Introduction
Acute lymphoblastic leukemia (ALL) is the most
commonly diagnosed malignancy in adolescents and young adults, and
represents almost one third of all cases of cancer in children
(1,2). ALL is a heterogeneous disease, and
multiple subtypes have been identified based on recurrent copy
number alterations and structural chromosomal rearrangements
(3–5). Due to genetic variations, the
incidence of ALL in children varies across regions, and is
determined by ethnicity (1). In
China, the incidence of childhood ALL is 4/100,000 children aged
<10 years (6). Although the
5-year event-free survival rate of children and adolescents with
ALL is ~80%, the remaining 20% of patients relapse, the outcome of
which remains poor (7–9). Therefore, identifying ALL-associated
differentially expressed genes is important for improving
therapeutic methods and extending patient survival rates in
childhood ALL.
Multiple technologies have been used to identify the
differentially expressed genes associated with childhood ALL. For
example, gene expression microarray analyses of differentially
expressed genes in ALL subtypes (10) have identified 80-300 genes as
marker genes, which are necessary for discriminating the subtypes.
In addition, sequencing-based methods, including serial analysis of
gene expression (SAGE), have been used to measure absolute gene
expression levels in ALL subtypes (11). However, as hybridization-based
methods are subject to technical limitations, certain genes may be
overlooked during the hybridization process in gene expression
microarray analysis. In addition, the cost of sequencing technology
hinders the widespread usage of SAGE for identifying differentially
expressed genes (12). Advances in
second-generation DNA sequencing technologies has enabled digital
gene expression (DGE) profiling to overcome the drawbacks of the
microarray analysis hybridization process and allow the detection
of differential expression of low-abundance transcripts on a
genome-wide scale (13). In the
present study, 3′ tag DGE, Gene Ontology (GO) analysis, and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) were
used to analyze the transcriptional profiles of bone marrow
mononuclear cells (BMMCs) from Chinese children with ALL and those
without ALL. The results of the analysis revealed numerous gene
expression changes attributable to pathogenesis. By investigating
changes in the expression of genes, various novel candidate genes
required for ALL were identified.
Materials and methods
Patient samples
A total of 10 bone marrow (BM) tissue samples were
obtained from children [median age at diagnosis, 8.1 years (range,
3–13.4 years); 5 males and 5 females] newly diagnosed with
childhood ALL at Xiangya Hospital (Changsha, China) between May and
August 2010. In addition, 10 non-ALL BM samples were obtained from
patients who did not have leukemia or other malignancies, but who
were undergoing BM aspiration as part of their clinical care at
Xiangya Hospital [median follow-up time, 6.75 years (range 4–13.9
years)]. ALL diagnosis was based on the French-American-British
classification standards, and the morphology, immunology,
cytogenetics and molecular biology classification (14). Complete remission, refractory
disease and BM relapse were defined, according to the National
Cancer Institute (15). The
patient clinical characteristics are listed in Table I. The primary BMMCs from the
patients with ALL were analyzed using flow cytometry. The
cell-surface antigen staining was performed using a
PerCP-conjugated anti-CD45 antibody (cat. no. 304028; BioLegend,
Inc., San Diego, CA, USA). For FACS analysis, 5×105
cells were acquired and scored using flow cytometer (Gallios™;
Beckman Coulter, Inc., Brea, CA, USA), and the data were analyzed
using FlowJo software (version 8.7; Tree Star, Inc., Ashland, OR,
USA). The BMMCs were isolated by Ficoll density gradient
centrifugation at 671 x g for 20 min at room temperature
(Ficoll-Paque was obtained from GE Healthcare Life Sciences;
Uppsala, Sweden). The cells at the interface were removed and
washed twice with 30 ml sterile phosphate-buffered saline
(Well-Biology Co., Ltd., Changsha, China) at 671 x g for 5 min. The
isolated cells were counted using the trypan blue (Well-Biology
Co., Ltd.,) exclusion method and an inverted phase contrast
microscope (IMT-2; Olympus Corporation, Tokyo, Japan). The cells
were pelleted and maintained at −80°C until RNA extraction. The RNA
was extracted from frozen cell pellets using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. In accordance with the
Xiangya Hospital Committee on Human Research Review Ethics
Committee, written informed consent was obtained from the patients,
or from the parents or guardians, as appropriate.
 | Table IClinical characteristics of patients
with ALL. |
Table I
Clinical characteristics of patients
with ALL.
| Sample ID | Immunophenotype | Genetic subtype | Age at diagnosis
(years) | WBC count at
diagnosis (109 cells/l) | Event |
|---|
| Patient 1 | B-ALL | t(12;21) | 3.0 | 114.2 | CR1 |
| Patient 2 | B-ALL | t(12;21) | 13.0 | 23.3 | CR1 |
| Patient 3 | B-ALL | t(12;21) | 4.2 | 6.4 | CR1 |
| Patient 4 | B-ALL | HeH | 13.4 | 78.3 | Remission |
| Patient 5 | B-ALL | HeH | 3.7 | 3.5 | CR1 |
| Patient 6 | B-ALL | t(9;22) | 10.2 | 33.6 | CR1 |
| Patient 7 | T-ALL | T-ALL | 6.0 | 121.5 | CR1 |
| Patient 8 | T-ALL | T-ALL | 13.2 | 67.8 | DCR1 |
| Patient 9 | T-ALL | T-ALL | 2.5 | 42.5 | CR1 |
| Patient 10 | T-ALL | T-ALL | 10.8 | 23.6 | Remission |
Preparation of sequencing libraries and
sequencing
The total RNA sample was digested using DNaseI
(M0303S; New England Biolabs, Inc., Ipswich, MA, USA) and purified
using oligo-dT beads (Invitrogen Dynabeads mRNA Purification kit;
Thermo Fisher Scientific, Inc.), then poly(A)-containing mRNA was
fragmented into 130 bp using First-Strand buffer (Thermo Fisher
Scientific, Inc.). First-strand cDNA was generated using N6 primer,
First Strand Master mix and Super Script II Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.) with the following
reaction conditions: 25°C for 10 min, 42°C for 40 min and 70°C for
15 min. The Invitrogen Second Strand Master mix (Thermo Fisher
Scientific, Inc.) was then added to synthesize the second-strand
cDNA (at 16°C for 1 h). The cDNA was purified with Agencourt AMPure
XP beads (Beckman Coulter, Inc.), combined with Invitrogen End
Repair mix (Thermo Fisher Scientific, Inc.) and incubated at 20°C
for 30 min. Invitrogen A-Tailing mix (Thermo Fisher Scientific,
Inc.) was added, followed by incubation at 37°C for 30 min. The
Adenylate 3′ Ends DNA (Agilent Technologies, Inc., Santa Clara, CA,
USA), Illumina adapter (sequence, CATGIAAAAA; Illumina, Inc., San
Diego, CA, USA) and ligation mix (Invitrogen; Thermo Fisher
Scientific, Inc.) were combined and the ligate reaction was
incubated at 20°C for 20 min. Fifteen rounds of PCR amplification
were performed with PCR Primer Cocktail and PCR Master mix (both
Takara Bio, Inc., Shiga, Japan) to enrich the cDNA fragments. Then
the PCR products were purified with Agencourt AMPure XP beads.
Sequencing libraries were prepared from 6 µg total RNA using
the NlaIII Digital Gene Expression Tag Profiling kit
(Illumina, Inc.). The bead-bound cDNA was digested with
NlaIII and ligated with the Illumina adaptor (sequence,
CATGIAAAAA) containing an MmeI recognition site. The
adapter-ligated cDNA was digested with MmeI to release the
cDNA from the bead, retaining a 17-bp sequence in the fragment.
Following removal of the 3′ fragments via magnetic bead
precipitation, a second Illumina adaptor was ligated to the 3′ end
of the fragment to form a tagged library. Following 15 cycles of
linear PCR amplification, 95-bp fragments were purified by 6%
Tris-borate-EDTA polyacrylamide gel electrophoresis (Thermo Fisher
Scientific, Inc.). Following denaturation, the single-chain
molecules were adhered onto an Illumina Sequencing Chip (Illumina,
Inc.). In situ amplification was performed to expand each
molecule into a single-molecule cluster sequencing template.
Subsequently, four color-labeled nucleotides (Thermo Fisher
Scientific, Inc.) were added, and sequencing was performed using
the sequencing by synthesis method (16,17).
Each tunnel generated millions of raw reads with 35-bp sequencing
lengths.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from the BM using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. The ensuing RT-qPCR was performed
using an Access RT-PCR system (ISOGEN; Nippon Gene, Tokyo, Japan),
according to the manufacturer's protocol. qPCR was performed using
a 25-µl reaction mixture containing 12.5 µl 2X Premix
Ex Taq™ (Takara Bio, Inc.), 5 pmol primer and 6.5 µl cDNA,
obtained as described above. Amplification was performed on an
Applied Biosystem PRISM 7700 system (Thermo Fisher Scientific,
Inc.) and the PCR conditions were as follows: 50°C for 2 min, 95°C
for 15 min, and 45 cycles at 95°C for 30 sec and 60°C for 1 min,
followed by 25°C for 2 min. Quantification was determined by the
standard curve and the 2−ΔΔCq method (4). All primer sets were designed, as
described previously, synthesized by Biosearch Technologies
(Novato, CA, USA) and are presented in Table II.
| Table IIPrimers used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table II
Primers used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Tm (°C) | Product size
(bp) |
|---|
| WT1 |
CTATTCGCAATCAGGGTTA |
AGGTGGCTCCTAAGTTCAT | 55 | 312 |
| RPS26 |
TCCGTGCCTCCAAGATGA |
ACGCATCGGGCACAGTTA | 54 | 103 |
| MSX1 |
CACAAGACGAACCGTAAGCC |
ACCATATCTTCACCTGCGTCT | 56 | 154 |
| CD70 |
TCTCCCGCCTCCCGTAGCAT |
TGTCCTGCCACCACTACGC | 56 | 310 |
| HOXC4 |
CAGACCTCCAGAAATGACG |
GGGTAGACTATGGGTTGCTT | 57 | 518 |
| HOXA5 |
GCAGCACCCACATCAGCA |
CTTCTGCGGGTCAGGTAA | 58 | 274 |
| HOXC6 |
TTCCTACTTCACTAACCCTTCC |
TGCCCTGCTCAGAACTAAA | 54 | 323 |
| β-actin |
GGGTCAGAAGGATTCCTGTG |
GGTCTCAAACATGATCTGGG | 54 | 219 |
Gene annotation
The human transcriptome (Ensembl version 58;
http://asia.ensembl.org/) was used as the
reference sequence for sequence read alignment and identification.
The DGE tags were annotated by mapping the reads to the
sequence-flanking NlaIII restriction sites on the coding
strands. Alignment and candidate gene identification were
performed, as reported previously (18). The total expression profile for
each gene was then calculated by summing all the tags mapped to the
same gene, including intronic tags.
Statistical analysis
Data were analyzed using GraphPad Prism Software
(version 5.0; GraphPad Software, Inc, San Diego, CA, USA). Data are
presented as means ± standard deviation or means ± standard error
of the mean. Statistical analyses were performed using two-tailed
Student's t-tests for the differences between two groups and
two-way analysis of variance for the difference between multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
mRNA sample preparation and Illumina
genome analyses
In the present study, BMMC samples were collected
from pediatric patients with ALL (Table I) and from non-ALL individuals. The
cellular mRNA was extracted and DGE sequencing libraries were
prepared for sequencing in an Illumina Genome Analyzer, which
obtained 15,200,000–20,400,000 quality-filtered sequence reads
(tags) per sample. Any tags with an abundance of <2 tags per
million (TPM), those that mapped to >1 gene, or those that did
not match the Ensembl database reference sequence were omitted.
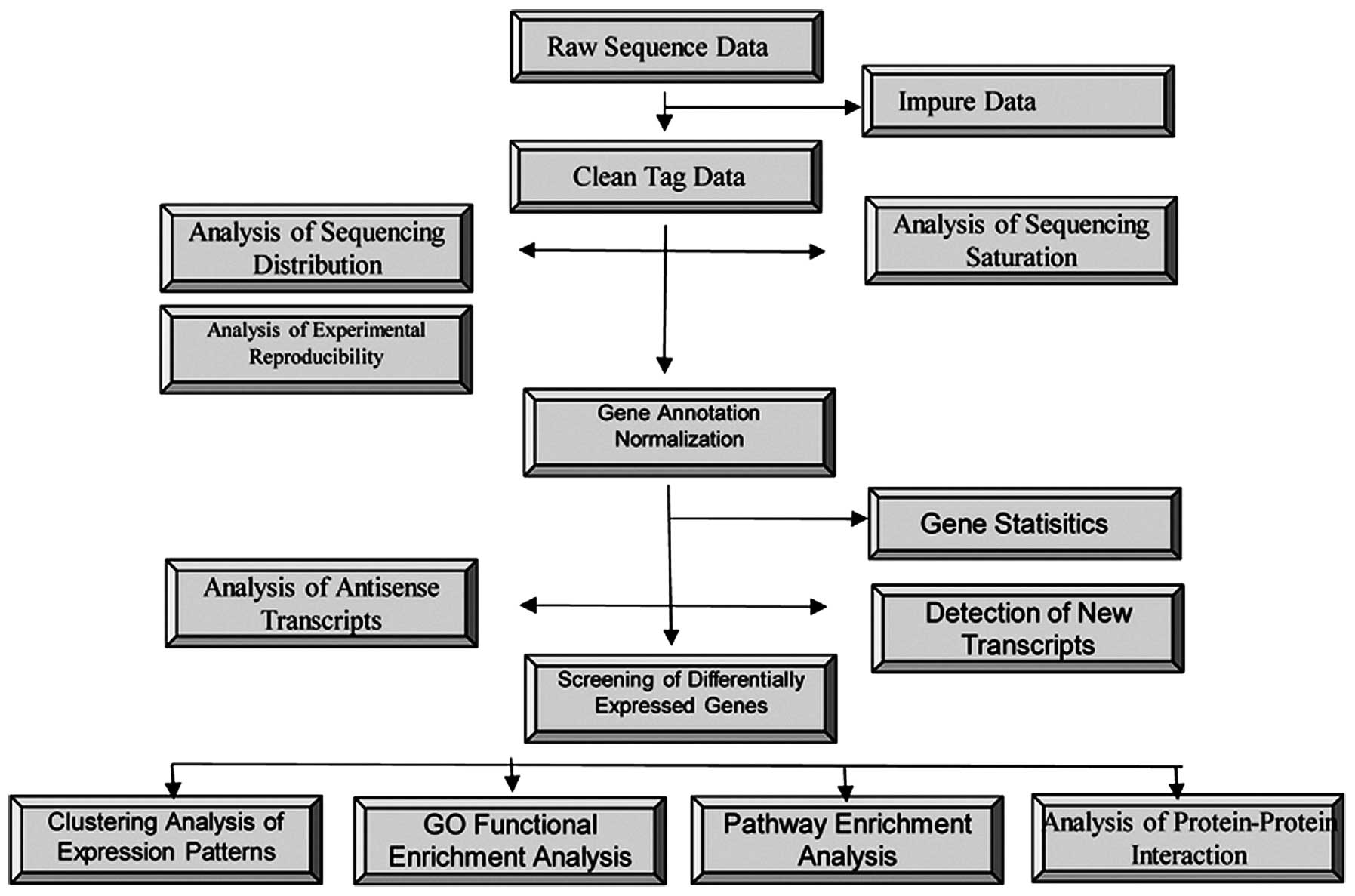
Following filtering, 25,000–53,000 unique nucleotide sequence tags
were obtained per library, which were mapped to the transcriptome
(Fig. 1).
Identification and functional
classification of differentially expressed genes
The differentially expressed genes were compared
between the patients with childhood ALL and the non-ALL
individuals. The selection criteria for putative differentially
expressed genes were as follows: i) Average fold change between ALL
and non-ALL groups ≥2; ii) single sample t-test false
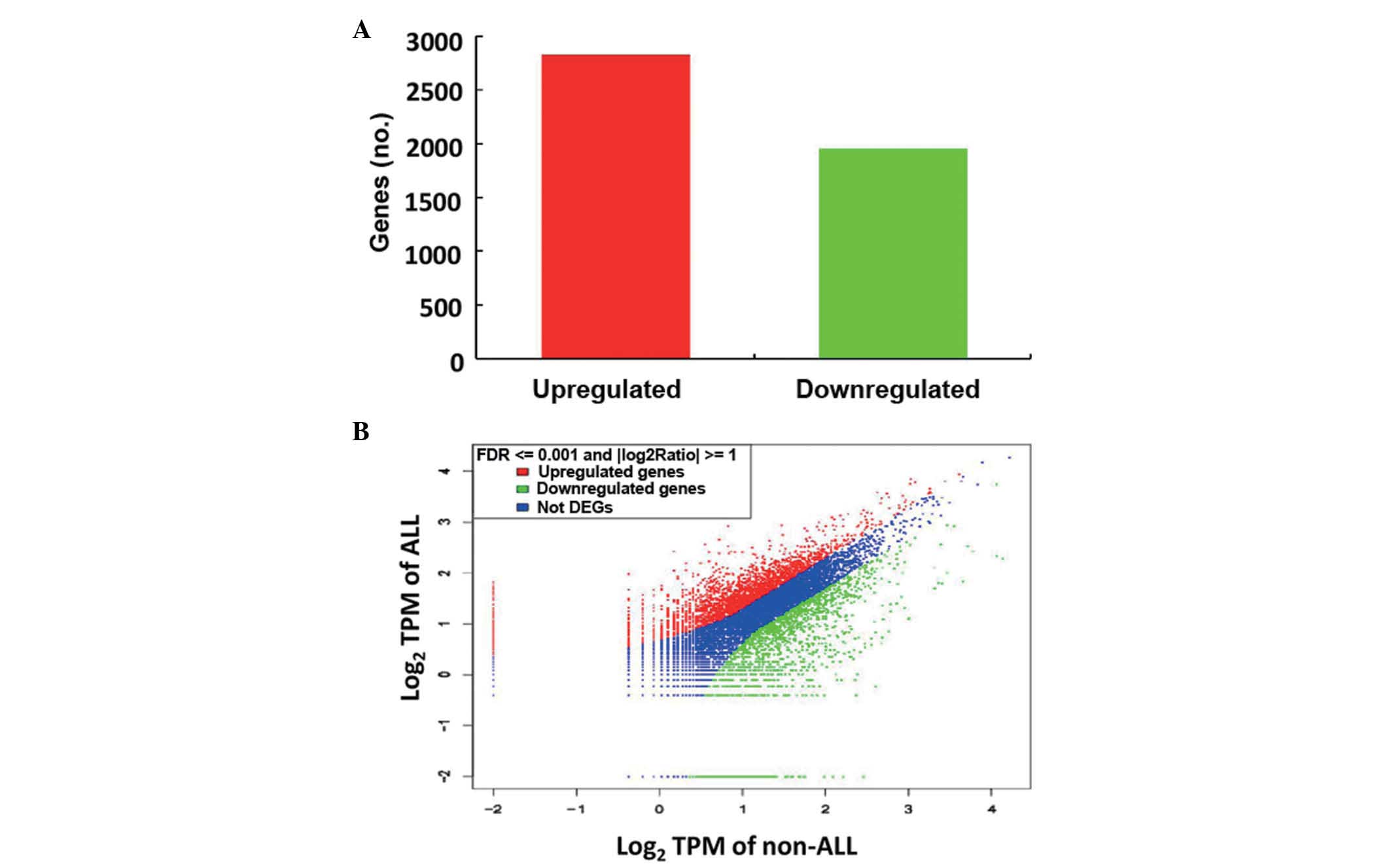
discovery rate <0.1%. In the ALL group, 2,825 genes were
upregulated and 1,952 genes were down-regulated (Fig. 2). Of the upregulated genes, 37 that
have been associated with the promotion of tumorigenesis, according
to Ensembl, were upregulated >10-fold in childhood ALL (Table III).
| Table IIICandidate genes upregulated 10-fold
in acute lymphoblastic leukemia. |
Table III
Candidate genes upregulated 10-fold
in acute lymphoblastic leukemia.
| Gene | log 2 ratio | Description | Function |
|---|
| HOXA11 | 12.69349 | Homeobox A11 | Endometrial
cancer |
| CCL1 | 12.50705 | Chemokine (C-C
motif) ligand 1 | CC chemokine
receptors, airway inflammation |
| WT1 | 12.05494 | Wilms' tumor 1 | Wilms' tumor,
Leukemia, uterine tumors |
| TWIST1 | 11.80292 | Twist homolog 1
(Drosophila) | Hepatocellular
carcinoma |
| SLITRK6 | 10.98513 | SLIT and NTRK-like
family, member 6 | Leading factor of
nerve growth |
|
HLA-DRB4 | 10.78627 | Major
histocompatibility complex, class II, DR β4 | Hepatitis,
leukocyte antigen, acute lymphoblastic leukemia |
| HOXC6 | 10.75322 | Homeobox C6 | Stem cell
differentiation for lymphocytes |
| ELN | 10.49685 | Elastin | Williams
syndrome |
| WIT1 | 10.20945 | Wilms' tumor
upstream neighbor 1 | Wilms' sarcoma
downstream factor |
| SYN1 | 10.00000 | synapsin I | Depression
neuroblastoma |
| CD70 | 9.911392 | CD70 molecule | Lymphocyte
antigen |
| CTHRC1 | 9.850187 | Collagen triple
helix repeat containing 1 | Gastric cancer,
liver cancer, colon cancer |
| GGT5 | 9.850187 |
γ-glutamyltransferase 5 | Liver cancer |
| PROCR | 9.575539 | Protein C receptor,
endothelial | Liver cancer |
| H19 | 9.370687 | H19, imprinted
maternally expressed transcript (non-protein coding) | Breast cancer,
cervical cancer, choriocarcinoma |
| HHIP | 9.326429 | Hedgehog
interacting protein | Pancreatic
cancer |
| LIF | 9.184875 | Leukemia inhibitory
factor (cholinergic differentiation factor) | Leukemia inhibitory
factor |
| HOXC4 | 9.184875 | Homeobox C4 | Stem cell
differentiation for lymphocytes |
| NCR3 | 9.134426 | Natural
cytotoxicity triggering receptor 3 | Multiple
myeloma |
| MSX1 | 9.134426 | Msh homeobox 1 | Apoptosis |
| ERBB2 | 9.027906 | v-erb-b2
erythroblastic leukemia viral oncogene homolog 2,
neuro/glioblastoma-derived oncogene homolog (avian) | Breast, stomach and
endometrial cancer, non-small cell lung cancer |
| LZTS1 | 8.851749 | leucine zipper,
putative tumor suppressor 1 | Primary esophageal
cancer |
| USP27X | 8.787903 | Ubiquitin specific
peptidase 27, X-linked | Ubiquitination |
|
HERV-FRD | 8.721099 | HERV-FRD provirus
ancestral Env polyprotein | Retrovirus |
| PMS2L1 | 8.573647 | Postmeiotic
segregation increased 2-like 1 pseudogene | Nasopharyngeal
carcinoma |
| IRX5 | 8.495855 | Iroquois homeobox
5 | Ovarian cancer |
| MKRN3 | 8.413628 | Makorin ring finger
protein 3 | Osteocarcinoma |
| IGFBP6 | 8.134426 | Insulin-like growth
factor binding protein 6 | Breast cancer |
| TRIM2 | 8.027906 | Tripartite
motif-containing 2 | Cancer cell
development |
| FZD7 | 7.912889 | Frizzled homolog 7
(Drosophila) | Wnt signal |
| NTRK1 | 7.912889 | Neurotrophic
tyrosine kinase, receptor, type 1 | Thyroid cancer |
| RPS26 | 7.912889 | Ribosomal protein
S26 | Non-Hodgkin's
lymphoma |
| MYCN | 6.665077 | v-myc
myelocytomatosis viral related oncogene, neuroblastoma derived
(avian) | Neuroblastoma |
| SPINK2 | 6.427893 | Serine peptidase
inhibitor, Kazal type 2 (acrosin-trypsin inhibitor) | Liver cancer |
| OLIG1 | 5.786659 | Oligodendrocyte
transcription factor 1 | Oligodendrocyte
tumor |
| HOXA7 | 5.578747 | Homeobox A7 | Epithelial ovarian
cancer |
| NAT14 | 5.022015 | N-acetyltransferase
14 (GCN5-related, putative) | Lung cancer |
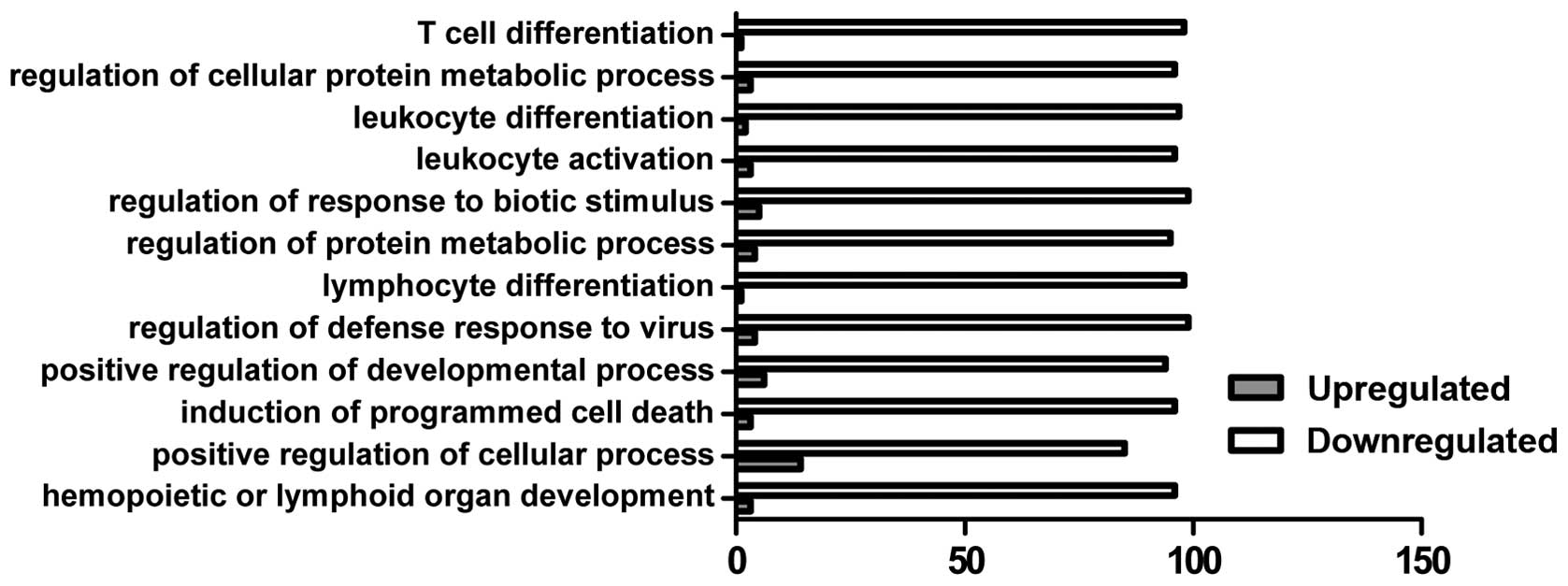
GO analysis
In the present study, GO analysis was performed by
mapping each differentially expressed gene into the GO database
(http://www.geneontology.org/). The
predominant functional group of upregulated genes was associated
with the positive regulation of the cellular process, whereas the
down-regulated genes were associated with multiple function groups
involved in immune cell differentiation, the metabolic process and
programmed cell death (Fig.
3).
RT-qPCR analysis of differentially
expressed genes
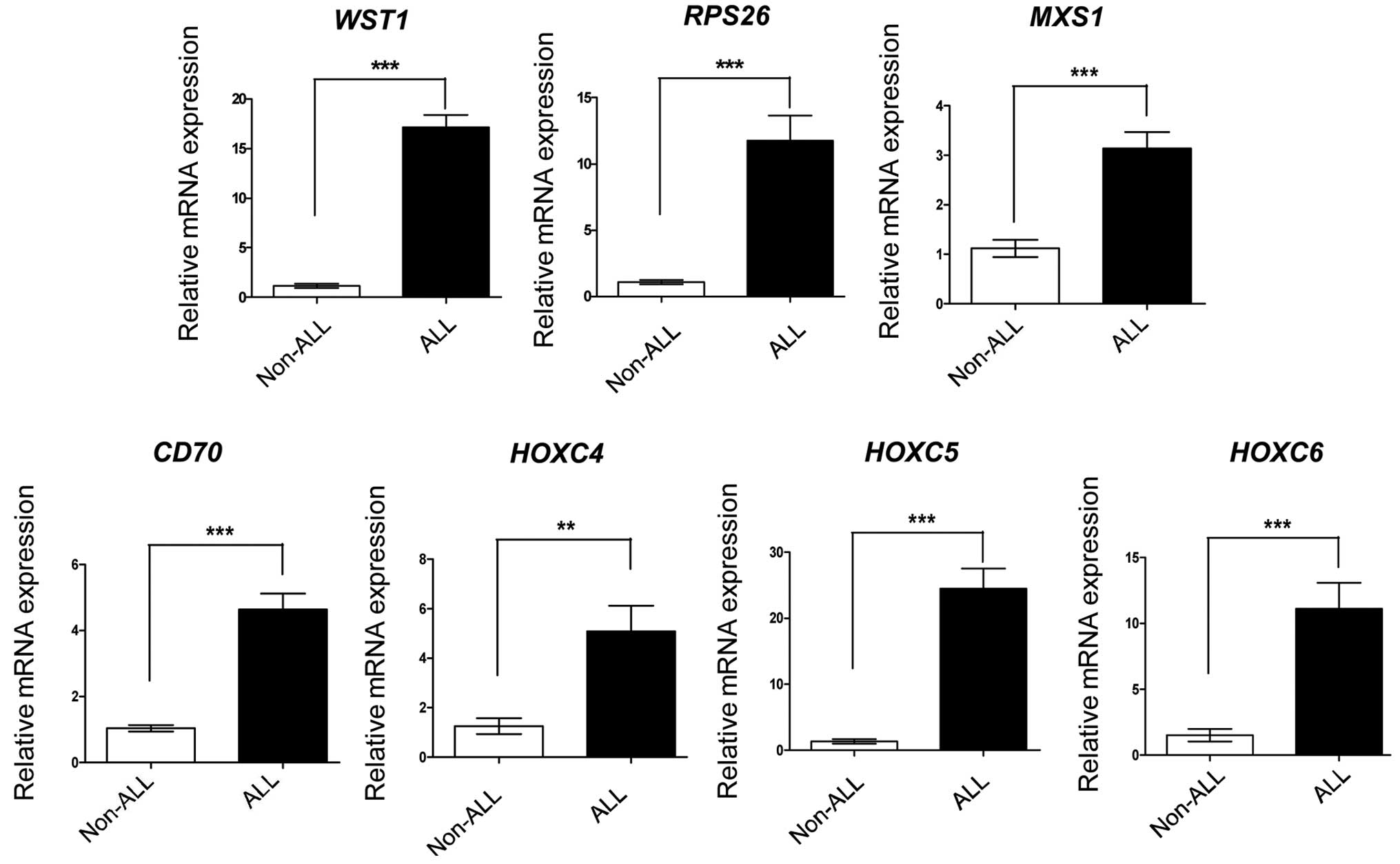
To further evaluate the DGE profiling reliability in
the present study, RT-qPCR analysis was performed for a subset of
seven genes, which had been determined by the DGE profiling as
upregulated >10-fold in the ALL group. The seven genes were
expressed at high levels in the ALL samples, and had the same
expression profiles as in the initial DGE profiling (Fig. 4). This result confirmed the
reliability of the DGE profiling.
Discussion
ALL is a multigenic disease with multiple subtypes;
recurrent copy number alterations and structural chromosomal
rearrangements characterize the disease. Although the clinical
perspective for the ALL subtypes is well established, the
underlying molecular mechanisms in the development of ALL
development remain to be fully elucidated Thus, char-acterizing
ALL-associated differentially expressed genes is important for
determining the molecular mechanisms of ALL and for early ALL
diagnosis. Several genomic-wide expression profiling approaches
have been used to identify the ALL-associated differentially
expressed genes (19). The
development of second-generation sequencing has enabled the use of
DGE profiling for determining genome-wide gene expression profiles
in ALL cell samples. For example, Nordlund et al used DGE
profiling to characterize the gene expression patterns in different
ALL subtypes (20), and found that
antisense tags expressed from the non-coding strand were also
expressed as a subtype-specific pattern. Additionally, other
studies have used microarray-based methods to profile ALL gene
expression patterns (10,11,21)
However, these previous studies were performed in Western
countries, and comparable information in Asian populations is
limited. To the best of our knowledge, the present study is the
first to use DGE profiling to determine gene expression patterns in
childhood ALL in a Chinese population.
The development of second-generation sequencing
technology has resulted in DGE profiling possessing several
advantages, compared with earlier methods of genome-wide expression
analysis (22) For example, it
requires smaller RNA samples and costs less than other
transcriptome sequencing methods, and it overcomes the limitations
of the hybridization process present in microarray-based methods.
In addition, computational calculation analysis of DGE data is less
challenging. In the present study, DGE profiling revealed that 37
genes were upregulated by >10-fold in the childhood ALL group,
compared with the non-ALL group. Notably, these genes are important
in tumorigenesis. For example, the wild-type WT1 is
expressed in breast cancer, renal cell cancer, ovarian cancer,
mesothelioma, lung cancer, melanoma and acute leukemia (23,24).
and high levels of WT1 are associated with poor prognosis in
ovarian cancer and leukemia (25,26)
Other genes, including HOXA11, HHIP, NCR3 and
ERBB2 are involved in the tumorigenesis of several types of
solid tumor (27–31). Thus, in addition to characterizing
the expression patterns of ALL-associated genes in the present
study, novel candidate genes have been identified, which may be
associated with ALL. In addition, GO analysis demonstrated that
these genes were predominantly involved in immune cell
differentiation, the metabolic process and programmed cell death,
suggesting the importance of these signaling pathways in ALL.
Future investigations to characterize the roles of these genes in
childhood ALL experimentally may further understanding.
In conclusion, DGE profiling was conducted to
determine the gene expression profile of childhood ALL in a Chinese
population, identifying 2,825 upregulated and 1,952 down-regulated
genes. Of these, 37 of the upregulated genes were upregulated by
>10-fold, and were found to be important in tumorigenesis. These
findings suggested that DGE profiling can provide novel genome-wide
information on gene expression in ALL.
Acknowledgments
The present study was supported by grants from The
National Natural Sciences Foundation of China (grant nos. 31171328
and 81370648 to Professor Li-Zhi Cao, 81270606 to Dr Yan Yu,
81100359 to Dr Ming-Hua Yang and 81400132 to Dr Shan Zhu) and the
Natural Science Foundation of Hunan Province, China (grant no.
2015JJ6118).
References
|
1
|
Biswas S, Chakrabarti S, Chakraborty J,
Paul PC, Konar A and Das S: Childhood acute leukemia in West
Bengal, India with an emphasis on uncommon clinical features. Asian
Pac J Cancer Prev. 10:903–906. 2009.
|
|
2
|
Burke PW and Douer D: Acute lymphoblastic
leukemia in adolescents and young adults. Acta Haematol.
132:264–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Awan T, Iqbal Z, Aleem A, Sabir N, Absar
M, Rasool M, Tahir AH, Basit S, Khalid AM, Sabar MF, et al: Five
most common prognos-tically important fusion oncogenes are detected
in the majority of Pakistani pediatric acute lymphoblastic leukemia
patients and are strongly associated with disease biology and
treatment outcome. Asian Pac J Cancer Prev. 13:5469–5475. 2012.
View Article : Google Scholar
|
|
4
|
Shaikh MS, Adil SN, Shaikh MU and Khurshid
M: Frequency of chromosomal abnormalities in Pakistani adults with
acute lymphoblastic leukemia. Asian Pac J Cancer Prev.
15:9495–9498. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao M, Yang M, Yang L, Yu Y, Xie M, Zhu
S, Kang R, Tang D, Jiang Z, Yuan W, et al: HMGB1 regulates
autophagy through increasing transcriptional activities of JNK and
ERK in human myeloid leukemia cells. BMB Rep. 44:601–606. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yeoh AE, Tan D, Li CK, Hori H, Tse E and
Pui CH; Asian Oncology Summit 2013: Management of adult and
paediatric acute lymphoblastic leukaemia in Asia:
Resource-stratified guidelines from the Asian Oncology Summit 2013.
Lancet Oncol. 14:e508–e523. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gaynon PS: Childhood acute lymphoblastic
leukaemia and relapse. Br J Haematol. 131:579–587. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kulkarni KP and Marwaha RK: Outcomes of
children with relapsed acute lymphoblastic leukemia in India. Asian
Pac J Cancer Prev. 12:1101–1102. 2011.PubMed/NCBI
|
|
9
|
Tharnprisan P, Khiewyoo J, Sripraya P and
Wiangnon S: Relapse-free rate with childhood acute lymphoblastic
leukemia treated under the thai national protocol. Asian Pac J
Cancer Prev. 14:1127–1130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soheila K, Hamid A, Farid Z, Mostafa RT,
Nasrin DN, Syyed-Mohammad T and Vahide T: Comparison of univariate
and multivariate gene set analysis in acute lymphoblastic leukemia.
Asian Pac J Cancer Prev. 14:1629–1633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou
X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, et al: Gene
expression profiling of pediatric acute myelogenous leukemia.
Blood. 104:3679–3687. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Unfried C, Burbach G, Korf HW and Von Gall
C: Melatonin receptor 1-dependent gene expression in the mouse pars
tuberalis as revealed by cDNA microarray analysis and in situ
hybridization. J Pineal Res. 48:148–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Zhang H, Li H and Miao X:
Second-generation sequencing supply an effective way to screen RNAi
targets in large scale for potential application in pest insect
control. PLoS One. 6:e186442011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bennett JM, Catovsky D, Daniel MT,
Flandrin G, Galton DA, Gralnick HR and Sultan C: Proposed revised
criteria for the classification of acute myeloid leukemia. A report
of the French-American-British Cooperative Group. Ann Intern Med.
103:620–625. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheson BD, Cassileth PA, Head DR, Schiffer
CA, Bennett JM, Bloomfield CD, Brunning R, Gale RP, Grever MR and
Keating MJ: Report of the National Cancer Institute-sponsored
workshop on definitions of diagnosis and response in acute myeloid
leukemia. J Clin Oncol. 8:813–819. 1990.PubMed/NCBI
|
|
16
|
't Hoen PA, Ariyurek Y, Thygesen HH,
Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ and
den Dunnen JT: Deep sequencing-based expression analysis shows
major advances in robustness, resolution and inter-lab portability
over five microarray platforms. Nucleic Acids Res. 36:e1412008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morrissy AS, Morin RD, Delaney A, Zeng T,
McDonald H, Jones S, Zhao Y, Hirst M and Marra MA: Next-generation
tag sequencing for cancer gene expression profiling. Genome Res.
19:1825–1835. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Ruan J and Durbin R: Mapping short
DNA sequencing reads and calling variants using mapping quality
scores. Genome Res. 18:1851–1858. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mullighan CG: Genomic profiling of
B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin
Haematol. 24:489–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nordlund J, Kiialainen A, Karlberg O,
Berglund EC, Göransson-Kultima H, Sønderkær M, Nielsen KL,
Gustafsson MG, Behrendtz M, Forestier E, et al: Digital gene
expression profiling of primary acute lymphoblastic leukemia cells.
Leukemia. 26:1218–1227. 2012. View Article : Google Scholar :
|
|
21
|
Figueroa ME, Chen SC, Andersson AK,
Phillips LA, Li Y, Sotzen J, Kundu M, Downing JR, Melnick A and
Mullighan CG: Integrated genetic and epigenetic analysis of
childhood acute lymphoblastic leukemia. J Clin Invest.
123:3099–3111. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mullighan CG: Genome sequencing of
lymphoid malignancies. Blood. 122:3899–3907. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Iranparast S, Assarehzadegan MA, Heike Y,
Hossienzadeh M and Khodadadi A: Wilms' tumor gene (WT1) expression
correlates with vascular epithelial growth factor (VEGF) in newly
acute leukemia patients undergoing chemotherapy. Asian Pac J Cancer
Prev. 15:9217–9223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang L, Han Y, Suarez Saiz F and Minden
MD: A tumor suppressor and oncogene: The WT1 story. Leukemia.
21:868–876. 2007.PubMed/NCBI
|
|
25
|
Bergmann L, Miething C, Maurer U, Brieger
J, Karakas T, Weidmann E and Hoelzer D: High levels of Wilms' tumor
gene (wt1) mRNA in acute myeloid leukemias are associated with a
worse long-term outcome. Blood. 90:1217–1225. 1997.PubMed/NCBI
|
|
26
|
Netinatsunthorn W, Hanprasertpong J,
Dechsukhum C, Leetanaporn R and Geater A: WT1 gene expression as a
prognostic marker in advanced serous epithelial ovarian carcinoma:
An immunohistochemical study. BMC Cancer. 6:902006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arteaga CL and Engelman JA: ERBB
receptors: From oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hwang JA, Lee BB, Kim Y, Park SE, Heo K,
Hong SH, Kim YH, Han J, Shim YM, Lee YS and Kim DH: HOXA11
hypermethylation is associated with progression of non-small cell
lung cancer. Oncotarget. 4:2317–2325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koch J, Steinle A, Watzl C and Mandelboim
O: Activating natural cytotoxicity receptors of natural killer
cells in cancer and infection. Trends Immunol. 34:182–191. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo LP, Han B, Yu XP, Chen XY, Zhou J,
Chen W, Zhu YF, Peng XL, Zou Q and Li SY: Anti-metastasis activity
of black rice anthocyanins against breast cancer: Analyses using an
ErbB2 positive breast cancer cell line and tumoral xenograft model.
Asian Pac J Cancer Prev. 15:6219–6225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song Y and Zuo Y: Occurrence of HHIP gene
CpG island methylation in gastric cancer. Oncol Lett. 8:2340–2344.
2014.PubMed/NCBI
|