cultured chondrocytes
(Fig. 1), which was indicative of
a chondrocyte model.
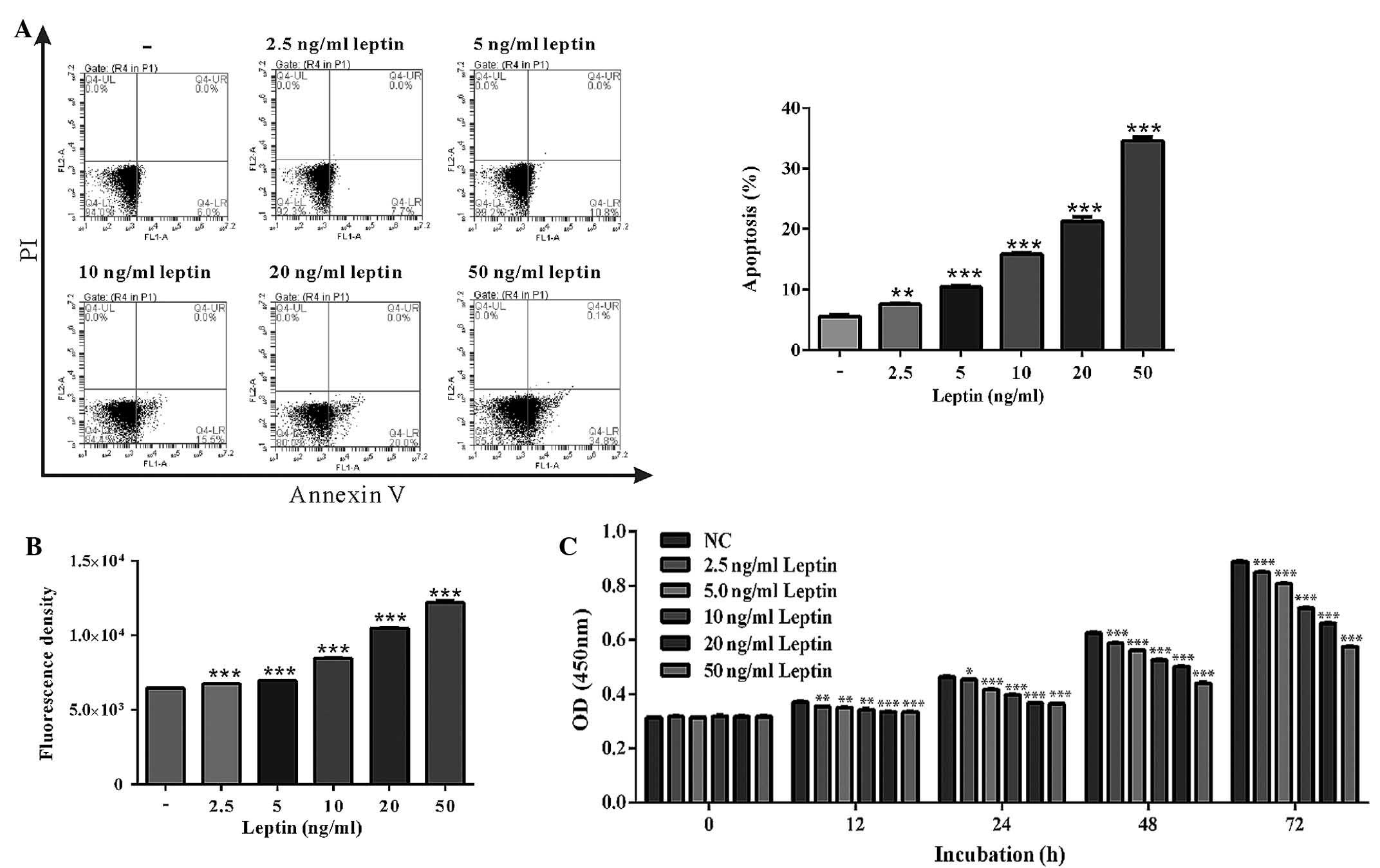
Following treatment with leptin for 48 h,
chondrocytes were stained with Annexin V for further analysis
(Fig. 2). The FACS results
indicated that chondrocytes underwent marked apoptosis following
administration of leptin in a dose-dependent manner (from 7.60 to
34.60%), as compared with the control group (5.63%) (Fig. 2A). Leptin is a proinflammatory
adipokine that is able to generate ROS production in a number of
disease models (14). In order to
address this in the current model, ROS production was measured in
injured chondrocytes. Treatment with various quantities of leptin
for 48 h markedly induced the generation of ROS by mitochondria
(Fig. 2B). Furthermore, the CCK-8
assay indicated that the viability of chondrocytes was reduced in
an increasingly dose-dependent manner over time in response to
leptin treatment (Fig. 2C).
Inhibition of JAK2-STAT3 signaling
ameliorates leptin-induced chondrocyte apoptosis and rescues
reduced cell viability
Leptin binding triggers the leptin receptor to
recruit cytoplasmic kinases, including JAK2, to initiate leptin
signaling (15). Previous studies
have indicated that selective inhibition of JAK signaling has a
beneficial effect in modulating leptin-associated diseases
(16,17). Following the administration of the
JAK2 inhibitor, AG490, injured chondrocytes were rescued from
leptin-induced apoptosis. As presented in Fig. 3A, incubation with AG490 alone for
48 h decreased the basal level of apoptosis in ACLT chondrocytes
(from 5.87 to 2.10%). Co-incubation with leptin and AG490
significantly reduced leptin-induced apoptosis almost to basal
levels (from 14.83 to 7.87%). In addition, AG490 reduced ROS
production induced by leptin (Fig.
3B). The production of ROS in the AG490 + leptin group was
decreased to almost the same level as the ACLT group.
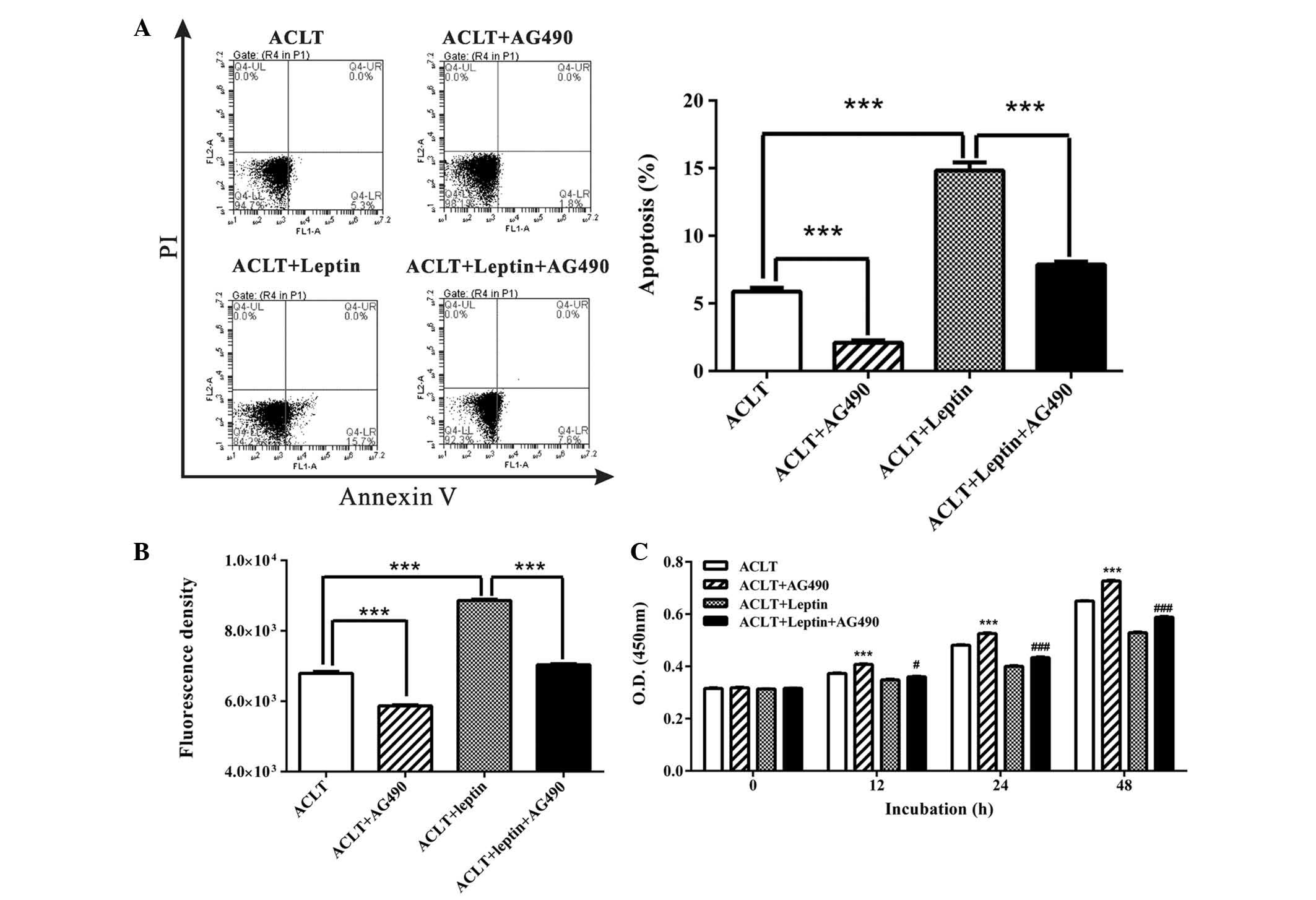 | Figure 3AG490 reduced leptin-induced
chondrocyte apoptosis or ROS production, and restored viability.
(A) Following a 48 h incubation, AG490 suppressed leptin-induced
chondrocyte apoptosis. IV gated cells indicated apoptosis.
***P<0.001. (B) Detection of intracellular ROS was
conducted using the DHE probe. Treated chondrocytes were stained
with DHE followed by analysis using a fluorescence-activated cell
sorting machine. ***P<0.001. (C) Viability of the
chondrocytes was tested using Cell Counting kit-8. A dose- and
time-dependent effect of leptin on the viability of chondrocytes
was observed. Data are presented as the mean ± standard deviation.
***P<0.001, ACLT + AG490 group vs. ACLT group;
#P<0.05, ###P<0.001, ACLT + leptin +
AG490 group vs. ACLT + leptin group. ROS, reactive oxygen species;
DHE, dihydroethidium; ACLT, anterior cruciate ligament transection;
PI, propidium iodide; OD, optical density. |
Furthermore, the CCK-8 assay demonstrated that AG490
restored chondrocyte viability in leptin-treated and -untreated
cells. After a 48 h incubation, AG490 increased chondrocyte
viability by 11.84 and 11.32% in leptin-untreated and -treated
cells, respectively (Fig. 3C).
These results suggest that as a proinflammatory
adipokine, leptin may result in damage to chondrocytes, and JAK2
inhibition may protect chondrocytes via clearance of ROS and
inhibition of apoptosis, thereby restoring cell viability.
Inhibition of the JAK2-STAT3 signaling
pathway alters protein expression associated with apoptosis and
autophagy in chondrocytes following leptin-induced damage
The present study demonstrated that selective
inhibition of the JAK2-STAT3 signaling pathway by AG490 may protect
chondrocytes from leptin-induced apoptosis. Therefore, certain
proteins associated with apoptosis and the JAK2-STAT3 signaling
pathway were detected (Fig. 4).
Upon environmental stimulus, leptin binds to its receptor and
recruits cytoplasmic JAK2 to initiate leptin signaling. Activation
of JAK2 by autophosphorylation initiates recruitment and
phosphorylation of STAT3 for additional effects (18). As shown in Fig. 4A and C, incubation with leptin for
3 h notably induced phosphorylation of JAK2 and STAT3, and
administration of 10 nM of AG490 significantly inhibited activities
of JAK2/STAT3 in the leptin-treated and -untreated groups. Leptin
has been reported to increase the production of MMPs, which belong
to a large family of proteases that degrade the extracellular
matrix and directly damage cartilage (10). In addition to reducing apoptosis,
the expression levels of MMP-13 were reduced following treatment
with AG490 for 24 h, indicating improved chondrocyte function
(Fig. 4B and D).
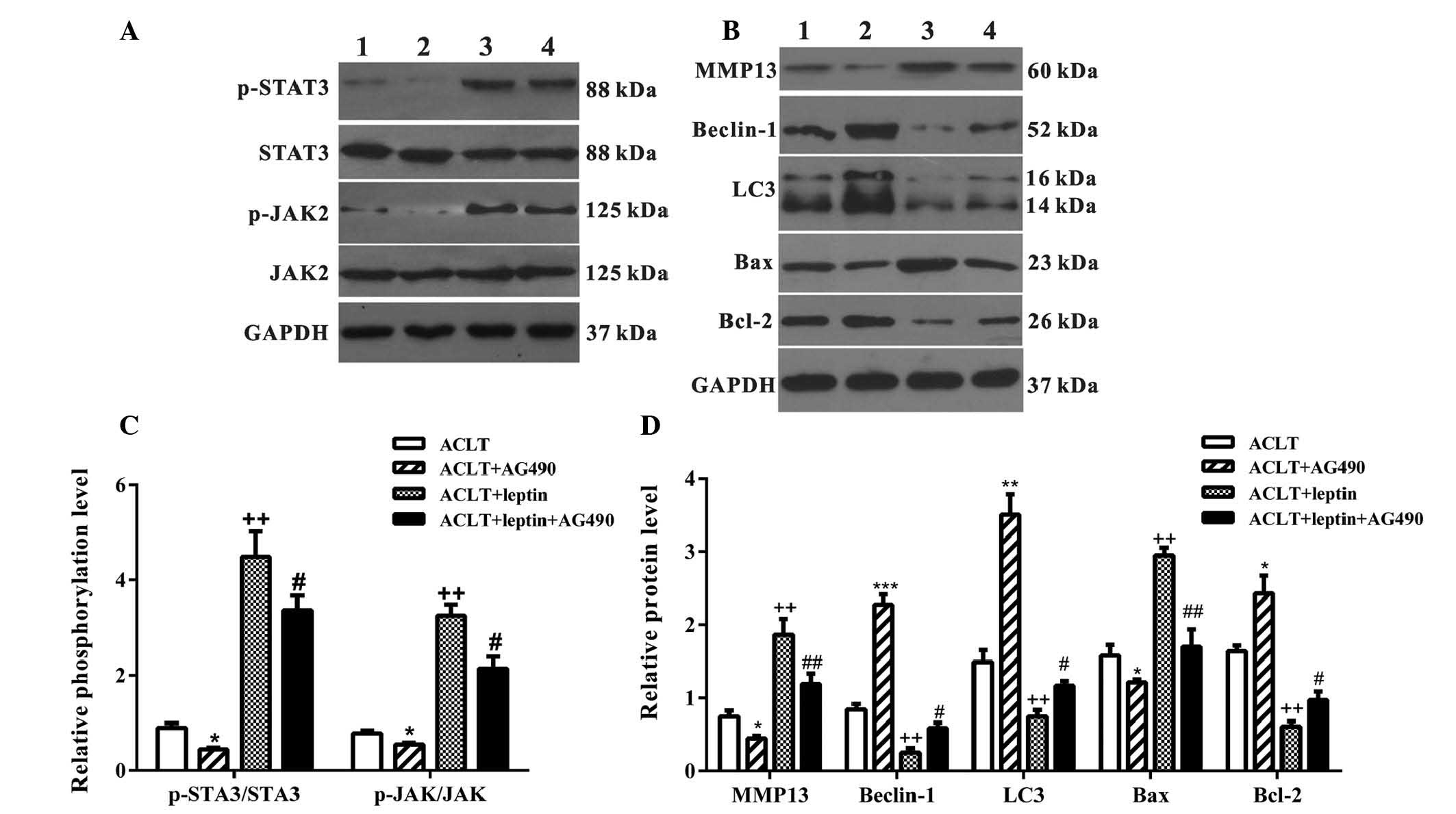 | Figure 4Western blot analysis of JAK-STAT
signaling activity and apoptosis/autophagy-associated protein
expression. (A) A 4 h treatment with AG490 or leptin + AG490
inhibited phosphorylation of STAT3 and JAK2. GAPDH served as an
internal loading control. (B) Following incubation for 24 h, a
number of autophagy key regulators, including Beclin-1 and LC3, and
apoptotic markers were detected. (C and D) Quantification of the
western blotting is presented. Lane 1, ACLT group; lane 2, ACLT +
AG490; lane 3, ACLT + leptin; lane 4, ACLT + leptin + AG490.
Molecular weight for each protein is labeled. Data are presented as
the mean ± standard deviation. *P<0.05, AG490 group
vs. ACLT group; +P<0.05, ++P<0.01, ACLT
+ leptin group vs. ACLT group; #P<0.05,
##P<0.01, ACLT + leptin + AG490 group vs. ACLT +
leptin group. JAK, Janus kinase; STAT, signal transducer and
activator of transcription; LC3, microtubule-associated protein
1A/1B-light chain 3; ACLT, anterior cruciate ligament transection;
MMP-13, matrix metalloproteinase-13; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
The Bax/Bcl-2 family is essential for the regulation
of apoptosis, and an increased ratio of Bax/Bcl-2 results in
caspase activation and downstream apoptosis (19). Leptin increased Bax expression and
suppressed Bcl-2 expression; however, this was reversed by AG490
(Fig. 4B).
Unlike apoptosis, autophagy in normal adult
articular cartilage is an important mechanism for cellular
homeostasis, and may be complicated depending on the stage of
development (20). Under the
majority of circumstances, autophagy promotes cell survival by
adapting cells to stress conditions. In an OA model, autophagy was
reduced alongside a decrease in the key regulators, including
autophagy protein (ATG)5, LC3 and Beclin-1 (21). In the present study, detection of
Beclin-1 and LC3 expression was conducted in ACLT chondrocytes;
leptin reduced the expression levels of Beclin-1 and LC3, which
were restored by AG490 (Fig. 4B and
D).
Discussion
Obesity is a global public health problem, which is
identified as a metabolic low-grade inflammatory condition. Obesity
is characterized by articular cartilage degradation, subchondral
bone sclerosis, osteophyte formation and synovial inflammation
(2). OA is strongly associated
with obesity, beyond the burden of excess body weight on
weight-bearing joints (22), for
example, a previous study reported a two-fold increase in hand OA
risk in obese individuals (3).
Increasing evidence has indicated that metabolic factors released
by WAT may be responsible for the high prevalence of OA among obese
people (4). Among the
adipose-derived molecules, referred to as adipokines, leptin has
been most investigated. Leptin is a proinflammatory adipokine, the
expression levels of which are positively correlated with body mass
index (BMI) (23). Obese
individuals generally exhibit a high level of circulating leptin,
which is associated with higher prevalence of knee OA, following
age, ethnicity and BMI adjustments (5,24).
In addition, leptin is observed in synovial fluid at three times
higher concentrations than in non-obese individuals (25). This leptin is released by local
chondrocytes (26), thus
indicating that local and systemic leptin may have distinct
functions in bone metabolism.
The expression of collagen II is used as a marker
for the healthy chondrocyte phenotype, and is also observed in
primary cultured rat chondrocytes (27). The expression of leptin receptors
in human chondrocytes was originally detected by Figenschau et
al (6), which indicated that
leptin directly participated in joint damage by initiating iNOS
production. In addition, this injury required other proinflammatory
cytokines, including interferon (IFN)-γ or interleukin (IL)-1
(9,11). The in vivo ACLT model
established in the present study provided an ideal inflammatory
environment for elucidating the function of leptin in vitro
without administration of more proinflammatory cytokines.
A previous study has reported that leptin triggers
the expression of iNOS, alone or in combination with IL-1β, to
potentially induce apoptosis in human or rodent chondrocytes
(7). In the present study, leptin
was demonstrated to induce apoptosis and reduce cell viability in a
dose- and time-dependent manner. Furthermore, damaged chondrocytes
themselves, which were used in the current system, may release
inflammatory cytokines that associate with leptin to trigger this
deterioration process. However, the specific effective cytokines
and detailed underlying mechanisms remain to be elucidated and
require further study in the future.
During cellular stress, mitochondrial membrane
potential may be altered by dysfunction of intracellular ionic
charge, leading to the generation of ROS, which is a key indicator
of cell death or injury. It has previously been demonstrated that
oxidative damage as a result of ROS contributes to chondrocyte
apoptosis (28), and treatment of
human chondrocytes with an anti-oxidative agent may result in a
prolonged replicative lifespan (29). However, to the best of our
knowledge, no previous studies have addressed the possible
involvement of leptin in ROS production, and the present study
reports for the first time that leptin contributes to ROS-induced
oxidative stress in chondrocytes.
Leptin exerts a proinflammatory effect via JAK2,
PI3K and MAPKs in chondrocytes, and selective blocking of
individual intracellular signaling pathways may exert beneficial
effects. Upon leptin binding, the leptin receptor recruits
cytoplasmic kinases, such as JAK2, and forms a receptor homodimer
to facilitate autophosphorylation and activation of JAK2 in order
to initiate leptin signaling. Furthermore, activation of JAK2
initiates recruitment of STAT members, including STAT3, for
subsequent signal transduction (15). Using the specific JAK2 inhibitor,
AG490, the JAK2 signaling pathway was revealed to be associated
with the effects of leptin on apoptosis of chondrocytes. Otero
et al (9,11) demonstrated that inhibition of the
JAK2-STAT3 signaling pathway with AG490 may result in the
suppression of leptin-induced iNOS expression with IL-1 or IFN-γ in
human chondrocytes. In the present study, in addition to protection
from apoptosis, AG490 increased cell viability and reduced the
production of ROS, suggesting its additional protective influence
on cell viability. Key factors in associated signaling pathways
also indicated the same outcomes; the phosphorylation levels of
JAK2 and STAT3 were reduced in the AG490 group with or without
leptin stimulation. Consistent with the results from the ROS
detection and apoptosis experiments, the ratio of Bcl-2/Bax was
upregulated, indicating a reduction in apoptosis.
The function of autophagy in chondrocytes is complex
and involves numerous mediators produced by chondrocytes. Autophagy
maintains cellular homeostasis in normal adult articular cartilage
and chondrocytes. Chondrocytes in the superficial zone exhibit a
marked expression of autophagy proteins, Beclin-1, ATG5 and LC3
(30), and during the damaging
process of OA, decreases in Beclin-1 and LC3 are accompanied by
increased apoptosis (30). In the
present study, AG490 reduced apoptosis in chondrocytes and restored
Beclin-1 and LC3 expression, which may be beneficial for improving
normal cellular functions.
In conclusion, as a proinflammatory adipokine,
leptin markedly reduced chondrocyte function, and induced
apoptosis, in an in vitro OA model. The JAK2-STAT3 signaling
pathway was considered the underlying molecular mechanism by which
leptin induces apoptosis, and blocking this signal transduction in
affected joints may be considered a promising therapeutic target
during OA therapy.
Acknowledgments
The present study was supported by the Natural
Science Foundation of China (grant no. 81372001) and the Science
and Technology Commission of Shanghai Municipality Fund (grant no.
13ZR1427000).
References
|
1
|
Felson DT: An update on the pathogenesis
and epidemiology of osteoarthritis. Radiol Clin North Am. 42:1–9.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gregor MF and Hotamisligil GS:
Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yusuf E, Nelissen RG, Ioan-Facsinay A,
Stojanovic-Susulic V, DeGroot J, van Osch G, Middeldorp S, Huizinga
TW and Kloppenburg M: Association between weight or body mass index
and hand osteoarthritis: A systematic review. Ann Rheum Dis.
69:761–765. 2010. View Article : Google Scholar
|
|
4
|
Pottie P, Presle N, Terlain B, Netter P,
Mainard D and Berenbaum F: Obesity and osteoarthritis: More complex
than predicted! Ann Rheum Dis. 65:1403–1405. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dumond H, Presle N, Terlain B, Mainard D,
Loeuille D, Netter P and Pottie P: Evidence for a key role of
leptin in osteoarthritis. Arthritis Rheum. 48:3118–3129. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Figenschau Y, Knutsen G, Shahazeydi S,
Johansen O and Svein-björnsson B: Human articular chondrocytes
express functional leptin receptors. Biochem Biophys Res Commun.
287:190–197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vuolteenaho K, Koskinen A, Kukkonen M,
Nieminen R, Päivärinta U, Moilanen T and Moilanen E: Leptin
enhances synthesis of proinflammatory mediators in human
osteoarthritic cartilage-mediator role of NO in leptin-induced
PGE2, IL-6 and IL-8 production. Mediators Inflamm. 2009:3458382009.
View Article : Google Scholar
|
|
8
|
Li D, Xie G and Wang W: Reactive oxygen
species: The 2-edged sword of osteoarthritis. Am J Med Sci.
344:486–490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Otero M, Lago R, Lago F, Reino JJ and
Gualillo O: Signalling pathway involved in nitric oxide synthase
type II activation in chondrocytes: Synergistic effect of leptin
with interleukin-1. Arthritis Res Ther. 7:R581–R591. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koskinen A, Vuolteenaho K, Nieminen R,
Moilanen T and Moilanen E: Leptin enhances MMP-1, MMP-3 and MMP-13
production in human osteoarthritic cartilage and correlates with
MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp
Rheumatol. 29:57–64. 2011.PubMed/NCBI
|
|
11
|
Otero M, Gomez Reino JJ and Gualillo O:
Synergistic induction of nitric oxide synthase type II: In vitro
effect of leptin and interferon-gamma in human chondrocytes and
ATDC5 chondrogenic cells. Arthritis Rheum. 48:404–409. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pickarski M, Hayami T, Zhuo Y and Duong
LT: Molecular changes in articular cartilage and subchondral bone
in the rat anterior cruciate ligament transection and
meniscectomized models of osteoarthritis. BMC Musculoskelet Disord.
12:1972011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tew SR, Murdoch AD, Rauchenberg RP and
Hardingham TE: Cellular methods in cartilage research: Primary
human chondrocytes in culture and chondrogenesis in human bone
marrow stem cells. Methods. 45:2–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hamada Y, Fujii H and Fukagawa M: Role of
oxidative stress in diabetic bone disorder. Bone. 45(Suppl 1):
S35–S38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ghilardi N and Skoda RC: The leptin
receptor activates janus kinase 2 and signals for proliferation in
a factor-dependent cell line. Mol Endocrinol. 11:393–399. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohba S, Lanigan TM and Roessler BJ: Leptin
receptor JAK2/STAT3 signaling modulates expression of Frizzled
receptors in articular chondrocytes. Osteoarthritis Cartilage.
18:1620–1629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mutabaruka MS, Aoulad Aissa M, Delalandre
A, Lavigne M and Lajeunesse D: Local leptin production in
osteoarthritis subchondral osteoblasts may be responsible for their
abnormal phenotypic expression. Arthritis Res Ther. 12:R202010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bahrenberg G, Behrmann I, Barthel A,
Hekerman P, Heinrich PC, Joost HG and Becker W: Identification of
the critical sequence elements in the cytoplasmic domain of leptin
receptor isoforms required for Janus kinase/signal transducer and
activator of transcription activation by receptor heterodimers. Mol
Endocrinol. 16:859–872. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adams JM and Cory S: Life-or-death
decisions by the Bcl-2 protein family. Trends Biochem Sci.
26:61–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lotz MK and Caramés B: Autophagy and
cartilage homeostasis mechanisms in joint health, aging and
osteoarthritis. Nat Rev Rheumatol. 7:579–587. 2011.PubMed/NCBI
|
|
21
|
Shapiro IM, Layfield R, Lotz M, Settembre
C and Whitehouse C: Boning up on autophagy: The role of autophagy
in skeletal biology. Autophagy. 10:7–19. 2014. View Article : Google Scholar :
|
|
22
|
Aspden RM: Obesity punches above its
weight in osteoarthritis. Nat Rev Rheumatol. 7:65–68. 2011.
View Article : Google Scholar
|
|
23
|
Friedman JM and Halaas JL: Leptin and the
regulation of body weight in mammals. Nature. 395:763–770. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gualillo O, Eiras S, Lago F, Diéguez C and
Casanueva FF: Elevated serum leptin concentrations induced by
experimental acute inflammation. Life Sci. 67:2433–2441. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Presle N, Pottie P, Dumond H, Guillaume C,
Lapicque F, Pallu S, Mainard D, Netter P and Terlain B:
Differential distribution of adipokines between serum and synovial
fluid in patients with osteoarthritis. Contribution of joint
tissues to their articular production. Osteoarthritis Cartilage.
14:690–695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Distel E, Cadoudal T, Durant S, Poignard
A, Chevalier X and Benelli C: The infrapatellar fat pad in knee
osteoarthritis: An important source of interleukin-6 and its
soluble receptor. Arthritis Rheum. 60:3374–3377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldring MB, Tsuchimochi K and Ijiri K:
The control of chondrogenesis. J Cell Biochem. 97:33–44. 2006.
View Article : Google Scholar
|
|
28
|
Hashimoto S, Takahashi K, Amiel D, Coutts
RD and Lotz M: Chondrocyte apoptosis and nitric oxide production
during experimentally induced osteoarthritis. Arthritis Rheum.
41:1266–1274. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henrotin Y and Kurz B: Antioxidant to
treat osteoarthritis: Dream or reality? Curr Drug Targets.
8:347–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Almonte-Becerril M, Navarro-Garcia F,
Gonzalez-Robles A, Vega-Lopez MA, Lavalle C and Kouri JB: Cell
death of chondrocytes is a combination between apoptosis and
autophagy during the pathogenesis of Osteoarthritis within an
experimental model. Apoptosis. 15:631–638. 2010. View Article : Google Scholar : PubMed/NCBI
|