Introduction
Atrial fibrillation (AF), a supraventricular
tachyarrhythmia with chaotic atrial electrical activation and
consequent ineffective atrial contraction, is the most common form
of sustained cardiac arrhythmia, accounting for approximately
one-third of hospitalizations for various types of cardiac rhythm
disturbances (1). The estimated
prevalence of AF is 1–2% in the worldwide population, and the
incidence increases rapidly with advancing age, rising from 6% in
individuals aged >65 years to 10% in individuals aged ≥80 years
(1–3). AF can result in a reduction in
quality of life, poor exercise tolerance, thromboembolic stroke,
congestive heart failure and increased rates of mortality (1). AF confers a 5-fold increase for the
risk of stroke, and a 2-fold increase for the risk of heart failure
and succumbing to ortality (1).
Therefore, AF represents a substantial socioeconomic burden, which
is likely to increase in the future due to the ageing population
(4,5). Despite significant morbidity and
mortality rates, the etiologies responsible for AF in a
considerable proportion of patients remain to be elucidated.
AF is frequently associated with various cardiac
disorders and noncardiac comorbidities (1), including valvular heart disease,
hypertensive heart disease, ischemic heart disease, renal failure,
thyroid dysfunction, diabetes and inflammation (6,7).
However, in 15–30% of patients, AF occurs in the absence of
previously associated pathology or predisposing factors, defined as
idiopathic AF, of which up to 15% exhibits familial clustering and
is termed familial AF (8). In
previous years, an increasing number of population-based studies
have demonstrated that genetic defects are pivotal in the
pathogenesis of AF, and mutations in >25 genes, including those
coding for ion channels, transcription factors and signaling
molecules, have been causally linked to AF (8,9–23).
However, AF is a genetically heterogeneous disorder, and the
genetic determinants underpinning AF in a significant number of
cases remain to be elucidated.
A previous study by Sinner et al (24) identified five novel AF
susceptibility loci by using a combination of genotyping,
expression quantitative trait loci mapping and functional analysis,
including a locus on chromosome 12q24 intronic to TBX5. The
AF-associated single nucleotide polymorphism (SNP) at the
TBX5 locus, rs10507248, which was also significantly
associated with ischemic stroke, was shown to modulate the
expression of TBX5 in human atrial tissues. In a genome-wide
association investigation, Holm et al (25) found that the SNP in TBX5 was
positively associated with the electrocardiograph PR interval, QRS
duration, QT interval, and with common arrhythmias, including AF
and advanced atrioventricular block. In addition, the associations
between the SNP in TBX5 and the electrocardiographic
parameters (PR interval, QRS duration and QT interval) and AF were
replicated independently in a Chinese Han population (26). As TBX5 is widely expressed
in the heart, including the atria, atrioventricular node and
ventricular bundle branches, and mutations in TBX5 have been
reported to underlie Holt-Oram syndrome, features of which include
forelimb malformations and congenital heart defects,
atrioventricular conduction abnormalities and AF, of which AF is
the predominant phenotype (27,28),
the present study hypothesized that genetically defective
TBX5 may predispose to AF in a subset of patients.
Therefore, the present study aimed to identify novel mutations in
TBX5 responsible for AF, which may have potential
implications for genetic counseling of AF patients.
Materials and methods
Study subjects
In the present study, subjects were recruited from
the Chinese Han population at the Shanghai Gongli Hospital and
Shanghai Chest Hospital (Shanghai, China), and included 190
unrelated patients with idiopathic AF (98 males; 92 females; age
range, 38–57 years) and 400 unrelated healthy individuals (206
males; 194 females; age range, 38–59 years), which were used as
controls. Whenever available, the index patient's first- and
second-degree relatives were also enrolled. All participants
underwent detailed clinical evaluation, including family history,
medical history, physical examination, routine biological tests, a
standard 12-lead electrocardiogram and a transthoracic
echocardiogram. Subjects with structural heart disease, ischemic
heart diseases, hypertension, diabetes, or any other known risk
factor for AF were excluded from the investigation. The patients
were clinically classified in accordance with the 2014 AHA/ACC/HRS
Atrial Fibrillation Guideline (1).
The classification was as follows: Idiopathic AF, AF occurring in
individuals without other cardiac or systemic diseases; familial
AF, idiopathic AF occurring in two or more first-degree relatives;
paroxysmal AF, AF that terminates spontaneously or with
intervention within 7 days of onset; persistent AF, AF with a
duration of >7 days; longstanding persistent AF, continuous AF
for a duration of >12 months; permanent AF, when a joint
decision was made by the patient and clinician to cease further
attempts to restore and/or maintain sinus rhythm. The present study
conformed to the principles of the Declaration of Helsinki
(29). The experimental protocol
was reviewed and approved by the ethics committee of Shanghai Chest
Hospital, Shanghai Jiao Tong University (Shanghai, China). Prior to
the investigation, all the participants provided written informed
consent.
Genetic screening for TBX5 mutations
Peripheral venous blood samples and clinical data
were collected from all participating subjects (Table I). Genomic DNA was isolated from
blood leukocytes using a Wizard Genomic DNA Purification kit
(Promega, Madison, WI, USA). The primers used for amplification of
the coding exons and splice junctions of TBX5 by polymerase
chain reaction (PCR) were designed, as described previously
(30), and manufactured by Sangon
Biotech Co., Ltd. (Shanghai, China). The primer sequences were as
follows: Exon 1 (428 bp) forward (F), 5′-GACGCCATAATCCTCTGGGC-3′
and reverse (R), 5′-AAGAGCTGCCTCCACCTACT-3′; exon 2 (598 bp) F,
5′-GTCATGATCTCCGCCGTGTC-3′ and R, 5′-GAACAGCGAAGGAGGCAGCG-3′; exon
3 (493 bp) F, 5′-AGGGCGAGGCCGAGTTTATG-3′ and R,
5′-ACGACCCTTGGAGTTGGGTC-3′; exon 4 (462 bp) F,
5′-GGCACTTTTAGGGTTCGCCC-3′ and R, 5′-TCTCCTCATCGGCACACCAG-3′; exon
5 (480 bp) F, 5′-GAGTCCAGGCCAGTGAGGTC-3′ and R,
5′-CCGCTTTTCCAGAGGCGTTG-3′; exons 6 and 7 (675 bp) F,
5′-TGGTGCGCTTCTCCTAACACT-3′ and R, 5′-CTCCGACGCCCCATGCGAGG-3′; exon
8a (487 bp) F, 5′-CCCTGATCCGACGTCTTTCC-3′ and R,
5′-AACACGACAACTCCATGTGC-3′; exon 8b (437 bp) F,
5′-CTGAGTGGGTGCACACTGGA-3′ and R, 5′-AGGGCTGGAGGATTCGCTTC-3′; and
exon 8c (676 bp) F, 5′-ACTTGGGGTCTCGGGCACGC-3′ and R,
5′-CGAACTTCGGGGCTGTGCAG-3′.
 | Table IBaseline characteristics of the
patients with idiopathic AF and control individuals. |
Table I
Baseline characteristics of the
patients with idiopathic AF and control individuals.
| Variable | Patients
(n=190) | Controls
(n=400) | P-value |
|---|
| Demographics | | | |
| Age (years) | 53±9 | 54±8 | 0.1738 |
| Male, n (%) | 98 (52) | 206 (52) | 0.9857 |
| BMI
(kg/m2) | 24±4 | 24±3 | 1.0000 |
| Positive family
history of AF (%) | 82 (43) | 0 (0) | <0.0001 |
| Type of AF | | | |
| Paroxysmal AF
(%) | 72 (38) | 0 (0) | <0.0001 |
| Persistent AF
(%) | 55 (29) | 0 (0) | <0.0001 |
| Long-standing
persistent AF (%) | 37 (19) | 0 (0) | <0.0001 |
| Permanent AF
(%) | 26 (14) | 0 (0) | <0.0001 |
| Echocardiographic
parameters | | | |
| LAD (mm) | 38±6 | 35±5 | <0.0001 |
| LVEF (%) | 63±5 | 63±6 | 1.0000 |
| Medical
history | | | |
| Stroke or TIA
(%) | 10 (5) | 0 (0) | <0.0001 |
| ICD (%) | 5 (3) | 0 (0) | 0.0033 |
| Treatment of
AF | | | |
| Catheter based
ablation (%) | 86 (45) | 0 (0) | <0.0001 |
| Pharmacological
cardioversion (%) | 51 (27) | 0 (0) | <0.0001 |
| Electrical
cardioversion (%) | 28 (15) | 0 (0) | <0.0001 |
| Follow-up (%) | 25 (13) | 0 (0) | <0.0001 |
Amplification of the genomic DNA fragment by PCR was
performed on a Veriti Thermal Cycler (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) using a 25 μl
reaction mixture consisting of 2 μl genomic DNA (100
ng/μl), 2.5 μl 10X Taq Buffer (Qiagen, Hilden,
Germany), 5 μl 5X Q Solution (Qiagen), 2 μl dNTP
Mixture (2.5 mM each; Takara Biotechnology Co., Ltd., Dalian,
China), 0.5 μl of each primer (20 mM each), 0.25 μl
HotStar TaqDNA polymerase (5 U/μl; Qiagen) and 12.25
μl deionized H2O. The thermal cycling conditions
were as follows: An initial pre-denaturation at 95°C for 15 min,
followed by 35 cycles of denaturation at 95°C for 1 min, annealing
at 62°C for 30 sec and extension at 72°C for 1 min, with a final
extension at 72°C for 5 min. Each amplicon was sequenced using a
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) under an ABI PRISM 3130
XL DNA analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The identified variant was validated by the resequencing of
a second PCR product, and queried in the SNP (http://www.ncbi.nlm.nih.gov/SNP), 1000 Genomes
(www.1000genomes.org), and Exome Variant
Server (EVS; http://evs.gs.washington.edu/EVS) databases to confirm
it as novel.
Alignment of multiple TBX5 protein
sequences across species
The amino acid sequences of multiple TBX5 proteins
from various species, including human (NP_000183.2), chimpanzee
(XP_001154140.2), monkey (XP_001111737.1), dog (XP_005636327.1),
cattle (NP_001179678.1), mouse (NP_035667.1), rat (NP_001009964.1),
fowl (NP_989504.1), zebrafish (NP_570990.1) and frog
(NP_001185697.1), were aligned using the online Multiple Sequence
Comparison by Log-Expectation program (http://www.ebi.ac.uk/Tools/msa/muscle/).
Expression plasmids and site-directed
mutagenesis
The TBX5-pcDNA3.1 expression plasmid was
constructed, as described previously (30). Briefly, the full-length wild-type
cDNA of the human TBX5 gene were amplified by PCR using the
cDNA prepared in our previous study (9), digested with EcoRI (Takara
Biotechnology Co., Ltd.) and NotI (Takara Biotechnology Co., Ltd.),
and subsequently inserted into the pcDNA3.1 vector (Invitrogen;
Thermo Fisher Scientific, Inc.). The mutant TBX5-pcDNA3.1
was generated by PCR-mediated site-directed mutagenesis using a
QuickChange II XL Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA), and verified by sequencing. The NK2 homeobox 5
(NKX2-5)-pEFSA expression plasmid and atrial natriuretic factor
(ANF)-luciferase (ANF-luc) reporter, which harbors the 2,600 bp
5′-flanking region of the ANF gene and expresses Firefly
luciferase, were provided by Dr Ichiro Shiojima (Chiba University
School of Medicine, Chiba, Japan).
Luciferase reporter gene assays
COS-7 cells (provided by the Cardiovascular
Laboratory at the Shanghai Chest Hospital) were plated in 12-well
Costar culture plates (BD Biosciences, Franklin Lakes, NJ, USA) at
a density of 1×105 cells/well, and maintained in
Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum.
Tranfections were performed on the second day of plating using
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The pGL4.75 internal control
vector, which expressed Renilla luciferase (hRluc/cytomegalovirus;
Promega), was used in the transient transfection assays to
normalize transfection efficiency. In each transfection experiment,
the same quantity (0.5 μg) of expression plasmid DNA
(wild-type TBX5-pcDNA3.1, NKX2-5-pEFSA or mutant
TBX5-pcDNA3.1) was used, either alone or in combination with
1.0 μg ANF-luc and 0.04 μg pGL4.75. The cells were
harvested 48 h following transfection, and the activities of
Firefly and Renilla luciferase were measured using the Dual-Glo
luciferase assay system (Promega). The activity of the ANF
promoter was determined and expressed as the fold activation of
Firefly luciferase relative to Renilla luciferase. The experiments
were repeated at least three times in triplicate.
Statistical analysis
Data are expressed as the mean ± standard deviation,
unless otherwise indicated. Student's unpaired t-test or
Fisher's exact test were used to determine significant differences.
Two-tailed P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical characteristics of the recruited
subjects
In the present study, a cohort of 190 unrelated
patients with idiopathic AF was clinically evaluated and compared
with 400 unrelated control individuals. All the patients had an
electrocardiogram-documented AF phenotype, without known secondary
causes of AF. The average age of the patients at initial diagnosis
of idiopathic AF was 46±9 years. The control individuals had normal
electrocardiographic results with no history of AF occurrence. No
significant differences were identified between the patient and
control groups in ethnicity, gender or age. The baseline clinical
characteristics of the subjects are summarized in Table I.
Identification of a novel TBX5
mutation
Through the use of sequencing, a heterozygous
missense mutation in TBX5 was identified in one of the 146
unrelated patients with AF, with a mutational prevalence of ~0.53%.
Specifically, a substitution of thymine (T) for cytosine (C) was
identified in the first nucleotide of codon 132 (c.394C>T),
predicting the change of proline (P) to serine (S) at amino acid
position 132 (p.P132S). This mutation was in an index patient, who
was diagnosed with lone AF at the age of 39 years. The DNA
sequencing chromatograms, showing the heterozygous mutation of
c.394C>T in TBX5 and its control sequence, are shown in
Fig. 1A. A schematic diagram of
TBX5, showing the T-box structural domain and location of the
mutation identified in the present study is presented in Fig. 1B. The missense mutation, which was
absent in the 200 control individuals, was not found in the SNP,
1000 Genome and EVS databases (accessed on May 9, 2015). Genetic
analyses of the proband's family members showed that the mutation
was present in all the affected living family members, but was
absent in the unaffected family members examined. Analysis of the
pedigree revealed that the mutation co-segregated with AF, and was
transmitted in an autosomal dominant pattern in the family with
complete penetrance. Additionally, the proband's sister (II-8) had
mild bilateral forelimb deformities, a secundum atrial septal
defect and atrioventricular conduction block, a phenotype of
atypical Holt-Oram syndrome. The pedigree structure of the family
is shown in Fig. 1C, and the
phenotypic characteristics of the affected living family members
are presented in Table II.
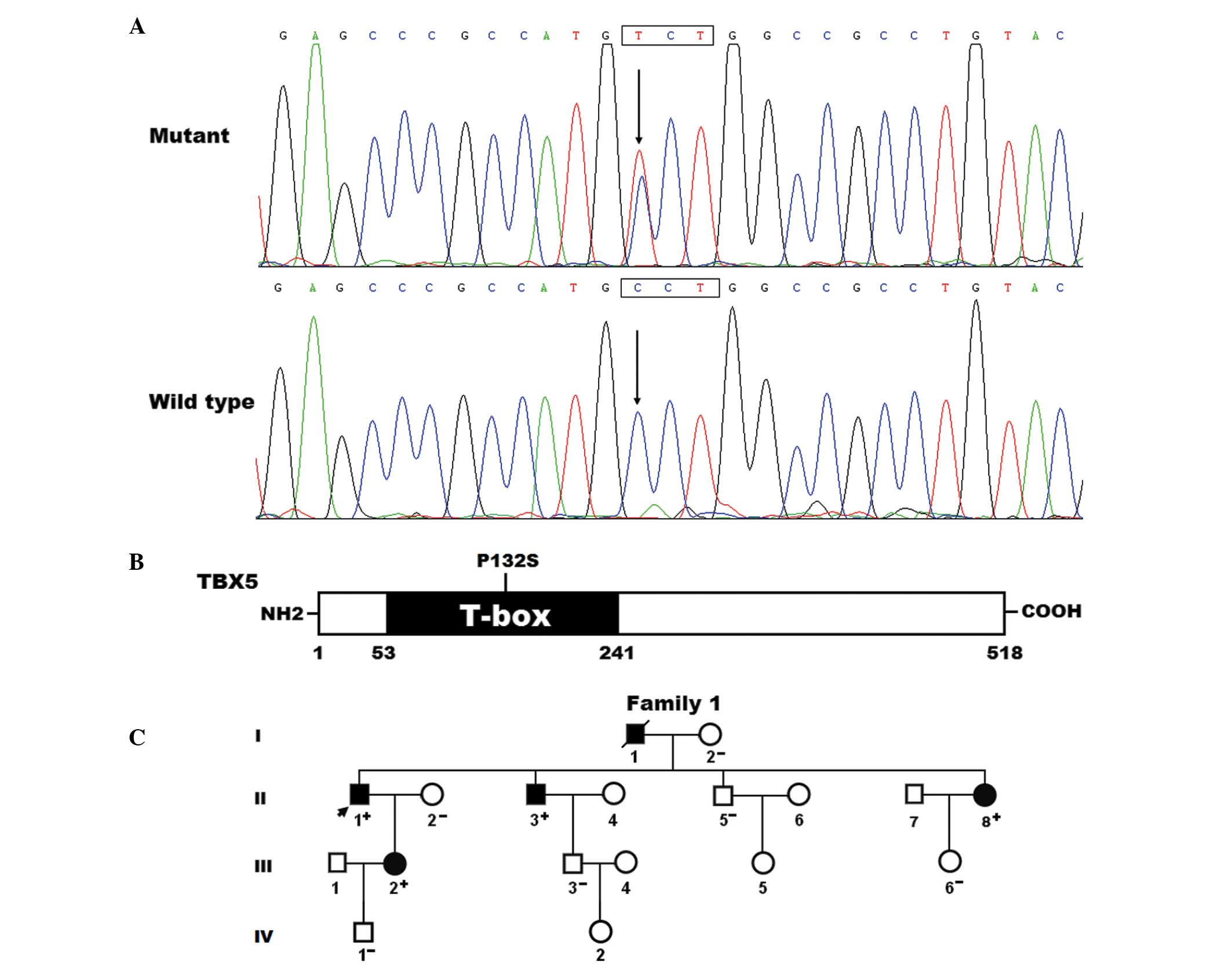 | Figure 1Novel TBX5 mutation is associated
with familial AF. (A) Electropherogram output showing the
heterozygous TBX5 mutation and its wild-type control. The
arrows point to the heterozygous nucleotides of C/T in the proband
(mutant) and the homozygous nucleotides of C/C in the control
individual (wild-type). The rectangle indicate the nucleotides,
which comprise the codon of TBX5. (B) Schematic diagram of
the TBX5 protein structure with the AF-associated mutation shown.
The mutation identified in patients with familial AF is shown above
the T-box structural domain. NH2 indicates an amino-terminus and
COOH indicates a carboxyl-terminus. (C) Pedigree of a family
containing individuals with AF-associated TBX5 mutation. The family
was designated as family 1. Family members are identified by
generations and numbers. Square, male family member; circle, female
member; symbol with a slash, deceased member; closed symbols,
affected members; open symbols, unaffected members; arrows,
proband; +, carrier of TBX5 mutation; −, non-carrier. TBX, T-box;
AF, atrial fibrillation. |
| Table IIPhenotypic characteristics and status
of the TBX5 mutation of the affected living family members in the
pedigree. |
Table II
Phenotypic characteristics and status
of the TBX5 mutation of the affected living family members in the
pedigree.
Subject information
| Phenotype
| Electrocardiogram
| Echocardiogram
| Genotype
|
|---|
| Identity | Gender | Age at time of
study (years) | Age at diagnosis of
AF (years) | AF
(classification) | Heart rate
(bpm) | QRS interval
(ms) | QT/QTc | LAD (mm) | LVEF (%) | TBX5 mutation |
|---|
| Family 1 | | | | | | | | | | P132S |
| II-1 | M | 56 | 39 | Permanent | 69 | 100 | 412/441 | 42 | 58 | +/− |
| II-3 | M | 53 | 34 | Persistent | 63 | 96 | 376/384 | 36 | 64 | +/− |
| II-8 | F | 48 | 42 | Paroxysmal | 72 | 114 | 422/462 | 40 | 60 | +/− |
| III-2 | F | 30 | 30 | Paroxysmal | 75 | 90 | 420/469 | 32 | 62 | +/− |
Multiple alignments of TBX5 protein
sequences among various species
The alignment of multiple amino acid sequences of
TBX5 proteins across species, including human, chimpanzee, monkey,
dog, cattle, mouse, rat, fowl, zebrafish and frog, showed that the
altered proline at amino acid residue 132 of TBX5 was completely
conserved evolutionarily, suggesting that this amino acid is of
functional importance (Fig.
2).
Mutant TBX5 exhibits decreased
transcriptional activity
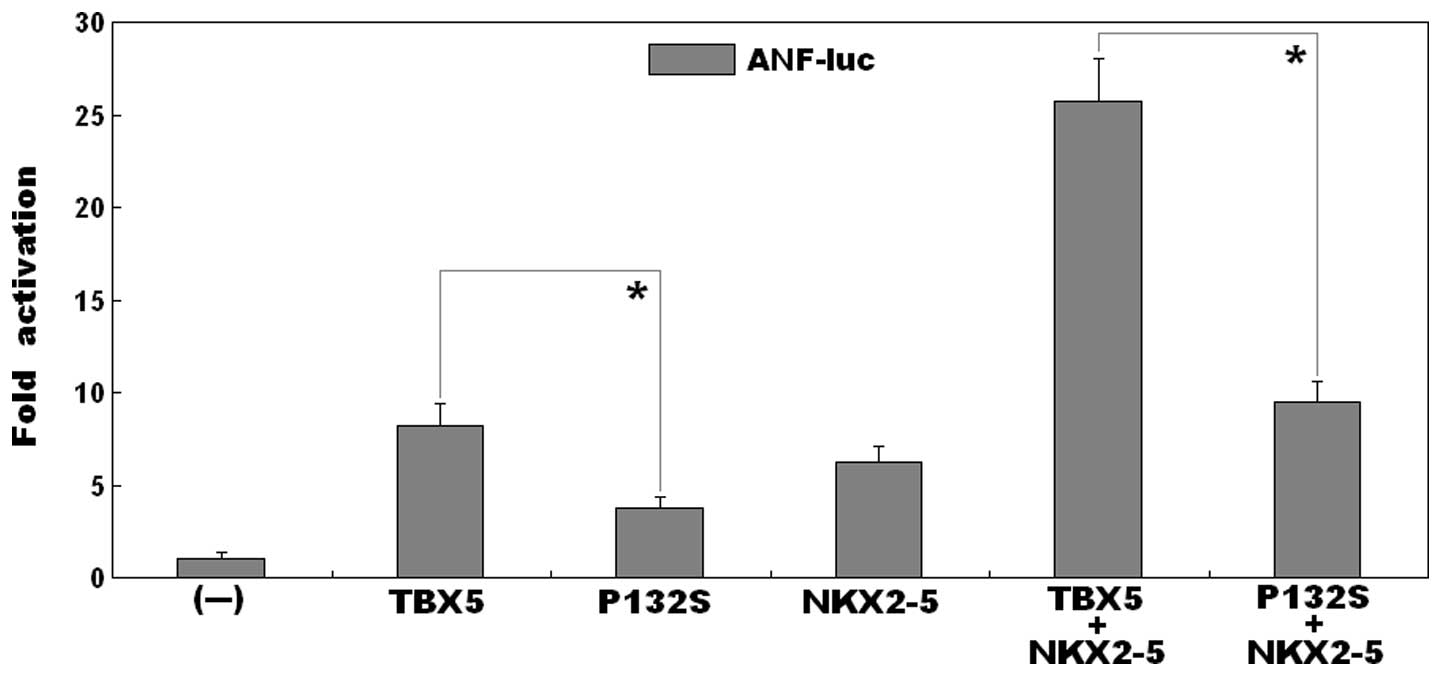
As shown in Fig. 3,
the same quantity (0.5 μg) of the wild-type and P132S-mutant
TBX5 constructs transcriptionally activated the ANF promoter
bŷ8-fold and ~4-fold, respectively (wild-type, vs. mutant:
t=6.0007, P=0.0039). This indicated that the P132S-mutant
TBX5 had significantly decreased transcriptional activity, compared
with its wild-type counterpart.
Synergistic activation is reduced between
mutant TBX5 and NKX2-5
As shown in Fig. 3,
in the presence of 0.5 μg of wild-type NKX2-5, the same
quantity (0.5 μg) of wild-type and P132S-mutant TBX5 induced
the activation of the ANF promoter by ~26-fold and ~9-fold,
respectively (wild type, vs. mutant: t=10.7419, P=0.0004),
suggesting that the mutant TBX5 had reduced synergistic
transcriptional activation with NKX2-5, compared with the
wild-type.
Discussion
In the present study, a novel heterozygous mutation
of p.P132S in TBX5 was identified in a family comprising
individuals diagnosed with AF. The missense mutation, which
co-segregated with AF in the family with complete penetrance, was
absent in the 800 reference chromosomes from a matched control
population. The alignment of multiple TBX5 protein sequences across
species revealed that the altered amino acid was completely
conserved evolutionarily. Functional analysis revealed that the
P132S-mutant TBX5 was associated with significantly decreased
transcriptional activation, which was shown when alone or in
synergy with NKX2-5. Therefore, it is likely that mutated TBX5
predisposes individuals carrying this mutation to AF.
As a member of the TBX transcription factor family,
TBX5 is located on human chromosome 12q24.1, encoding a
protein of 518 amino acids. The TBX5 protein contains a
functionally important structural domain, termed T-box, which is
essential for DNA-binding affinity and specificity, and for
protein-protein interactions (27). In the present study, the TBX5
mutation identified in the patients with AF was located in the
T-box, and biological analyses demonstrated that the mutation
impaired the transactivational function of TBX5 in the absence and
presence of NKX2-5. These findings suggested that
haploinsufficiency or dominant-negative effects resulting from the
TBX5 mutation may be an alternative pathological mechanism
of AF in a minority of patients.
The fact that the TBX5 loss-of-function mutation
confers enhanced susceptibility to AF may be partially attributed
to developmental defects of the heart. In humans and vertebrates,
TBX5 is expressed at high levels in the embryonic heart, with a
crucial role in cardiovascular development, including myocardial
cell proliferation, specification, differentiation, migration,
tissue patterning and morphogenesis (27). In mice, TBX5 is expressed in
the cardiac crescent, linear heart tube, common atrium, ventricles,
inferior and superior vena cavae, and throughout the central
conduction system, including the atrioventricular node and
ventricular bundle branches (31,32).
The homozygous deletion of TBX5 in mice leads to embryonic
death, predominantly as a result of failure of cardiac looping,
hypoplasia of sinuatria and left ventricle; whereas heterozygous
TBX5-null mice suffer from atrial septal defects,
ventricular septal defects, endocardial cushion defects, left heart
hypoplasia, and distinct morphological and functional defects in
the atrioventricular and bundle branch conduction systems, similar
to what has been observed in patients with Holt-Oram syndrome
(32,33). In humans, multiple longitudinal
studies have shown that abnormal cardiac conduction is an
independent risk factor of AF (28,34–37).
Taken together, these observational results indicate that
genetically compromised TBX5 increases the susceptibility of
humans to AF, most likely by causing hypoplasia of the heart, in
particular within the cardiac conduction system.
Previous studies have shown that TBX5 physically
interacts with other cardiac transcriptional factors, including
NKX2-5, GATA4, GATA5 and GATA6, and forms a transcriptional complex
to synergistically activate multiple downstream genes that are
crucial for cardiovascular development, including ANF and
CX40 (27,38–41).
In addition, loss-of-function mutations in several
transcriptionally cooperative partners and target molecules of
TBX5, including NKX2-5, GATA4, GATA5, GATA6, ANF and CX40, have
been implicated in the pathogenesis of AF in humans (8–14,42–44).
Therefore, functionally impaired TBX5 may contribute to AF by
reducing the expression of target genes.
Of note, Postma et al (28) reported that a gain-of-function
mutation in TBX5 resulted in atypical Holt-Oram syndrome and
AF, which was similar to prior reports showing that cytogenetic
abnormalities, which produced TBX5 duplication with presumed
overexpression of TBX5, caused Holt-Oram syndrome-associated
anomalies (45–48). In addition, several studies in
experimental models have demonstrated that the biologic
consequences in cells with reduced and augmented expression levels
of TBX5 are similar (45,49–52).
Taken together, these previous findings and those from the present
study indicate that the dose of TBX5 requires fine regulation to
avoid cardiovascular pathology (53).
In conclusion, to the best of our knowledge, the
present study was the first to report the association of a TBX5
loss-of-function mutation with AF, which provides a novel insight
into the molecular mechanisms underlying AF and has potential
implications for the development of novel therapeutic strategies
for AF. Furthermore the present study demonstrated that AF may be
the only clinical presentation of Holt-Oram syndrome associated
with a TBX5 mutation, thus suggesting that there is a
requirement for electrocardiographic monitoring in patients with
Holt-Oram syndrome.
Acknowledgments
The present study was supported by grants from the
National Natural Science Fund of China (grant nos. 81270161,
81370301, 81470372 and 81400244), the Key Program for Basic
Research of Shanghai, China (grant no. 14JC1405500), the Natural
Science Fund of Shanghai, China (grant nos. 13ZR1438400,
14ZR1438000 and 15ZR1438100), the Experimental animal Project of
Shanghai, China (grant nos. 14140903600 and 12140902800), and the
Key Project of Shanghai Chest Hospital, China (grant nos.
2014YZDH10102 and 2014YZDH20500).
References
|
1
|
January CT, Wann LS, Alpert JS, Calkins H,
Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD,
Field ME, et al: 2014 AHA/ACC/HRS guideline for the management of
patients with atrial fibrillation: A report of the American College
of Cardiology/American Heart Association Task Force on practice
guidelines and the Heart Rhythm Society. Circulation.
130:e199–e267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS,
Bailey KR, Abhayaratna WP, Seward JB and Tsang TS: Secular trends
in incidence of atrial fibrillation in olmsted county, minnesota,
1980 to 2000 and implications on the projections for future
prevalence. Circulation. 114:119–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duncan ME, Pitcher A and Goldacre MJ:
Atrial fibrillation as a cause of death increased steeply in
England between 1995 and 2010. Europace. 16:797–802. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coyne KS, Paramore C, Grandy S, Mercader
M, Reynolds M and Zimetbaum P: Assessing the direct costs of
treating nonvalvular atrial fibrillation in the United States.
Value Health. 9:348–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ball J, Carrington MJ, McMurray JJ and
Stewart S: Atrial fibrilation: Profile and burden of an evolving
epidemic in the 21st century. Int J Cardiol. 167:1807–1824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mazurek T, Kiliszek M, Kobylecka M,
Skubisz-Głuchowska J, Kochman J, Filipiak K, Królicki L and Opolski
G: Relation of proinflammatory activity of epicardial adipose
tissue to the occurrence of atrial fibrillation. Am J Cardiol.
113:1505–1508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu G, Yan YP, Zheng XX, Xu YL, Lu J, Hui
RT and Huang XH: Meta-analysis of nonsteroidal anti-inflammatory
drug use and risk of atrial fibrillation. Am J Cardiol.
114:1523–1529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hong K and Xiong Q: Genetic basis of
atrial fibrillation. Curr Opin Cardiol. 29:220–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.
|
|
10
|
Shi HF, Yang JF, Wang Q, Li RG, Xu YJ, Qu
XK, Fang WY, Liu X and Yang YQ: Prevalence and spectrum of GJA5
mutations associated with lone atrial fibrillation. Mol Med Rep.
7:767–774. 2013.PubMed/NCBI
|
|
11
|
Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG,
Tan HW, Liu X, Fang WY and Bai D: Novel germline GJA5/connexin40
mutations associated with lone atrial fibrillation impair gap
junctional intercellular communication. Hum Mutat. 34:603–609.
2013.PubMed/NCBI
|
|
12
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: A novel NKX2.5 loss-of-function mutation responsible for
familial atrial fibrillation. Int J Mol Med. 31:1119–1126.
2013.PubMed/NCBI
|
|
13
|
Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang
WY, Liu X and Yang YQ: Prevalence and spectrum of Nkx2.5 mutations
associated with idiopathic atrial fibrillation. Clinics (Sao
Paulo). 68:777–784. 2013. View Article : Google Scholar
|
|
14
|
Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang
J, Li RG, Xu L, Jiang WF, Qiu XB, et al: Mutational spectrum of the
NKX2-5 gene in patients with lone atrial fibrillation. Int J Med
Sci. 11:554–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu
X, Fang WY and Yang YQ: Mutations of the SCN4B-encoded sodium
channel β4 subunit in familial atrial fibrillation. Int J Mol Med.
32:144–150. 2013.PubMed/NCBI
|
|
16
|
Beavers DL, Wang W, Ather S, Voigt N,
Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, et al:
Mutation E169K in junctophilin-2 causes atrial fibrillation due to
impaired RyR2 stabilization. J Am Coll Cardiol. 62:2010–2019. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olesen MS, Refsgaard L, Holst AG, Larsen
AP, Grubb S, Haunsø S, Svendsen JH, Olesen SP, Schmitt N and Calloe
K: A novel KCND3 gain-of-function mutation associated with
early-onset of persistent lone atrial fibrillation. Cardiovasc Res.
98:488–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang YQ, Xu YJ, Li RG, Qu XK, Fang WY and
Liu X: Prevalence and spectrum of PITX2c mutations associated with
familial atrial fibrillation. Int J Cardiol. 168:2873–2876. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou YM, Zheng PX, Yang YQ, Ge ZM and Kang
WQ: A novel PITX2c loss-of-function mutation underlies lone atrial
fibrillation. Int J Mol Med. 32:827–834. 2013.PubMed/NCBI
|
|
20
|
Wang J, Zhang DF, Sun YM and Yang YQ: A
novel PITX2c loss-of-function mutation associated with familial
atrial fibrillation. Eur J Med Genet. 57:25–31. 2014. View Article : Google Scholar
|
|
21
|
Qiu XB, Xu YJ, Li RG, Xu L, Liu X, Fang
WY, Yang YQ and Qu XK: PITX2C loss-of-function mutations
responsible for idiopathic atrial fibrillation. Clinics (Sao
Paulo). 69:15–22. 2014. View Article : Google Scholar
|
|
22
|
Wang J, Zhang DF, Sun YM, Li RG, Qiu XB,
Qu XK, Liu X, Fang WY and Yang YQ: NKX2-6 mutation predisposes to
familial atrial fibrillation. Int J Mol Med. 34:1581–1590.
2014.PubMed/NCBI
|
|
23
|
Macri V, Mahida SN, Zhang ML, Sinner MF,
Dolmatova EV, Tucker NR, McLellan M, Shea MA, Milan DJ, Lunetta KL,
et al: A novel trafficking-defective HCN4 mutation is associated
with early-onset atrial fibrillation. Heart Rhythm. 11:1055–1062.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sinner MF, Tucker NR, Lunetta KL, Ozaki K,
Smith JG, Trompet S, Bis JC, Lin H, Chung MK, Nielsen JB, et al:
Integrating genetic, transcriptional, and functional analyses to
identify 5 novel genes for atrial fibrillation. Circulation.
130:1225–1235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holm H, Gudbjartsson DF, Arnar DO,
Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA,
Jonasdottir A, Mathiesen EB, Njølstad I, et al: Several common
variants modulate heart rate, PR interval and QRS duration. Nat
Genet. 42:117–122. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zang X, Zhang S, Xia Y, Li S, Fu F, Li X,
Wang F, Zhang R, Tian X, Gao L, et al: SNP rs3825214 in TBX5 is
associated with lone atrial fibrillation in Chinese Han population.
PLoS One. 8:e649662013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Greulich F, Rudat C and Kispert A:
Mechanisms of T-box gene function in the developing heart.
Cardiovasc Res. 91:212–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Postma AV, van de Meerakker JB, Mathijssen
IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne
Deprez RH and Moorman AF: A gain-of-function TBX5 mutation is
associated with atypical Holt-Oram syndrome and paroxysmal atrial
fibrillation. Circ Res. 102:1433–1442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
World Medical Association: World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. J Postgrad Med. 48:206–208.
2002.PubMed/NCBI
|
|
30
|
Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM,
Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al: TBX5 loss-of-function
mutation contributes to familial dilated cardiomyopathy. Biochem
Biophys Res Commun. 459:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bruneau BG, Logan M, Davis N, Levi T,
Tabin CJ, Seidman JG and Seidman CE: Chamber-specific cardiac
expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev
Biol. 211:100–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moskowitz IP, Pizard A, Patel VV, Bruneau
BG, Kim JB, Kupershmidt S, Roden D, Berul CI, Seidman CE and
Seidman JG: The T-Box transcription factor Tbx5 is required for the
patterning and maturation of the murine cardiac conduction system.
Development. 131:4107–4116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bruneau BG, Nemer G, Schmitt JP, Charron
F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE
and Seidman JG: A murine model of Holt-Oram syndrome defines roles
of the T-box transcription factor Tbx5 in cardiogenesis and
disease. Cell. 106:709–721. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng S, Keyes MJ, Larson MG, McCabe EL,
Newton-Cheh C, Levy D, Benjamin EJ, Vasan RS and Wang TJ: Long-term
outcomes in individuals with prolonged PR interval or first-degree
atrioventricular block. JAMA. 301:2571–2577. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Soliman EZ, Prineas RJ, Case LD, Zhang ZM
and Goff DC Jr: Ethnic distribution of ECG predictors of atrial
fibrillation and its impact on understanding the ethnic
distribution of ischemic stroke in the Atherosclerosis Risk in
Communities (ARIC) study. Stroke. 40:1204–1211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schnabel RB, Sullivan LM, Levy D, Pencina
MJ, Massaro JM, D'Agostino RB Sr, Newton-Cheh C, Yamamoto JF,
Magnani JW, Tadros TM, et al: Development of a risk score for
atrial fibrillation (Framingham Heart Study): A community-based
cohort study. Lancet. 373:739–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Macfarlane PW, Murray H, Sattar N, Stott
DJ, Ford I, Buckley B, Jukema JW, Westendorp RG and Shepherd J: The
incidence and risk factors for new onset atrial fibrillation in the
PROSPER study. Europace. 13:634–639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hiroi Y, Kudoh S, Monzen K, Ikeda Y,
Yazaki Y, Nagai R and Komuro I: Tbx5 associates with Nkx2-5 and
synergistically promotes cardiomyocyte differentiation. Nat Genet.
28:276–280. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, et al: GATA4 mutations cause human
congenital heart defects and reveal an interaction with TBX5.
Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Linhares VL, Almeida NA, Menezes DC,
Elliott DA, Lai D, Beyer EC, Campos de Carvalho AC and Costa MW:
Transcriptional regulation of the murine Connexin40 promoter by
cardiac factors Nkx2-5, GATA4 and Tbx5. Cardiovasc Res. 64:402–411.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maitra M, Schluterman MK, Nichols HA,
Richardson JA, Lo CW, Srivastava D and Garg V: Interaction of Gata4
and Gata6 with Tbx5 is critical for normal cardiac development. Dev
Biol. 326:368–377. 2009. View Article : Google Scholar :
|
|
42
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
43
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
44
|
Hodgson-Zingman DM, Karst ML, Zingman LV,
Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett
JC Jr and Olson TM: Atrial natriuretic peptide frameshift mutation
in familial atrial fibrillation. N Engl J Med. 359:158–165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McDermott DA, Hatcher CJ and Basson CT:
Atrial fibrillation and other clinical manifestations of altered
TBX5 dosage in typical Holt-Oram syndrome. Circ Res. 103:e962008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Patel C, Silcock L, McMullan D, Brueton L
and Cox H: TBX5 intragenic duplication: A family with an atypical
Holt-Oram syndrome phenotype. Eur J Hum Genet. 20:863–869. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kimura M, Kikuchi A, Ichinoi N and Kure S:
Novel TBX5 duplication in a Japanese family with Holt-Oram
syndrome. Pediatr Cardiol. 36:244–247. 2015. View Article : Google Scholar
|
|
48
|
Al-Qattan MM and Abou Al-Shaar H:
Molecular basis of the clinical features of Holt-Oram syndrome
resulting from missense and extended protein mutations of the TBX5
gene as well as TBX5 intragenic duplications. Gene. 560:129–136.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hatcher CJ, Kim MS, Mah CS, Goldstein MM,
Wong B, Mikawa T and Basson CT: TBX5 transcription factor regulates
cell proliferation during cardiogenesis. Dev Biol. 230:177–188.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hatcher CJ, Diman NY, Kim MS, Pennisi D,
Song Y, Goldstein MM, Mikawa T and Basson CT: A role for Tbx5 in
proepicardial cell migration during cardiogenesis. Physiol
Genomics. 18:129–140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liberatore CM, Searcy-Schrick RD and
Yutzey KE: Ventricular expression of tbx5 inhibits normal heart
chamber development. Dev Biol. 223:169–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Al-Qattan MM and Abou Al-Shaar H:
Molecular basis of the clinical features of Holt-Oram syndrome
resulting from missense and extended protein mutations of the TBX5
gene as well as TBX5 intragenic duplications. Gene. 560:129–136.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mori AD, Zhu Y, Vahora I, Nieman B,
Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE,
Chen XJ, et al: Tbx5-dependent rheostatic control of cardiac gene
expression and morphogenesis. Dev Biol. 297:566–586. 2006.
View Article : Google Scholar : PubMed/NCBI
|