Introduction
As an inflammatory disease of the arteries with
characteristic plaque formation, atherosclerosis remains a
complicated disease associated with varied risk factors (1). Amongst them, oxidized low-density
lipoprotein (Ox-LDL) is key in the pathogenesis of atherosclerosis
via the transformation of macrophages and smooth muscle cells into
foam cells (2). The intracellular
accumulation of Ox-LDL within foam cells activates the NACHT, LRR
and PYD domains-containing protein 3 (NLRP3) inflammasome via a
possible route of reduced reactive oxygen species (ROS) generation
(3) and leads to the release of
other damage-associated molecular pattern molecule (DAMP) signals
into the extracellular environment. The DAMP signals then further
accelerate the deterioration of atherosclerotic plaque.
Numerous studies have described a directly
implicating role of the NLRP3 inflammasome in atherosclerosis
(3–5). Once activated, the NLRP3 inflammasome
cleaves pro-caspase-1 into caspase-1, facilitating further
activation of cytokines, interleukin (IL)-1β and IL-18. Pyroptosis,
a recently identified form of cell death, has been reported to be
directly involved in the non-apoptotic cell death of
atherosclerosis (2). Unlike
apoptosis, pyroptosis is solely dependent on activation of
caspase-1, which will in turn cleave inflammatory cytokines, IL-1β
and IL-18, into their active forms (6). Recently, Lin et al (2) reported that pyroptosis was the
primary form of cell death in Ox-LDL-treated macrophage and
atherosclerotic lesions. Yu et al (7) reported that the NLRP3 and absent in
melanoma 2 (AIM2) inflammasomes participated in mitochondrial
damage in a caspase-1-dependent manner. While activation of the two
types of inflammasome may lead to robust ROS generation, the NLRP3
inflammasome exhibits a more potent ability to cause loss of
mitochondrial membrane potential (7).
Mitochondrial (mt)DNA-depleted cells, defined as
rho0 cells, have become a valuable model for investigating the
influence of mitochondria on a variety of cell functions (8–10).
Due to their involvement in apoptosis and necrosis, rho0 cells are
used to investigate the cell death mechanism (11). Recently, studies have reported that
rho0 cells possess a significant resistance to apoptosis that is
induced by different agents (12,13).
However, to the best of our knowledge, the effects of mtDNA loss on
pyroptosis have not yet been elucidated.
The aim of the present study was to investigate the
susceptibility of rho0 cells to Ox-LDL-induced pyroptosis in murine
macrophages. Furthermore, the underlying mechanism of rho0 cells
mitigating Ox-LDL-induced macrophage pyroptosis was
investigated.
Materials and methods
Cell culture and mtDNA-depleted
cells
The J774A.1 murine macrophage cell line was
purchased from the China Center for Type Culture Collection
(Nanjing, China), and maintained in an atmosphere of 5%
CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal calf serum (FCS). The rho0 cell line (J774A.1 rho0) was
generated by incubation of J774A.1 in DMEM containing 10% FCS, 1 mM
pyruvate, 50 µg/ml uridine and 50 ng/ml ethidium bromide
(EB), all from Sigma-Aldrich (St. Louis, MO, USA), for 90 days at
37°C, as described previously (14). Rho0 cell auxotrophy for uridine and
pyruvate was assessed by performance of a Cell Counting Kit-8 assay
(Guangzhou Yiyuan Biotech Co. Ltd, Guangzhou, China) to confirm the
impairment of growth in the absence of these nutrients.
Polymerase chain reaction (PCR)
mtDNA from J774A.1 cells (China Center for Type
Culture Collection, Wuhan, China) was semi-quantified by PCR to
evaluate the remaining mtDNA in the rho0 cells. Total DNA was
isolated using DNAzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and semi-quantified by PCR was performed on a
thermal cycler (2720; Thermo Fisher Scientific, Inc.) using PCR
Master Mix (2X) (Thermo Fisher Scientific, Inc.) in a final volume
of 50 µl. The conditions for PCR were as follows: 94°C for
10 sec, 25 cycles at 94°C for 45 sec, 55°C for 60 sec and 72°C for
45 sec. The primer sequences used for PCR were as follows: mtCOI, F
5′-GCCCCAGATATAGCATTCCC-3′, R 5′-GTTCATCCTGTTCCTGCTCC-3′. 18S rRNA,
F 5′-TAGAGGGACAAGTGGCGTTC-3′, R 5′-CGCTGAGCCAGTCAGTGT-3′. All
primers were synthesized by Shanghai Sangon Biological Engineering
Technology & Services Co., Ltd. (Shanghai, China). 18s rRNA was
used as a refernce gene. The clones containing less mtDNA were then
selected and cultured for 12 weeks in the aforementioned medium,
but without ethidium bromide.
Western blot analysis
J774A.1 normal and rho0 cells were plated in 6-well
plates and grown to 50~60% confluence. Following stimulation with
100 µl Ox-LDL, the cells were harvested and lysed using 1 mM
phenylmethylsulfonyl fluoride (Sigma-Aldrich). The cell lysates
were centrifuged at 14,000 × g for 30 min at 4°C. Then the protein
concentrations in the supernatants were determined using a
bicinchoninic acid kit (Beyotime Institute of Biotechnology,
Haimen, China). Proteins (20 mg) from the homogenized samples were
separated by 15% SDS-PAGE for 1 h at 200 V and transferred onto
polyvinylidene fluoride membranes (EMD Millipore, Boston, MA, USA).
After blocking with Tris-buffered saline and Tween-20 (TBST)
containing 5% non-fat milk powder for 2 h at room temperature,
membranes were treated with the following primary antibodies:
Rabbit polyclonal anti-pro-caspase 1 (1:500; cat. no. BS1730;
Bioworld Technology, Inc., St Louis Park, MN, USA); rabbit
polyclonal anti-cleaved caspase 1 (1:500; cat. no. BS1730; Bioworld
Technology, Inc.); rabbit polyclonal anti-IL-1β (1:500; cat. no.
BS6067; Bioworld Technology, Inc.) and rabbit polyclonal
anti-β-actin (1:1,000; cat. no. BS-0061R; BIOSS, Beijing, China) at
4°C overnight. Next, they were washed with TBST three times and
incubated for 1 h with horseradish peroxidase-conjugated goat
anti-rabbit antibody (1:10,000; cat. no. A0208; Beyotime Institute
of Biotechnology) at room temperature. Immunoreactivities were
detected using an enhanced chemiluminescence detection system (Fast
Western Blot kit, ECL substrate; Thermo Fisher Scientific, Inc.)
and images were obtained using an automatic digital gel image
analysis system (4500SF; Tanon Science & Technology Co., Ltd.,
Shanghai, China).
Detection and quantification of lipid
aggregation in the macrophages
J774A.1 normal and J774A.1 rho0 cells were seeded in
6-well plates and grown to 50~60% confluence. 0, 25, 50 and 100
µg/ml Ox-LDL was added to the supernatants for 24 h. The
cells were washed with phosphate-buffered saline (PBS) and fixed
with formaldehyde. The fixed cells were stained with Oil Red O dye
(Nanjing Jiancheng Biotechnology Institute Co., Ltd., Nanjing,
China) for 30 min at 37°C. The status of intracellular lipid
accumulation was detected by microscopy (Nikon Eclipse 80i; Nikon
Corporation, Tokyo, Japan) to assess lipid aggregation.
Following stimulation with Ox-LDL, cells were
harvested and sonicated on ice for 5 sec, with 15 sec intervals
using an ultrasonic homogenizer (JY88-IIN; Ningbo Scientz
Biotechnology Co., Ltd., Ningbo, China). Following sonication for
10 min, the cells were centrifuged at 8,000 × g for 10 min at 4°C.
The supernatants were then collected and used for total cholesterol
quantification using the CHOD-PAP method and a Total Cholesterol
Assay kit (Nanjing Jiancheng Biotechnology Institute Co., Ltd.),
according to the manufacturer's instructions.
Cell death assay
Cell death was assessed by lactate dehydrogenase
(LDH) release (Beyotime Institute of Biotechnology), and EB and
acridine orange (AO) staining assays (Sigma-Aldrich).
LDH is a cytosolic enzyme that is released from
damaged cells, the presence of which can be detected by the
conversion of lactate to pyruvate with a parallel reduction of
nicotinamide adenine dinucleotide (NAD) to NADH. For LDH release,
50 µl supernatants of J774A.1 normal and rho0 cells
following treatment with Ox-LDL were collected and measured using
the LDH Cytotoxicity Assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
EB in combination with AO (EB/AO) is widely used to
evaluate cell death (15). While
AO is acidophilic green dye that preferentially stains the
lysosomes and nuclei of live cells, EB emits a red fluorescence
upon binding to DNA. For EB/AO staining (Sigma-Aldrich), suspended
and adherent cells in 96-well plates were collected by
centrifugation at 400 × g for 5 min and dyed with 8 µl EB/AO
mix per well (50 µg/ml v/v). Experiments were performed in
triplicate and images were acquired using an inverted fluorescence
microscopy (Nikon Eclipse 80i; magnification, ×400).
Detection of intracellular ROS production
by flow cytometry
The quantity of ROS generated in 100 µg/ml
Ox-LDL-treated J774A.1 normal and rho0 cells for 3 h was measured
by fluorescence-activated cell sorting (BD FACSVerse; BD
Biosciences, Franklin Lakes, NJ, USA) using fluorescent
2′,7′-dichlorodihy-drofluorescein diacetate (DCFH-DA;
Sigma-Aldrich).
Assessement of mitochondrial membrane
potential by confocal microscopy
Following treatment with 100 µg/ml Ox-LDL for
24 h, J774A.1 normal and rho0 cells were triple-stained with
MitoTracker Deep Red, MitoTracker Green and DAPI probes. The change
in mitochondrial membrane potential was detected by observing the
red signal under a confocal laser scanning microscope (Leica TCS
SP5; Leica, Mannheim, Germany).
Statistical analysis
Data are presented as means ± standard deviation and
were analyzed by SPSS 13.0 software (SPSS Inc., Chicago, IL, USA).
Statistical analysis was performed using Student's t test
for comparison between two groups and P<0.05 was considered to
indicate a statistically significant difference.
Results
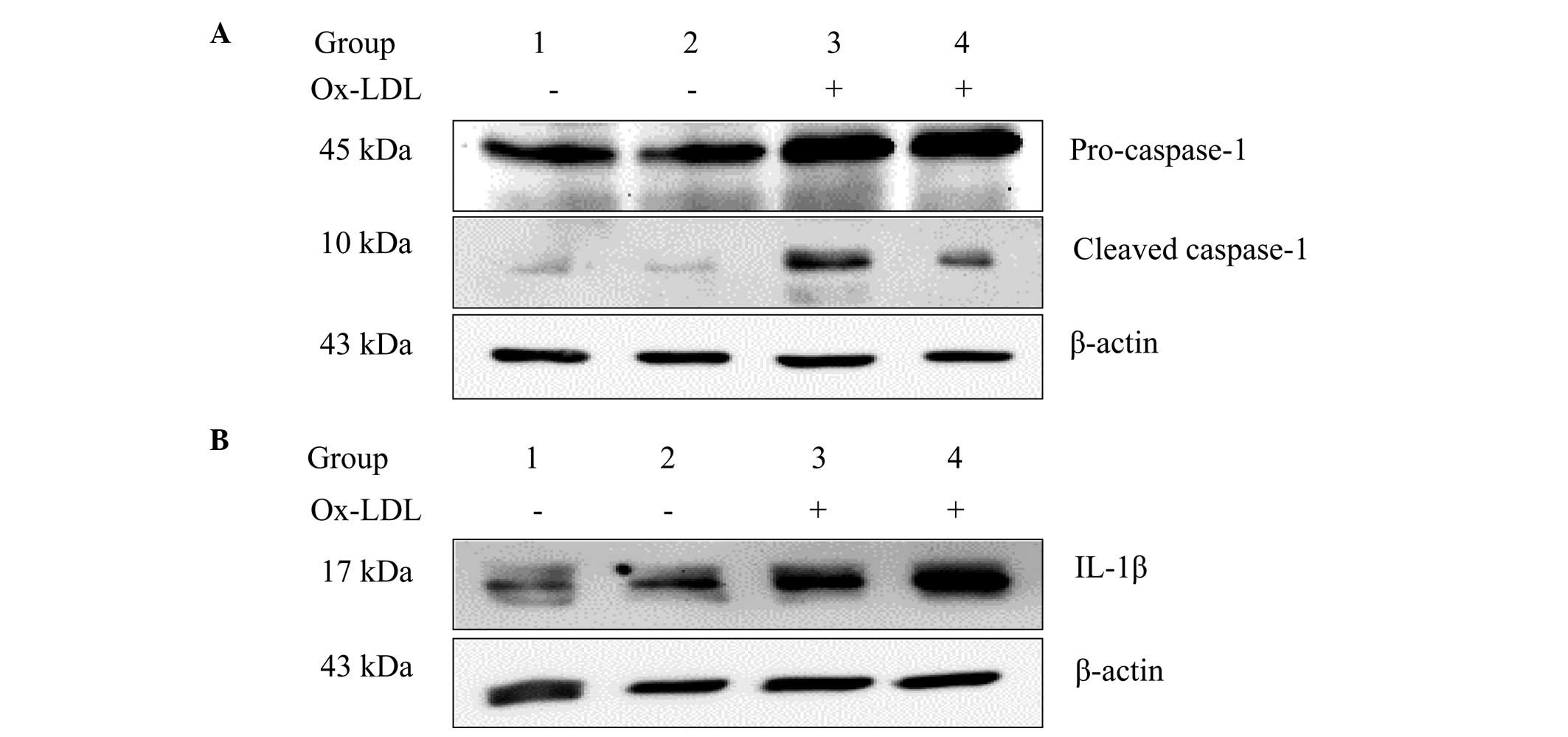
mtDNA depletion attenuates Ox-LDL-induced
caspase-1 activation and IL-1β expression
Ox-LDLs have been previously shown to trigger
caspase-1 activation in human macrophages (2). The effect of mtDNA depletion on
Ox-LDL-induced caspase-1 activation was examined in the present
study in murine macrophages. Cleaved caspase-1, which is
proteolytically active, promotes the expression of proinflammatory
cytokines, such as IL-1β and IL-18 (4). It was found that Ox-LDL stimulation
did not increase the expression levels of cleaved caspase-1 and
IL-1β in rho0 cells. As shown in Fig.
1, cleaved caspase-1 and IL-1β expression levels were markedly
reduced in J774A.1 rho0 cells when compared with the J774A.1 normal
cell group following treatment with 100 µg/ml Ox-LDL for 24
h.
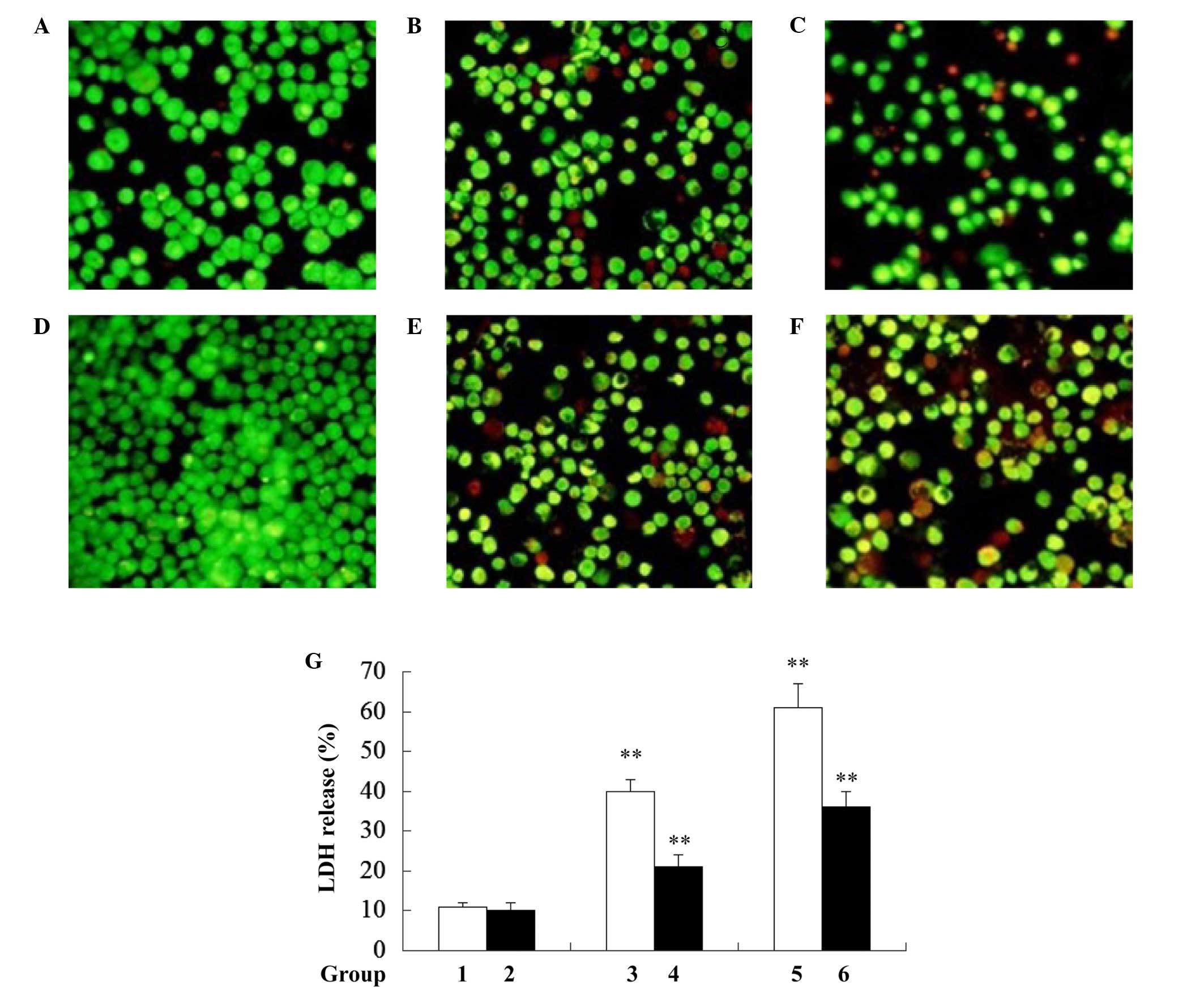
Rho0 cells are not sensitive to
Ox-LDL-induced macro-phage pyroptosis
Rho0 cells have been reported to resist apoptosis
(12). Whether mtDNA-depleted
cells are resistant to Ox-LDL-induced macrophage pyroptosis was
investigated in the present study. As hypothesized, it was found
that mtDNA-depleted J774A.1 (rho0) cells exhibited a decreased
ratio of dead cells when compared with J774A.1 normal cells
following treatment with 50 and 100 µg/ml Ox-LDL for 24 h
(Fig. 2A–F). Consistent with the
results from EB/AO staining, a significant reduction in LDH release
was observed in the rho0 cells when compared with the normal
J774A.1 cells following treatment with 50 and 100 µg/ml
Ox-LDL for 24 h (Fig. 2G).
Together, these results indicate that mtDNA-depleted J774A.1 cells
are resistant to Ox-LDL-induced cell death.
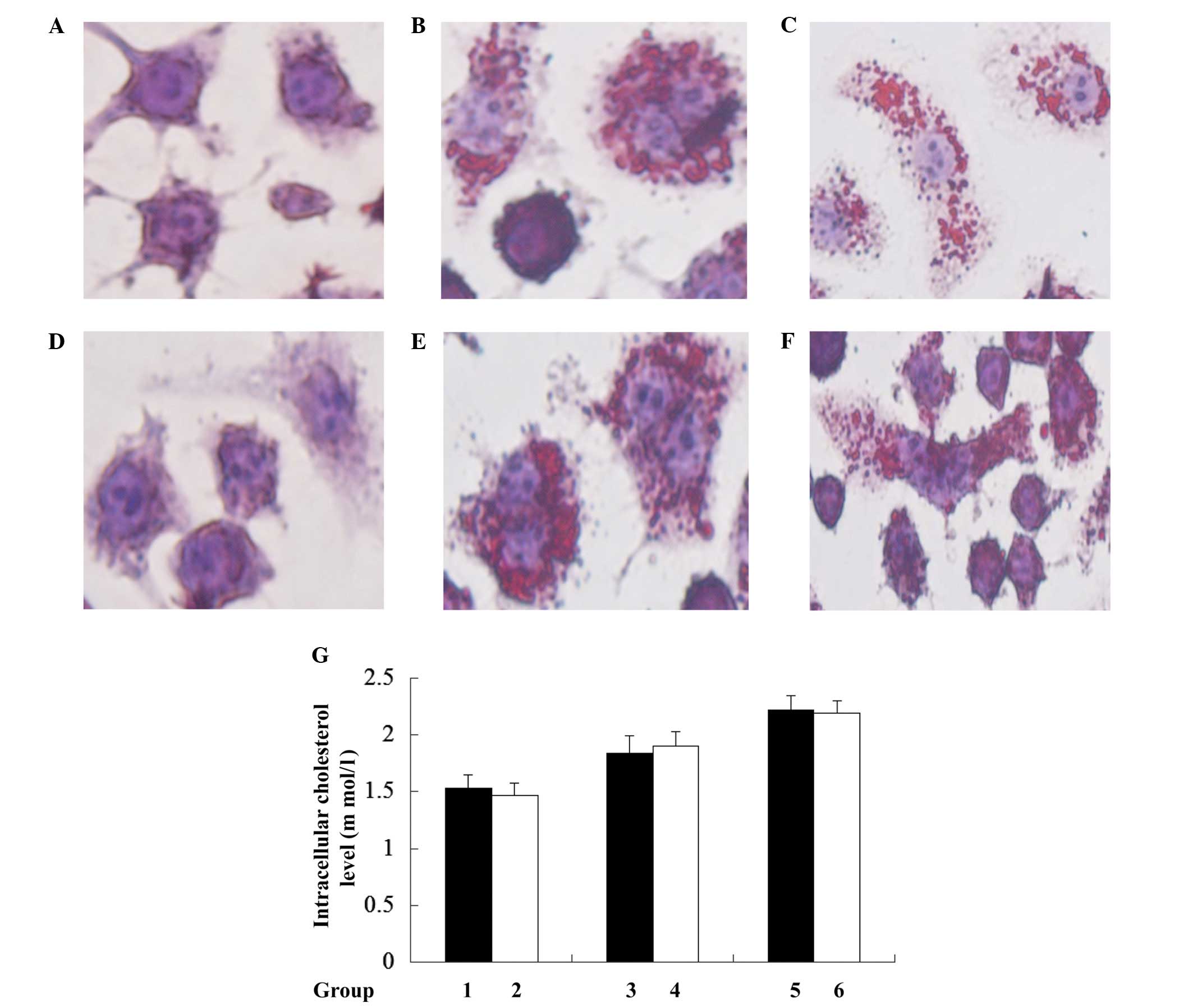
Loss of mtDNA does not alter
Ox-LDL-induced cholesterol accumulation in macrophages
To investigate whether J774A.1 rho0 cell resistance
to ox-LDL-induced cell death was caused by disruption of
intracellular cholesterol accumulation, lipid droplets (the
existing form of cholesterol in cells) were evaluated by Oil Red O
staining. This indicated no observable difference between J774A.1
rho0 and J774A.1 normal cells (Fig.
3). Further quantification of the intracellular cholesterol
results using the CHOD-PAP method indicated that mtDNA depletion
did not lead to significant disruption in intracellular total
cholesterol following treatment with 0, 50 and 100 µg/ml
Ox-LDL for 24 h when the J774A.1 rho0 cells were compared with the
J774A.1 normal cells. These results indicate that rho0 cells do not
affect Ox-LDL-induced cholesterol accumulation in murine
macrophages.
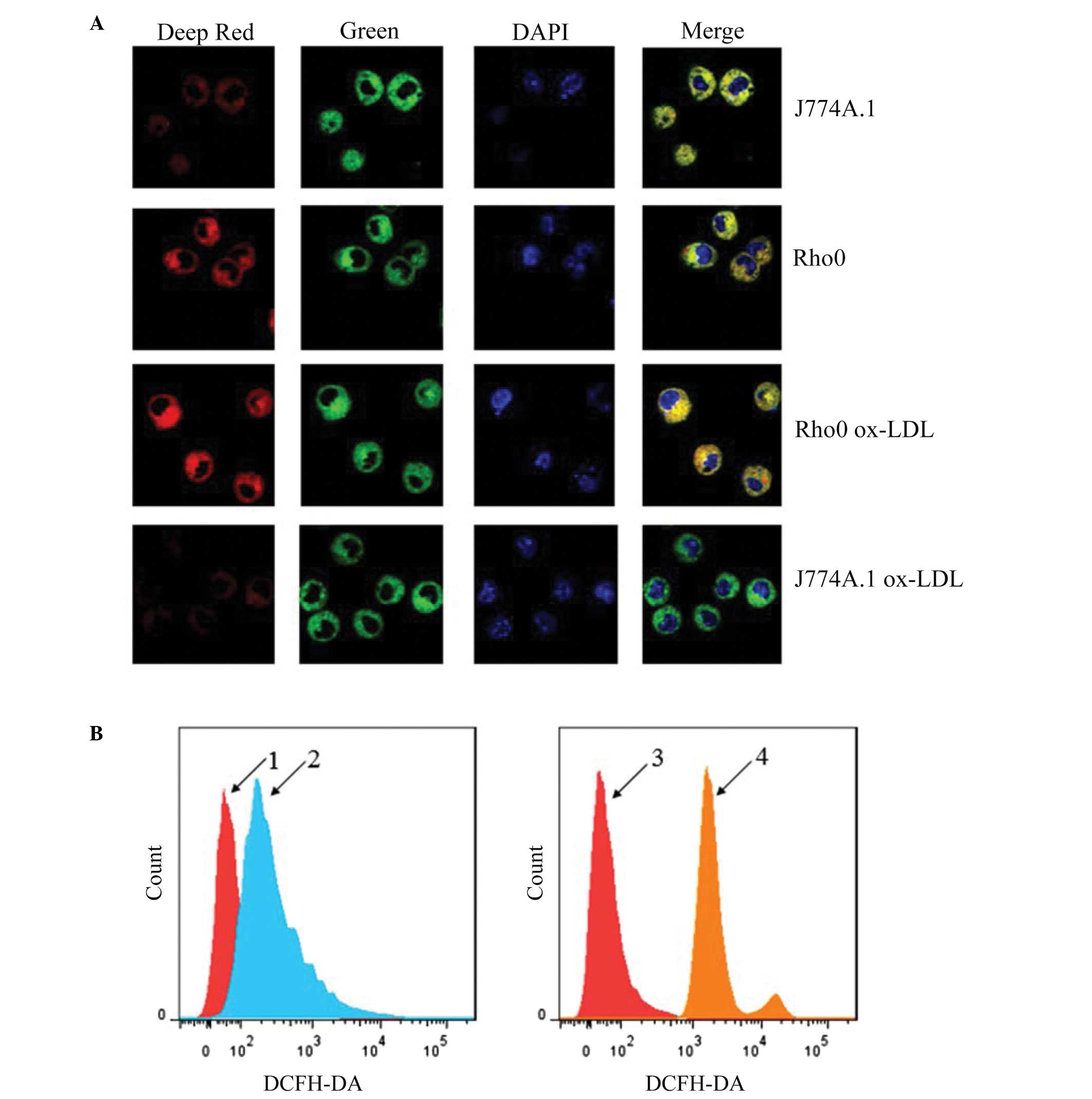
Rho0 cells maintain mitochondrial
membrane potential and exhibit reduced ROS production following
Ox-LDL treatment
The impact of mtDNA depletion on ROS and
mitochondrial membrane potential was determined using MitoTracker
Deep Red, MitoTracker Green and DAPI triple staining, and H2DCF-DA
probes for ROS labeling with a confocal microscope and flow
cytometer, respectively. As shown in Fig. 4A, J774A.1 rho0 cells displayed a
similar mitochondrial membrane potential to that of J774A.1 normal
cells. Following Ox-LDL treatment, a considerable decrement in
mitochondrial membrane potential was observed in the J774A.1 normal
cells, but not in the J774A.1 rho0 cells. As shown in Fig. 4B, the J774A.1 normal cells
displayed a higher ROS level than that of the J774A.1 rho0 cells
following Ox-LDL treatment. These results are consistent with the
hypothesis that the mitochondrial respiratory chain is the
predominant source of Ox-LDL-induced ROS production in J774A.1
cells, and loss of mtDNA enables J774A.1 rho0 cells to maintain the
mitochondrial membrane potential.
Discussion
In recent years, studies have described the
apoptosis resistance of rho0 cells (12,16).
Although the exact mechanism remains controversial, the notion that
the loss of mtDNA, and its triggering function in apoptotic cell
death, leads to oxidant injury and DNA damage (which are exhibited
in rho0 cells) is widely held. In an attempt to study the role of
mitochondria in pyroptosis, J774A.1 rho0 cells were established by
subjecting J774A.1 murine macrophages to chronic EB exposure,
according to a previous study (17). The mtDNA-depleted cell was
confirmed by PCR and an auxotrophy test (CCK-8; data not shown). In
the present study, J774A.1 rho0 cells were reported to be resistant
to Ox-LDL-induced macrophage pyroptosis. The results demonstrated
that intact, functional mitochondria are essential to the execution
of pyroptosis.
Pyroptosis is a recently identified novel form of
cell death, which is uniquely depended on caspase-1 activation
(18). There exists a number of
inflammasomes, which upon activation cleave pro-caspase-1 into
active caspase-1 and, in turn, initiate the pyroptosis process
(19). Among them, the NLRP3
inflammasome has been extensively investigated and identified to be
involved in Ox-LDL-induced macrophage pyroptosis in atherosclerosis
(20). A number of DAMPs of
mitochondrial origin, notably ROS and mtDNA, activate the NLRP3
inflammasome (5). Lin et al
(2) demonstrated that Ox-LDL
induced macrophage pyroptosis via activation of the NLRP3
inflammasome. Yu et al (7)
reported that various inflammasomes, including NLRP3 and AIM2,
cause mitochondrial damage and block the mitophagy process via
caspase-1 activation. Consistent with Sousa and Soares (21), the results from the present study
demonstrated that ROS production was diminished in rho0 cells.
However, whether rho0 cells attenuate Ox-LDL-induced macrophage
pyroptosis by decreasing ROS production, which subsequently
interferes with NLRP3 inflammasome activation, remains to be
elucidated. Furthermore, as rho0 cells are devoid of mtDNA, it is
also possible that Ox-LDL treatment reduces, or arrests, mtDNA
release from mitochondria as a danger signal, which triggers the
reduced activation of NLRP3 and AIM2 inflammasomes. Therefore,
further investigation is required into the roles of NLRP3 and AIM2
inflammasomes in Ox-LDL-induced rho0 macrophage pyroptosis.
Ox-LDL and oxidative stress are closely associated
with the pathogenesis of atherosclerosis (2). Previous studies described that
lectin-like Ox-LDL receptor 1 (22), toll-like receptor 4 (23) and cluster of differentiation 36
(24) participated in the
processes of binding and uptake of Ox-LDL, and foam cell formation.
In the current study, J774A.1 rho0 and J774A.1 normal cells were
treated with Ox-LDL, and the Oil Red O staining and quantification
of intracellular cholesterol results indicated that there was no
significant difference in Ox-LDL-induced lipid accumulation between
J774A.1 rho0 and J774A.1 normal cells. The results indicate that
cells continue uptake of Ox-LDL and form intracellular lipid
droplets without functional mitochondria.
In conclusion, the present study demonstrates that
J774A.1 rho0 cells are resistant to Ox-LDL-induced pyroptosis,
potentially via decreased ROS production and, thus, a weaker
activation of the NLRP3 inflammasome. In addition, the current data
implied that the loss of mtDNA did not affect Ox-LDL-induced
intracellular lipid accumulation and mitochondrial membrane
potential. However, the exact mechanism of rho0 cell resistance to
pyroptosis remains unclear and requires further investigation.
Acknowledgments
The present study was supported by National Natural
Science Foundation of China Grants (grant no. 30972791) awarded to
Baojun Huang.
References
|
1
|
Conti P and Shaik-Dasthagirisaeb Y:
Atherosclerosis: A chronic inflammatory disease mediated by mast
cells. Cent Eur J Immunol. 40:3–386. 2015.
|
|
2
|
Lin J, Shou X, Mao X, Dong J, Mohabeer N,
Kushwaha KK, Wang L, Su Y, Fang H and Li D: Oxidized low density
lipoprotein induced caspase-1 mediated pyroptotic cell death in
macrophages: Implication in lesion instability? PLoS One.
8:e621482013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rajamäki K, Lappalainen J, Oörni K,
Välimäki E, Matikainen S, Kovanen PT and Eklund KK: Cholesterol
crystals activate the NLRP3 inflammasome in human macrophages: A
novel link between cholesterol metabolism and inflammation. PLoS
One. 5:e117652010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Wang M, Huang K, Zhang Z, Shao N,
Zhang Y, Wang W and Wang S: Oxidized low-density lipoprotein
induces secretion of interleukin-1β by macrophages via reactive
oxygen species-dependent NLRP3 inflammasome activation. Biochem
Biophys Res Commun. 425:121–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar
|
|
6
|
Dostert C, Pétrilli V, Van Bruggen R,
Steele C, Mossman BT and Tschopp J: Innate immune activation
through Nalp3 inflammasome sensing of asbestos and silica. Science.
320:674–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu J, Nagasu H, Murakami T, Hoang H,
Broderick L, Hoffman HM and Horng T: Inflammasome activation leads
to Caspase-1-dependent mitochondrial damage and block of mitophagy.
Proc Natl Acad Sci USA. 111:15514–15519. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park SY, Choi B, Cheon H, Pak YK, Kulawiec
M, Singh KK and Lee MS: Cellular aging of mitochondrial
DNA-depleted cells. Biochem Biophys Res Commun. 325:1399–1405.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arduino DM, Esteves AR, Cortes L, Silva
DF, Patel B, Grazina M, Swerdlow RH, Oliveira CR and Cardoso SM:
Mitochondrial metabolism in Parkinson's disease impairs quality
control autophagy by hampering microtubule-dependent traffic. Hum
Mol Genet. 21:4680–4702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prigione A and Cortopassi G: Mitochondrial
DNA deletions and chloramphenicol treatment stimulate the
autophagic transcript ATG12. Autophagy. 3:377–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park SY, Chang I, Kim JY, Kang SW, Park
SH, Singh K and Lee MS: Resistance of mitochondrial DNA-depleted
cells against cell death: Role of mitochondrial superoxide
dismutase. J Biol Chem. 279:7512–7520. 2004. View Article : Google Scholar
|
|
12
|
Ferraresi R, Troiano L, Pinti M, Roat E,
Lugli E, Quaglino D, Taverna D, Bellizzi D, Passarino G and
Cossarizza A: Resistance of mtDNA-depleted cells to apoptosis.
Cytometry A. 73:528–537. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brar SS, Meyer JN, Bortner CD, Van Houten
B and Martin WJ II: Mitochondrial DNA-depleted A549 cells are
resistant to bleomycin. Am J Physiol Lung Cell Mol Physiol.
303:L413–L424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kirii H, Niwa T, Yamada Y, Wada H, Saito
K, Iwakura Y, Asano M, Moriwaki H and Seishima M: Lack of
interleukin-1beta decreases the severity of atherosclerosis in
ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 23:656–660.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ribble D, Goldstein NB, Norris DA and
Shellman YG: A simple technique for quantifying apoptosis in
96-well plates. BMC Biotechnol. 5:122005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee MS, Kim JY and Park SY: Resistance of
rho(0) cells against apoptosis. Ann N Y Acad Sci. 1011:146–153.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
King MP and Attardi G: Isolation of human
cell lines lacking mitochondrial DNA. Methods Enzymol. 264:304–313.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fink SL and Cookson BT: Apoptosis,
pyroptosis and necrosis: Mechanistic description of dead and dying
eukaryotic cells. Infect Immun. 73:1907–1916. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
21
|
Sousa CA and Soares EV: Mitochondria are
the main source and one of the targets of Pb (lead)-induced
oxidative stress in the yeast Saccharomyces cerevisiae. Appl
Microbiol Biotechnol. 98:5153–5160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nowicki M, Müller K, Serke H, Kosacka J,
Vilser C, Ricken A and Spanel-Borowski K: Oxidized low-density
lipoprotein (oxLDL)-induced cell death in dorsal root ganglion cell
cultures depends not on the lectin-like oxLDL receptor-1 but on the
toll-like receptor-4. J Neurosci Res. 88:403–412. 2010. View Article : Google Scholar
|
|
23
|
Serke H, Vilser C, Nowicki M, Hmeidan FA,
Blumenauer V, Hummitzsch K, Lösche A and Spanel-Borowski K:
Granulosa cell subtypes respond by autophagy or cell death to
oxLDL-dependent activation of the oxidized lipoprotein receptor 1
and toll-like 4 receptor. Autophagy. 5:991–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Serke H, Bausenwein J, Hirrlinger J,
Nowicki M, Vilser C, Jogschies P, Hmeidan FA, Blumenauer V and
Spanel-Borowski K: Granulosa cell subtypes vary in response to
oxidized low-density lipoprotein as regards specific lipoprotein
receptors and antioxidant enzyme activity. J Clin Endocrinol Metab.
95:3480–3490. 2010. View Article : Google Scholar : PubMed/NCBI
|