Introduction
Hyperuricemia is a common metabolic disorder. Recent
studies showed that it is also an independent risk factor for
chronic kidney disease (1,2). Elevated serum uric acid (UA) levels
not only decrease the glomerular filtration rate, but also induce
tubulointerstitial injury. However, the underlying mechanisms have
remained to be fully elucidated (2,3).
Previous studies indicated that high levels of UA promoted reactive
oxygen species (ROS) generation to increase oxidative stress in
several cell types, including mesangial cells, adipocytes and
vascular smooth muscle cells (4–6). ROS
are considered to be crucial mediators of cell apoptosis (7–9). In
kidney disease, apoptosis is one of the main causes of tubular cell
loss (10). Therefore, the present
study hypothesized that high levels of UA induce oxidative stress
within renal tubular cells and lead to apoptosis, which eventually
causes renal dysfunction.
Nicotinamide adenine dinucleotide phosphate oxidase
4 (Nox4) is the predominant Nox in the kidney (8,11).
It generates ROS by transferring an electron to molecular oxygen.
Nox4-derived ROS have an important role in cell signaling as
secondary messengers, mediating numerous biological processes,
including cell apoptosis (7,8).
However, it has remained elusive whether UA-mediated Nox4
expression represents an endogenous source of ROS in tubular
cells.
The present study confirmed that elevated UA
promotes apoptosis by upregulating the expression of Nox4 in renal
tubular cells. In addition, the underlying mechanisms were
examined, including ROS generation and the activation of
extracellular signal-regulated kinase (ERK)1/2 as well as P38.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM)/F-12,
fetal bovine serum (FBS), penicillin and streptomycin were obtained
from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), diphenylene
iodonium (DPI), bovine serum albumin (BSA) and uric acid were
purchased from Sigma-Aldrich (St. Louis, MO, USA)Polyclonal rabbit
anti-human Nox4 (cat. no. BS6796) was obtained from Bioworld
Technology, Inc. (St. Louis Park, MN, USA). Monoclonal rabbit
anti-human phosphorylated (p)-P38 (cat. no. 4631), p-ERK1/2 (cat.
no. 4376), P38 (cat. no. 8690), ERK1/2 (cat. no. 4695), B-cell
lymphoma 2 (Bcl-2; cat. no. 2870), Bcl-2-associated X protein (Bax;
cat. no. 5023) and β-actin (cat. no. 4970) primary antibodies, and
horseradish peroxidase-labeled goat anti-rabbit immunoglobulin G
(IgG) secondary antibody (cat. no. 7074) were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). A bicinchoninic acid
(BCA) protein assay kit was obtained from Thermo Fisher Scientific.
A polyvinylidene difluoride (PVDF) membrane and SuperECL Plus
hypersensitivity luminous fluid were purchased from Millipore
(Billerica, MA, USA). All chemical reagents were of analytical
grade.
Cell culture and treatment
HK-2 cells were obtained from the American Type
Culture Collection (Manassas, VA, USA). Cells were cultured at 37°C
in a humidified atmosphere containing 5% CO2 in
DMEM/F-12, which contained 10% FBS, 100 IU/ml penicillin and 100
μg/ml streptomycin. When the cell density reached 70–80%,
the cells were maintained in serum-free medium overnight in order
to synchronize the cell cycle and eliminate the influence of serum
on cellular functions. HK-2 cells were then incubated with various
concentrations of UA (4, 8 or 16 mg/dl) for 24, 48 and 72 h. To
inhibit the function of Nox, 10 μM DPI was applied for 30
min prior to incubation with UA. The negative control (NC) group
was not treated with UA or DPI.
Cell viability assay
The cell viability/cytotoxicity was determined using
a 3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2Htetrazolium bromide
(MTT) assay). HK-2 cells were seeded into 96-well culture plates at
the density of 2,000 cells per well and then treated with various
concentrations of UA. After 24, 48 or 72 h of incubation, the assay
was performed by adding 20 μl of MTT solution [5 mg/ml in
phosphate-buffered saline (PBS); Sigma-Aldrich] to each well,
followed by incubation for 4 h. Subsequently, 100 μl
dimethyl sulfoxide (DMSO; Sigma-Aldrich) was added to the culture
medium in each well to dissolve the formazan crystals and the
absorbance of each well was measured at 492 nm using a SpectraMax
M5 plate reader (Molecular Devices, LLC, Sunnyvale, CA, USA)
Measurement of ROS generation
ROS generation was detected using the DCFH-DA assay.
Suspended HK-2 cells (1×106 cells/well) were added to
6-well plates, then treated with 10 μmol/l DCFH-DA in
serum-free DMEM/F-12 for 20 min at 37°C. After two washes with PBS,
cells were collected and resuspended at 1×106 cells/ml.
DCF fluorescence was detected using a FACSCalibur flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA) at an excitation
wavelength of 488 nm and an emission wavelength of 535 nm.
Annexin V apoptosis assay
The number of apoptotic cells was evaluated by flow
cytometry using an FITC Annexin V Apoptosis Detection kit (BD
Biosciences) according to the manufacturer's instructions. In
brief, HK-2 cells were seeded in six-well plates at
2×105 cells per well and treated as described above.
Cells were washed twice with PBS and re-suspended in 200 μl
binding buffer. After addition of 5 μl Annexin V conjugate
and incubation for 10 min, the samples were resuspended in 200
μl binding buffer and 5 μl propidium iodide (PI). The
cells were examined using the FACSCalibur flow cytometer with ten
thousand events collected for each sample.
Western blot analysis
HK-2 cells were added to 6-well plates at a density
of 1×106 cells/well. Following serum starvation
overnight, cells were co-cultured with UA (4, 8 or 16 mg/dl) for 48
h. In the UA + DPI group, 10 μM DPI was applied for 30 min
prior to incubation with 16 mg/dl UA. Following two washes with
PBS, cells were collected and centrifuged at 14,860 × g for 5
minutes. With the supernatant removed, the cells were mixed with 80
μl lysis buffer (Beyotime Institute of Biotechnology,
Haimen, China). The lysate was incubated at 4°C for 15 min then
centrifuged for 20 min at 20,800 × g. The supernatant was collected
and quantified for protein content using the BCA method. Equal
amounts of total protein (50 μg) were loaded and separated
using 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis
then electrophoretically transferred onto a PVDF membrane. The
membrane was incubated with primary antibodies (dilution, 1:1,000)
at 4°C overnight followed by incubation with secondary antibody
(dilution, 1:5,000) at room temperature for 1 h. Proteins were
visualized by chemiluminescence using SuperECL Plus
hypersensitivity luminous fluid. The bands were observed with a
G:Box gel-imaging system (Syngene, Frederick, MD, USA) β-actin was
used as the internal control. The intensity of protein bands was
quantified using Quantity One software (version 4.62; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using SPSS
software 18.0 (SPSS, Inc., Chicago, IL, USA). Values are expressed
as the mean ± standard deviation. Differences were assessed by
Student's t-test or analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
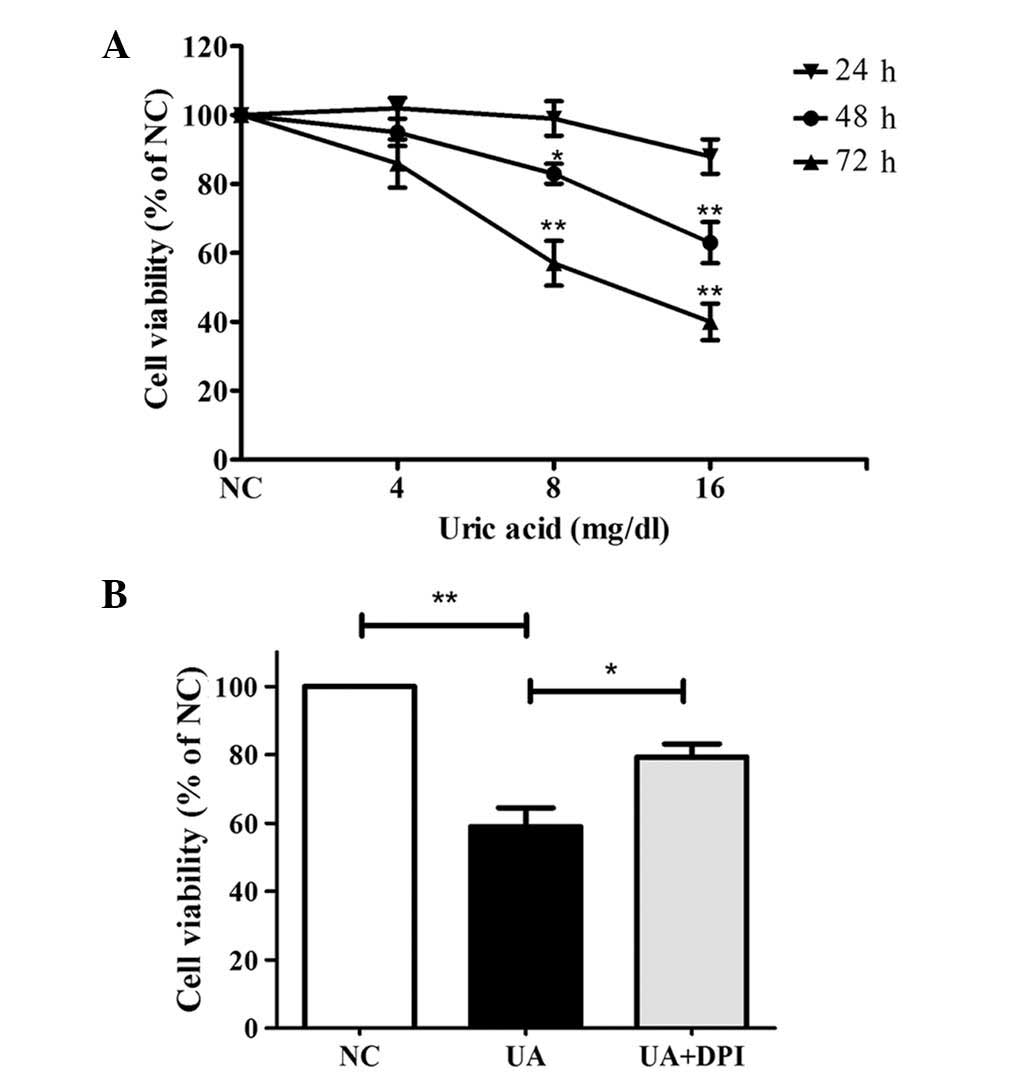
UA reduces kidney cell viability via
Nox
To evaluate the effects of UA on cell viability,
HK-2 cells were treated with 0, 4, 8 or 16 mg/dl UA for 24–72 h and
subjected to an MTT assay. As shown in Fig. 1A, UA decreased HK-2 cell viability
in a time- and dose-dependent manner. Treatment with 16 mg/dl UA
for 48 and 72 h significantly reduced the number of viable cells to
63±6% and 40±5%, respectively, of that of the control (P<0.01).
Therefore, 16 mg/dl UA for 48 h were determined as the ideal
conditions for further assessing the involvement of Nox. As shown
in Fig. 1B, Nox inhibition by
pre-treatment with DPI (10 μM for 30 min) significantly
attenuated the cytotoxic effects of UA (79±5.7% vs. 59±9.2%,
P<0.05).
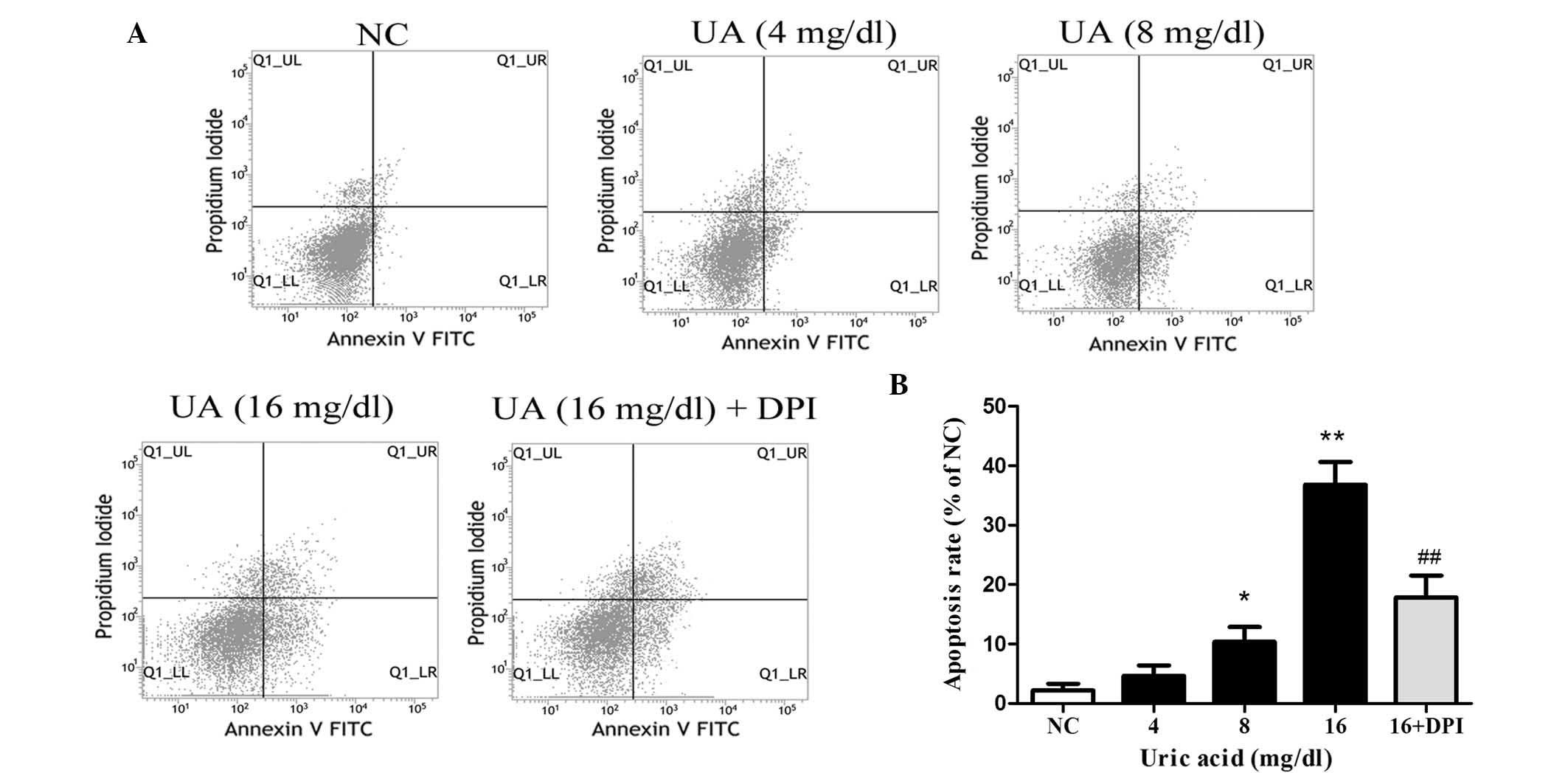
UA promotes kidney cell apoptosis via
Nox
To examine whether UA promotes apoptosis, HK-2 cells
were incubated with various doses of UA for 48 h and subjected to
flow cytometric analysis following Annexin V/PI double staining
(Fig. 2). UA was found to
concentration-dependently induce apoptosis in HK-2 cells, with 16
mg/dl UA producing an apoptotic rate of 36.8±4.4% compared with
2.2±0.8% in the NC group (P<0.01). In order to evaluate the
involvement of Nox, HK-2 cells were incubated with DPI (10
μM for 30 min) prior to treatment with 16 mg/dl UA for 48 h.
This Nox inhibition significantly inhibited the apoptotic effects
of UA, as indicated by a reduction of the apoptotic rate to 17.9±3%
(P<0.01 vs. UA at 16 mg/dl).
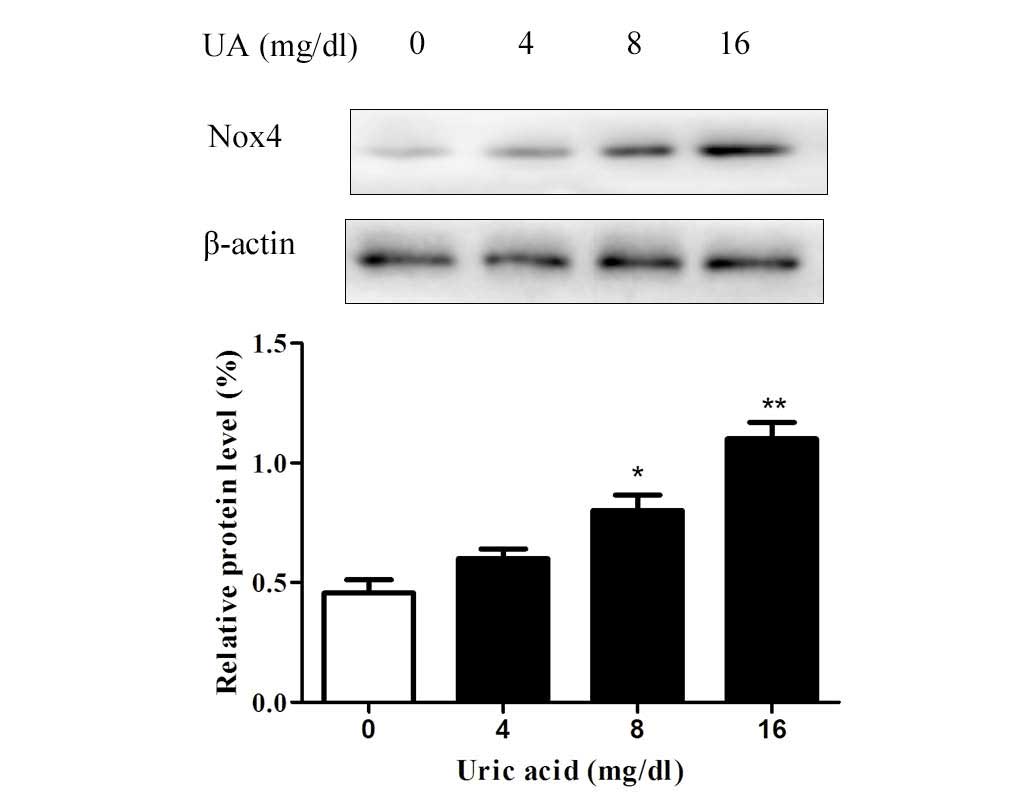
UA upregulates the expression of Nox4 in
HK-2 cells
Since the cytotoxic effects of UA on renal tubular
cells were observed to be inhibited by DPI, it was indicated that
UA exerts its effects via Nox. To examine the effects of UA on Nox,
western blot analysis of Nox4, the predominant Nox in renal tubular
cells, was performed. The results revealed that the protein levels
of Nox4 were upregulated by UA in a dose-dependent manner after
incubation for 48 h (Fig. 3).
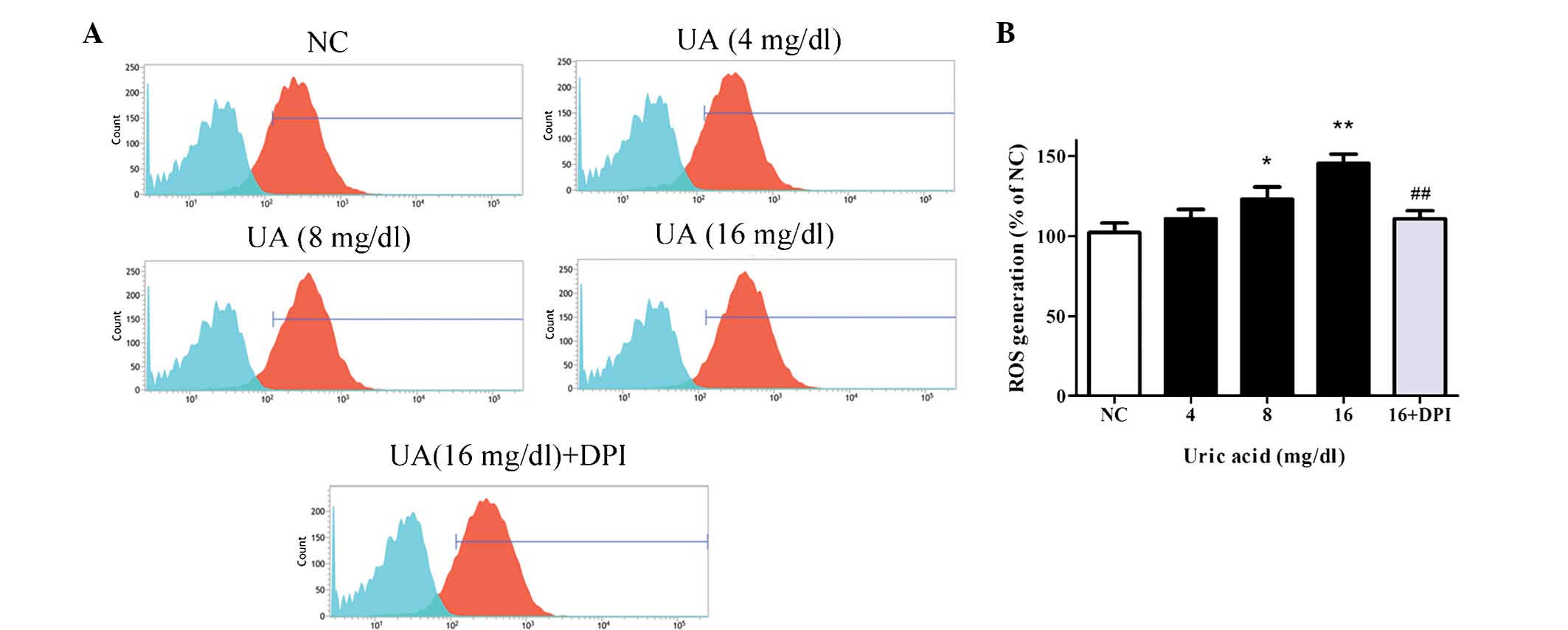
UA induces oxidative stress in HK-2 cells
via Nox
As Nox4 is known to regulate ROS production
(7,8), the effects of UA on the levels of ROS
in HK-2 cells were then assessed. Following treatment of cells with
various concentrations of UA for 48 h, ROS were detected by DCFH-DA
staining and flow cytometric analysis. As demonstrated in Fig. 4A, the blue populations indicate the
unstained cells (baseline control) and the red populations were
cells stained by DCFH-DA. The horizontal axis presents the mean
fluorescence intensity of the cells, which indicates the quantity
of ROS generation, and the vertical axis presents the number of
fluorescent cells, which was almost equal in each group. As
demonstrated by the rightward-shift of the red peak, the generation
of ROS was significantly enhanced by UA compared with control,
reaching 145.7±4.3% at 16 mg/dl UA (P<0.01). Additionally,
pretreatment with DPI significantly reduced the UA-induced
elevation of ROS production (110.9±5%) compared with 16 mg/dl UA
treatment (145.7±4.3%; P<0.01; Fig.
4B).
P38 and ERK1/2 signaling are involved in
UA-induced apoptosis
It has been demonstrated that mitogen-activated
protein kinase (MAPK) pathways are activated by upregulation of
intracellular ROS (12–14). P38 and ERK1/2 are important members
of the MAPK family, which regulates cell growth and apoptosis
(15–17). The present study examined the
effects of UA on the activation of P38 and ERK1/2 to investigate
the possible mechanism of UA-induced apoptosis. As shown in
Fig. 5A, the phosphorylation of
P38 and ERK1/2 significantly increased following treatment of HK-2
cells with 16 mg/dl UA for 48 h. Furthermore, inhibition of Nox
with DPI attenuated the UA-induced phosphorylation of P38 and
ERK1/2. These findings indicated UA treatment leads to the
activation of P38/ERK signaling in kidney cells, possibly by
upregulation of Nox4 expression and subsequent induction of
ROS.
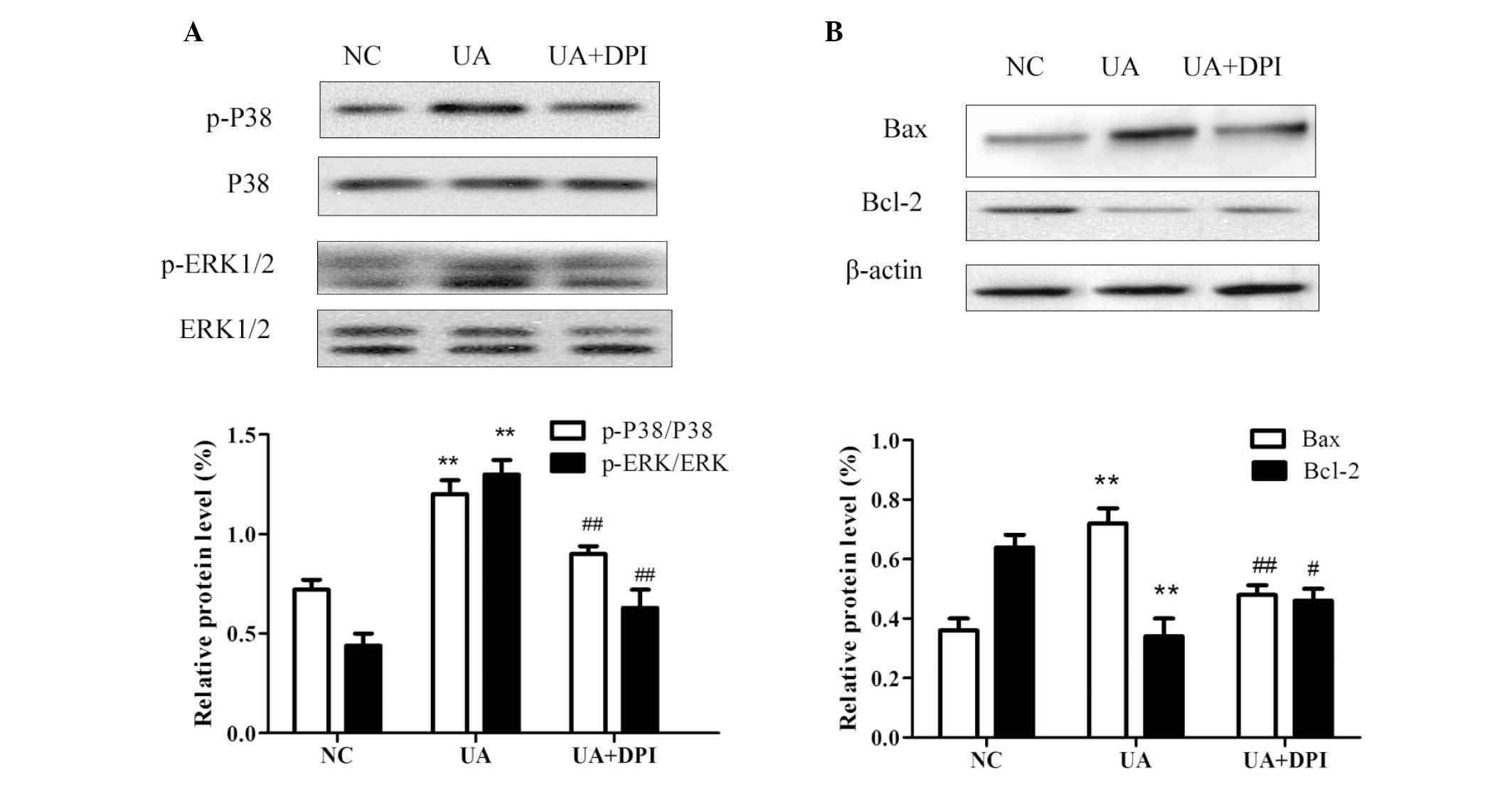 | Figure 5Western blot analysis of HK-2 cells
revealed that UA (16 mg/dl for 48 h) (A) significantly activated
the phosphorylation of P38 and ERK1/2, which was inhibited by
nicotinamide adenine dinucleotide phosphate oxidase inhibitor DPI
and (B) induced apoptosis signaling by increasing Bax and
decreasing Bcl-2, and that the balance of these apoptotic proteins
was restored by DPI. GAPDH and β-actin were used as internal
reference. Values are expressed as the mean ± standard deviation
from at least three independent experiments. n=3,
**P<0.01 vs. NC; #P<0.05,
##P<0.01 vs. UA. UA, uric acid; NC, negative control;
p-ERK, phosphorylated extracellular signal-regulated kinase; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-2-associated X protein; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase. |
To investigate the underlying mechanisms of cell
apoptosis, the effects of UA on the expression of Bcl-2 and Bax
were assessed by western blot analysis. While UA treatment caused a
significant increase in the protein levels of Bax and a decrease in
Bcl-2 in HK-2 cells, the Bcl-2/Bax balance was neutralized by
pre-treatment with DPI (Fig.
5B).
Discussion
The present study revealed that UA treatment led to
the upregulation of Nox4 expression in HK-2 cells in a
dose-dependent manner. Nox4 then promoted the generation of ROS,
leading to the induction of apoptosis. The phosphorylation of P38
and ERK1/2 and the imbalance of Bax/Bcl-2 were also demonstrated to
be involved in this process. These results suggested that elevated
UA promotes mitochondrial apoptosis through the P38/ERK pathway in
renal tubular cells, which is induced by upregulation of Nox4 and
production of ROS.
Uric acid is closely linked with chronic kidney
disease (CKD) (18). In the
pathophysiology of CKD, a decrease of the glomerular filtration
rate increases the serum levels of UA, which in turn promote CKD
progression (2); however, the
underlying mechanisms have remained to be fully elucidated. UA has
been demonstrated to trigger the upregulation of ROS in several
cell types. ROS are able to regulate or induce multiple cellular
processes, including epithelial-mesenchymal transition (19), growth (20), cell differentiation (21) and apoptosis (7–9,13).
The present study indicated that UA induced apoptosis in renal
tubular cells by inducing ROS, which may cause cell loss and
tubular dysfunction during the progression of CKD.
Of note, the present study demonstrated that the
effects of UA were inhibited by DPI, which is indicative of the
involvement of Nox. Nox has seven isoforms: Nox1, Nox2 (gp91phox),
Nox3, Nox4, Nox5, Duox1 and Duox2 (22). In the kidney, Nox4 represents the
predominant form and is abundantly expressed as a source of ROS
(8,11,23).
In tubular cells, Nox4 was shown to be upregulated by a variety of
metabolic factors, including high glucose, angiotensin II and
bilirubin (19,24,25).
The present study also confirmed that UA was able to promote the
expression of Nox4 in a dose-dependent manner. This may be the
primary origin of intracellular ROS.
Furthermore, the present study assessed the
potential mechanisms of ROS-induced apoptosis. As is known, the
MAPK family are key factors in numerous cellular processes. The
main MAPKs are P38, ERK1/2 and c-Jun N-terminal kinase 1/2, whose
activity is stimulated by phosphorylation. The ROS-induced
activation of MAPKs has been reported in several kidney diseases
(26–28), while the association between UA and
MAPKs has largely remained elusive; however, previous studies have
indicated the participation of MAPKs in UA-induced tubular cell
apoptosis (29,30). As the underlying mechanisms
required further elucidation, the present study examined the
phosphorylation of P38 and ERK1/2 and found them significantly
activated following exposure to UA. Furthermore, the present study
revealed that pre-treatment with DPI was able to suppress the above
effect, indicating that Nox4-derived ROS may be involved in the
mechanism of UA-induced renal cell injury. In addition, the
expression of Bax and Bcl-2, which can be regulated by MAPKs, was
detected, revealing that high levels of UA caused a Bax/Bcl-2
imbalance, suggesting that apoptosis was induced through the
mitochondrial pathway. A study by Verzola et al (30) also observed that UA promotes
apoptosis in renal tubular cells by activating Nox4. Compared with
this report, the present study provided further evidence to support
their findings. Taken together, the crucial role of Nox4-ROS-MAPK
pathway was clearly demonstrated by the current and previous
studies.
In conclusion, the present study demonstrated that
elevated UA promoted ROS-induced mitochondrial apoptosis by
upregulating Nox4 expression in HK-2 cells. The mechanism was shown
to involve the activation of P38 and ERK1/2. These findings
provided a possible mechanism by which UA promotes the progression
of chronic kidney disease. Furthermore, inhibition of Nox4 was
demonstrated to prevent tubular cells from apoptosis and may
therefore represent a therapeutic strategy chronic kidney
disease.
Acknowledgments
This study was supported by the Science and
Technology Development Program of Guangdong Province (no.
2012B031800081) and Young Teacher Foundation of Sun Yat-Sen
University (no. 12ykpy31).
References
|
1
|
Obermayr RP, Temml C, Gutjahr G,
Knechtelsdorfer M, Oberbauer R and Klauser-Braun R: Elevated uric
acid increases the risk for kidney disease. J Am Soc Nephrol.
19:2407–2413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson RJ, Nakagawa T, Jalal D,
Sánchez-Lozada LG, Kang DH and Ritz E: Uric acid and chronic kidney
disease: Which is chasing which? Nephrol Dial Transplant.
28:2221–2228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang
H and Yang XF: Uric acid, hyperuricemia and vascular diseases.
Front Biosci (Landmark Ed). 17:656–669. 2012. View Article : Google Scholar
|
|
4
|
Convento MS, Pessoa E, Dalboni MA, Borges
FT and Schor N: Pro-inflammatory and oxidative effects of
noncrystalline uric acid in human mesangial cells: Contribution to
hyperuricemic glomerular damage. Urol Res. 39:21–27. 2011.
View Article : Google Scholar
|
|
5
|
Corry DB, Eslami P, Yamamoto K, Nyby MD,
Makino H and Tuck ML: Uric acid stimulates vascular smooth muscle
cell proliferation and oxidative stress via the vascular
renin-angiotensin system. J Hypertens. 26:269–275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang JX, Zhang YP, Wu QN and Chen B: Uric
acid induces oxidative stress via an activation of the
renin-angiotensin system in 3T3-L1 adipocytes. Endocrine.
48:135–142. 2015. View Article : Google Scholar
|
|
7
|
Ago T, Kuroda J, Pain J, Fu C, Li H and
Sadoshima J: Upregulation of Nox4 by hypertrophic stimuli promotes
apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ
Res. 106:1253–1264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sedeek M, Nasrallah R, Touyz RM and Hébert
RL: NADPH oxidases, reactive oxygen species, and the kidney: Friend
and foe. J Am Soc Nephrol. 24:1512–1518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brown DI and Griendling KK: Regulation of
signal transduction by reactive oxygen species in the
cardiovascular system. Circ Res. 116:531–549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Havasi A and Borkan SC: Apoptosis and
acute kidney injury. Kidney Int. 80:29–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharma K: Obesity, oxidative stress, and
fibrosis in chronic kidney disease. Kidney Int Suppl. 4:113–117.
2014. View Article : Google Scholar
|
|
12
|
Park J, Min JS, Kim B, Chae UB, Yun JW,
Choi MS, Kong IK, Chang KT and Lee DS: Mitochondrial ROS govern the
LPS-induced pro-inflammatory response in microglia cells by
regulating MAPK and NF-kB pathways. Neurosci Lett. 584:191–196.
2015. View Article : Google Scholar
|
|
13
|
Zhao ZY, Luan P, Huang SX, Xiao SH, Zhao
J, Zhang B, Gu BB, Pi RB and Liu J: Edaravone protects HT22 neurons
from H2O2-induced apoptosis by inhibiting the MAPK signaling
pathway. CNS Neurosci Ther. 19:163–169. 2013. View Article : Google Scholar
|
|
14
|
Changchien JJ, Chen YJ, Huang CH, Cheng
TL, Lin SR and Chang LS: Quinacrine induces apoptosis in human
leukemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and
suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol Appl
Pharmacol. 284:33–41. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu W, Ning R, Chen RN, Huang XF, Dai QS,
Hu JH, Wang YW, Wu LL, Xiong J, Hu G, et al: Aspafilioside B
induces G2/M cell cycle arrest and apoptosis by up-regulating H-Ras
and N-Ras via ERK and p38 MAPK signaling pathways in human hepatoma
HepG2 cells. Mol Carcinog. Feb 14–2015.Epub ahead of print.
|
|
16
|
Shi M, He X, Wei W, Wang J, Zhang T and
Shen X: Tenascin-C induces resistance to apoptosis in pancreatic
cancer cell through activation of ERK/NF-kB pathway. Apoptosis.
20:843–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Y, Zhang S, Su D, Liu J, Cheng Y, Zou
L, Li W and Jiang Y: Inhibiting (pro)renin receptor-mediated p38
MAPK signaling decreases hypoxia/reoxygenation-induced apoptosis in
H9c2 cells. Mol Cell Biochem. 403:267–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boban M, Kocic G, Radenkovic S, Pavlovic
R, Cvetkovic T, Deljanin-Ilic M, Ilic S, Bobana MD, Djindjic B,
Stojanovic D, et al: Circulating purine compounds, uric acid, and
xanthine oxidase/dehydrogenase relationship in essential
hypertension and end stage renal disease. Ren Fail. 36:613–618.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He T, Guan X, Wang S, Xiao T, Yang K, Xu
X, Wang J and Zhao J: Resveratrol prevents high glucose-induced
epithelial-mesenchymal transition in renal tubular epithelial cells
by inhibiting NADPH oxidase/ROS/ERK pathway. Mol Cell Endocrinol.
402:13–20. 2015. View Article : Google Scholar
|
|
20
|
Zhang Y, Yamamoto T, Hisatome I, Li Y,
Cheng W, Sun N, Cai B, Huang T, Zhu Y, Li Z, et al: Uric acid
induces oxidative stress and growth inhibition by activating
adenosine monophosphate-activated protein kinase and extracellular
signal-regulated kinase signal pathways in pancreatic β cells. Mol
Cell Endocrinol. 375:89–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mouche S, Mkaddem SB, Wang W, Katic M,
Tseng YH, Carnesecchi S, Steger K, Foti M, Meier CA, Muzzin P, et
al: Reduced expression of the NADPH oxidase NOX4 is a hallmark of
adipocyte differentiation. Biochim Biophys Acta. 1773:1015–1027.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawahara T, Quinn MT and Lambeth JD:
Molecular evolution of the reactive oxygen-generating NADPH oxidase
(Nox/Duox) family of enzymes. BMC Evol Biol. 7:1092007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sedeek M, Callera G, Montezano A, Gutsol
A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM
and Hébert RL: Critical role of Nox4-based NADPH oxidase in
glucose-induced oxidative stress in the kidney: Implications in
type 2 diabetic nephropathy. Am J Physiol Renal Physiol.
299:F1348–F1358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim SM, Kim YG, Jeong KH, et al:
Angiotensin II-induced mitochondrial Nox4 is a major endogenous
source of oxidative stress in kidney tubular cells. PLoS One.
7:e397392012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oh SW, Lee ES, Kim S, Na KY, Chae DW, Kim
S and Chin HJ: Bilirubin attenuates the renal tubular injury by
inhibition of oxidative stress and apoptosis. BMC Nephrol.
14:1052013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qi W, Niu J, Qin Q, Qiao Z and Gu Y:
Glycated albumin triggers fibrosis and apoptosis via an NADPH
oxidase/Nox4-MAPK pathway-dependent mechanism in renal proximal
tubular cells. Mol Cell Endocrinol. 405:74–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao X, Wu J, Qian Y, et al: Oxidized
high-density lipoprotein impairs the function of human renal
proximal tubule epithelial cells through CD36. Int J Mol Med.
34:564–572. 2014.PubMed/NCBI
|
|
28
|
Jaiman S, Sharma AK, Singh K and Khanna D:
Signalling mechanisms involved in renal pathological changes during
cisplatin-induced nephropathy. Eur J Clin Pharmacol. 69:1863–1874.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Quan H, Peng X, Liu S, Bo F, Yang L, Huang
Z, Li H, Chen X and Di W: Differentially expressed protein profile
of renal tubule cell stimulated by elevated uric acid using SILAC
coupled to LC-MS. Cell Physiol Biochem. 27:91–98. 2011.PubMed/NCBI
|
|
30
|
Verzola D, Ratto E, Villaggio B, Parodi
EL, Pontremoli R, Garibotto G and Viazzi F: Uric acid promotes
apoptosis in human proximal tubule cells by oxidative stress and
the activation of NADPH oxidase NOX 4. PLoS One. 9:e1152102014.
View Article : Google Scholar : PubMed/NCBI
|