Introduction
Hypertension is a common clinical condition that is
associated with high morbidity and mortality. The characteristics
of hypertension-associated cardiac remodeling include inflammation,
hypertrophy and fibrosis. Cardiac function deteriorates as the
fibrosis progresses, ultimately resulting in heart failure
(1). As inflammatory cells,
platelets are involved in the physiological and pathological
processes of numerous cardiovascular diseases. Following
activation, platelets promote cardiac inflammation and fibrosis in
conditions of angiotensin II (Ang II)-induced hypertension, and
these effects may be prevented by a purinergic receptor P2Y12
antagonist that inhibits platelet activation (2). However, the mechanisms that underlie
the role of platelet activation in initiating this process remain
to be elucidated.
Platelets are generally known as central mediators
in thrombosis and platelets are key in inflammation and immunity
(3). Platelets interact with
various types of leukocytes, including monocytes and neutrophils,
to induce a systemic inflammatory response (3,4).
Upon activation, platelets alter their shape and gene expression
pattern, expressing specific adhesion molecules (including
P-selectin, cluster of differentiation 40L, glycoprotein IIb and
tumor necrosis factor superfamily member 14) (4), and secreting numerous inflammatory
cytokines and chemokines [including interleukin (IL)-1, platelet
factor 4 and chemokine (C-C motif) ligand 5] (5). P-selectin is a key adhesion molecule,
it binds to its receptor P-selectin glyco-protein ligand-1
(PSGL-1), which is expressed on the surface of blood monocytes, and
mediates monocyte recruitment to sites of inflammation. The
monocytes become activated, and release numerous inflammatory
mediators (including monocyte chemoattractant protein-1, tumor
necrosis factor-α, IL-1β and IL-6), which in turn stimulate
platelets to secrete more activating factors (3). This activation loop repeats and
amplifies the inflammatory response. The present study hypothesized
that P-selectin secreted by activated platelets may be involved in
the inflammatory response associated with Ang II-induced
hypertensive cardiac inflammation and fibrosis.
Materials and methods
Ethics statement
The animals used in the present study were bred and
maintained in the Laboratory of Animal Experiments of Shanxi
Medical University. The mice were fed a standard diet and were used
in accordance with the US National Institutes of Health Guide for
the Care and Use of Laboratory Animals (6). The present study was approved by the
Institutional Animal Care and Use Committee of Shanxi Medical
University.
Animal models
Male P-sel knockout (KO) mice and wild-type (WT)
C57BL/6 littermates (age, 8 weeks; weight, 23–25 g; Jackson
Laboratory, Sacramento, CA, USA) were used for the experiments in
the present study. The mice were maintained under conditions of 50%
relative humidity, a 12/12 h light/dark cycle and 22°C, housed
separately with ad libitum access to food and water. The
mice were randomized into groups (10 mice/group in P-selectin
expression experiemtns, 6 mice/group in all other experiments) that
were subjected to different experimental conditions. A sodium
pentobarbital (50 mg/kg; Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) solution was delivered intraperitoneally
to anesthetize the animals. Ang II (Sigma-Aldrich, St. Louis, MO,
USA) was dissolved in a 0.01 N acetic acid saline solution (Tianjin
Chemical Experiment Plant, Tianjin, China), and osmotic minipumps
(Alzet Model 1007D; DURECT Corporation, Cupertino, CA, USA) infused
with Ang II or vehicle were inserted subcutaneously into the back
of the mice to deliver Ang II at a release rate of 1,500 ng/kg/min
for 7 days. All the treatments were well tolerated by the mice.
Blood pressure measurement
Systolic blood pressure (SBP) data were collected
using a computerized mouse tail-cuff system (BP-98A; Softron Co.,
Ltd., Tokyo, Japan). SBP was measured prior to the initiation of
the experiment and on days 4–7 of the Ang II infusion period. The
mean of ten repeated values was calculated for each analysis
point.
Echocardiography
The mice were anesthetized using isoflurane
inhalation (Sigma-Aldrich). Cardiac function was analyzed using the
Vevo 770 system (VisualSonics, Inc., Toronto, ON, Canada). The
heart images were acquired in 2D mode in the parasternal short-axis
view. The echocardiographic parameters were acquired in triplicate
in M-mode for all the mice.
Soluble P-selectin enzyme-linked
immunosorbent assay (ELISA)
The concentration of plasma, which was obtained by
centrifugation (1760 × g; 10 min; 4°C) of soluble P-selectin was
measured using a mouse P-selectin ELISA kit (ELM-Pselectin)
obtained from RayBiotech, Inc. (Norcross, GA, USA) according to the
manufacturer's protocols. The optical densities were read at a
wavelength of 450 nm using an ultraviolet spectrophotometer (Varian
Cary® 50; Agilent Technologies, Inc., Santa Clara, CA,
USA).
Bleeding time measurement
The tail transection method was used to measure the
bleeding time (7). Briefly, to
record the maximum bleeding time in 900 sec, the end of bleeding
was considered as the terminal point. Furthermore, the bleeding
time was continuously recorded when a new arrest lasting >30 sec
occurred and when the bleeding restarted within 30 sec.
Platelet isolation
Whole blood was collected from the hearts of
anesthetized donor mice and mixed with acid citrate dextrose (ACD;
Sigma-Aldrich) at a 9:1 ratio. The ACD-anticoagulated blood was
centrifuged at 200 × g for 7 min at room temperature to obtain
platelet-rich plasma (PRP). The PRP was centrifuged at 180 × g for
a further 5 min at room temperature. The platelets were washed,
centrifuged at 850 × g for 12 min at room temperature, and
resuspended in suspension buffer (minimal essential medium; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal calf serum (10099-141; Gibco; Thermo Fisher Scientific, Inc.)
and 300 ng/ml prostaglandin I2 (N5160; Sigma-Aldrich) at a final
concentration of 1×108 platelets/ml.
Platelet transplantation
The 8-week-old male recipient mice were
immunosuppressed using intraperitoneal busulfan, which also reduced
platelet count, for 5 days (40 mg/kg/day; Sigma-Aldrich). The
bleeding time of the recipients was recorded as described above.
The day prior to the operation, donor mouse blood samples (8 ml)
from the inner canthus were collected. A complete blood cell count
of the blood samples collected in EDTA-containing tubes was
performed using an automatic cell counter, and 1.5 ml
(108/ml) platelets were injected into the recipient mice
via the tail vein (8).
The division of the group in platelet
transplantation
The mice were divided into 5 groups of 6 mice, as
follows: i) WT recipient mice transplanted with WT platelets and
infused with Ang II (WT-WT, positive control); ii) P-sel KO
recipient mice transplanted with WT platelets and infused with Ang
II (KO-WT, to assess the role of platelet-derived P-selectin); iii)
WT recipient mice transplanted with P-sel KO platelets and infused
with Ang II (WT-KO, to assess the role of endothelium-derived
P-selectin); iv) P-sel KO recipient mice transplanted with P-sel KO
platelets and infused with Ang II (KO-KO, positive control); 5) WT
recipient mice transplanted with WT platelets and infused with
saline (WT-WT', negative control).
Tissue and histology preparation
All the mice were sacrificed on day 7 with
pentobarbital, and the hearts were collected, washed with a
heparin-containing saline solution (Sigma-Aldrich), fixed in 10%
formalin (Sigma-Aldrich), and embedded in paraffin (Sigma-Aldrich).
The paraffin-embedded fixed tissues were then cut into
5-µm-thick sections, placed on polylysine-coated glass
slides (HL-H05-2; Nantong Hailun Bio-Medical Apparatus
Manufacturing Co., Ltd., Haimen, China) and stained with
hematoxylin (Sigma-Aldrich) and Masson's trichrome reagent (Beijin
Solarbio Science & Technology Co., Ltd.). The fibrotic areas
were quantitated as the ratio of the area that had stained blue to
the total section area using the NIS-Elements analysis program
(Nikon Corporation, Tokyo, Japan). Immunohistochemistry was
performed using a standard procedure as previously described
(2). The sections were incubated
overnight at 4°C with antibodies against transforming growth factor
β1 (TGF-β1; rabbit polyclonal; sc-146; 1:200; Santa Cruz
Biotechnology, Inc., CA, USA), α-smooth muscle actin (α-SMA; rabbit
polyclonal; ab66133; 1:300; Abcam, Cambridge, MA, USA), and Mac-2
(galectin-3; rabbit polyclonal; sc-20157; 1:200; Santa Cruz
Biotechnology, Inc.). The images were captured using a microscope
equipped with a camera (ECLIPSE 80i/90i, Nikon Corporation).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (qPCR)
analysis
Total RNA was extracted from heart tissue in the 3D
peptide hydrogel using TRIzol according to the manufacturer's
protocols (Invitrogen; Thermo Fisher Scientific, Inc.). RNA was
quantified using the NanoDrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.). DNase I (18068-015; Invitrogen; Thermo Fisher
Scientific, Inc.) was then applied to 2 µg RNA prior to
reverse-transcription with MMLV reverse transcriptase from the
SuperScript® III First-Strand Synthesis System and oligo
(dT) primers (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. qPCR reactions were performed
on an iQ5 Real-Time PCR Detection system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) using SYBR Green I (Takara Bio, Inc.,
Otsu, Japan) with GAPDH serving as a control. The cycling
conditions were as follows: 95°C for 2 min, followed by 35 cycles
of 95°C for 30 sec and 60°C for 30 sec. The following primers
(9) were used to amplify the
fragments: Sense, 5′-GAGCGGAGAGTACTGGATCG-3′ and antisense,
5′-TACTCGAACGGGAATCCATC-3′ for collagen I; sense,
5′-GCCCTGGACACCAACTATTGC-3′ and antisense,
5′-GGAGCGCACGATCATGTTGG-3′ for TGF-β1; sense,
5′-GCAAACAGGAATACGACGAAGC-3′ and anti-sense,
5′-GCTTTGGGCAGGAATGATTTG-3′ for α-SMA; and sense,
5′-CCTGGAGAAACCTGCCAAGTATGA-3′ and antisense,
5′-AAGCAGGAATGAGAAGAGGCTGAG-3′ for GAPDH. The experiments were
repeated 3 times. The quantification cycle values (Cq values) were
used to calculate the fold differences using the 2−ΔΔCq method
(10).
Western blotting analysis
Protein was extracted from the heart tissue and
analyzed by western blotting as previously described (11). In brief, fresh hearts were lysed
with radioimmunoprecipitation assay lysis buffer and 1 mM
phenylmethane sulfonyl fluoride (Wuhan Boster Biological
Technology, Ltd., Wuhan, China). Protein samples (60 µg)
were separated by 10% sodium dodecyl sulfate-polyacrylimide gel
electrophoresis (100 mV, 90 min). Nonspecific proteins were blocked
by incubating the membrane with 5% non-fat dried milk in
Tris-buffered saline containing 0.1% Tween 20 for 1 h at room
temperature with agitation. Proteins were transferred from the gel
to nitrocellulose membranes, which were incubated overnight at 4°C
with primary antibodies against GAPDH (rabbit polyclonal; CW0101M;
1:5,000; Beijing ComWin Biotech Co., Ltd., Beijing, China), α-SMA
(1:1,000; Abcam), or TGF-β1 (1:1,000; Santa Cruz Biotechnology,
Inc.). They were then incubated at room temperature for 1 h with
mouse anti-rabbit HRP-conjugated IgG secondary antibodies (sc-2357;
1:5,000; Santa Cruz Biotechnology, Inc.). Images were captured and
quantified using a Bio-Rad ChemiDoc XRS system (Bio-Rad
Laboratories, Inc.) with Image Lab software, version 2.2 (Bio-Rad
Laboratories, Inc.), and protein expression levels were normalized
to GAPDH expression.
Statistical analyses
The unpaired Student's t-test was utilized to
compare two groups and analysis of variance was performed to
compare several groups of animals from the transplant experiments.
The Mann-Whitney U test was applied to analyze the bleeding time
data. SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA) was used to
analyze the data. The data are presented as the mean ± standard
error of the mean and P<0.05 was considered to indicate a
statistically significant difference.
Results
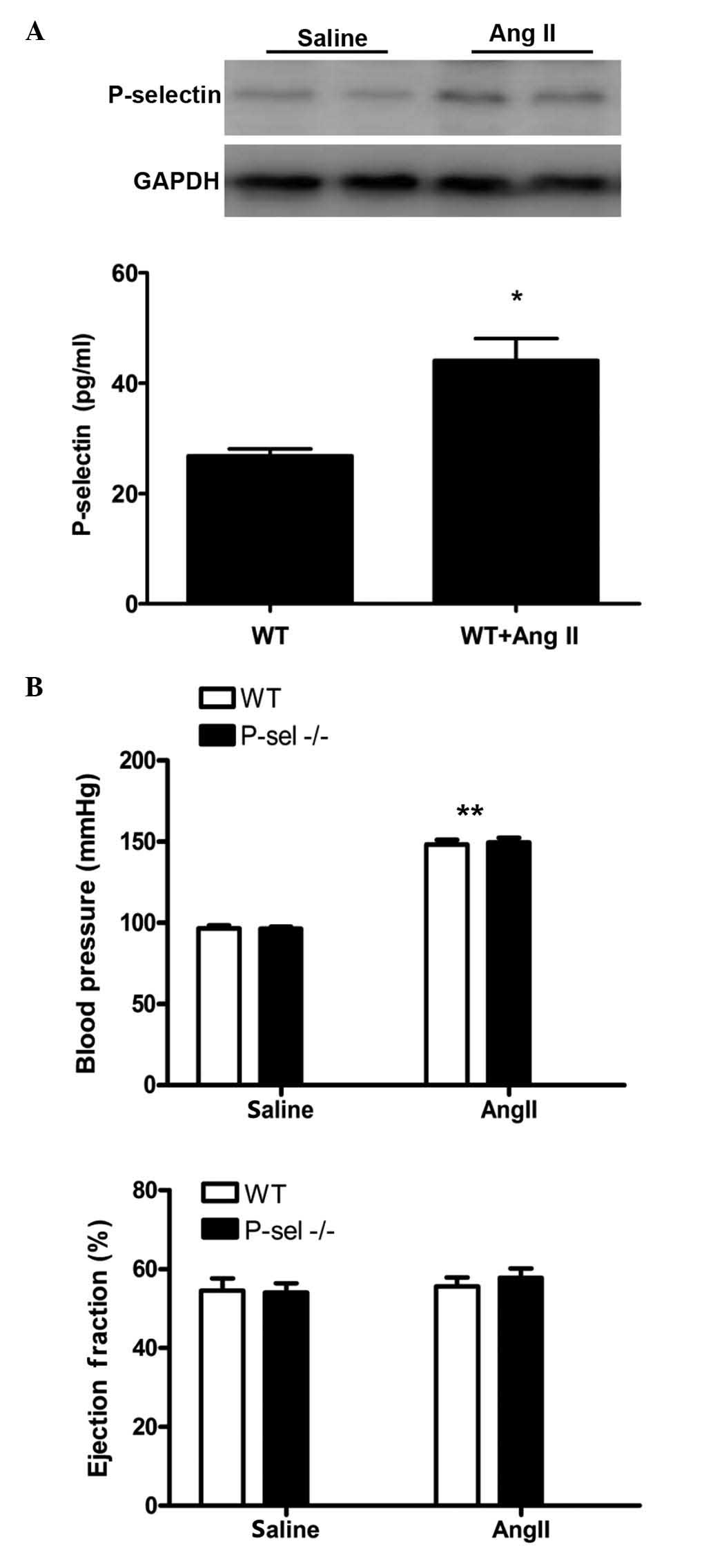
Increased P-selectin expression levels
were observed in the myocardium and plasma of Ang II-induced
hypertensive mice
To investigate the regulation of P-selectin
expression in Ang II-induced hypertension, WT mice were infused
with Ang II at a dose of 1,500 ng/kg/min for 7 days (12,13).
P-selectin expression was assessed by western blotting of protein
from myocardial samples and by ELISA for plasma samples. P-selectin
expression levels were markedly increased in the myocardium and
significantly increased in plasma samples following Ang II infusion
compared with saline infusion (P<0.01; Fig. 1A). The SBP was elevated in the WT
and P-sel KO groups following Ang II infusion compared with saline
infusion (P<0.001). However, no differences in the SBP were
observed between the WT and P-sel KO mice. The left ventricular
ejection fraction in the two groups (Fig. 1B) remained unchanged following Ang
II infusion. Thus, P-selectin deficiency did not influence SBP or
cardiac function after 7 days of Ang II infusion.
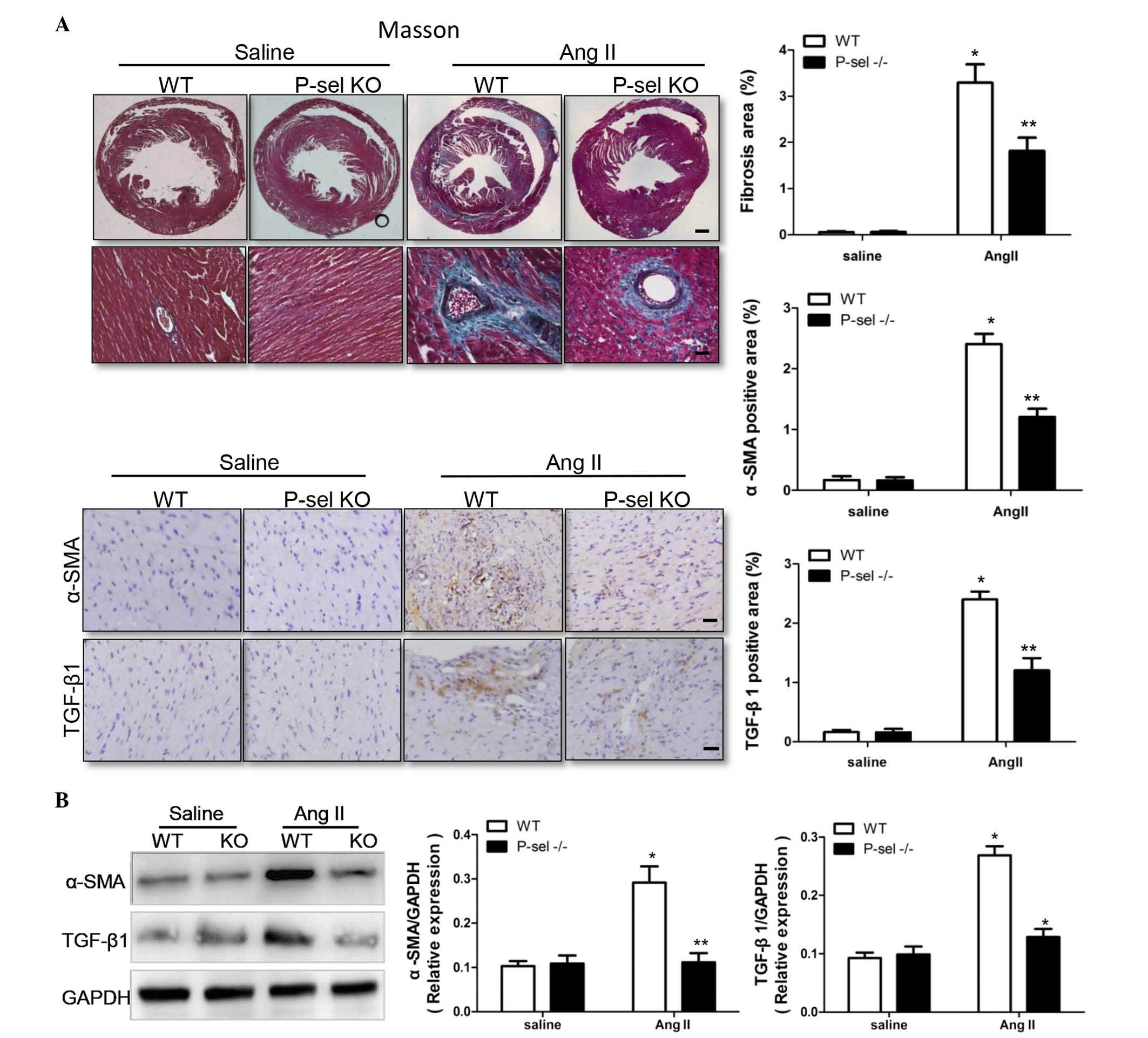
P-selectin deficiency decreases cardiac
fibrosis
P-sel KO mice were used to examine the effect of
P-selectin on Ang II-induced cardiac fibrosis, as assessed using
Masson's trichrome staining. Compared with saline infusion, the
fibrotic areas were significantly (P<0.05) larger in the WTP-sel
KO hearts following Ang II infusion (Fig. 2A). TGF-β1 is an important signal
transduction molecule in cardiac fibrosis (14,15),
and α-SMA increases during the differentiation of fibroblasts into
myofibroblasts. TGF-β1 and α-SMA levels were markedly higher in WT
hearts compared with P-sel KO hearts following Ang II infusion, as
indicated by immunohistochemistry (Fig. 2A) and by western blotting (Fig. 2B). Thus, P-selectin is critical in
cardiac fibrosis in response to Ang II infusion.
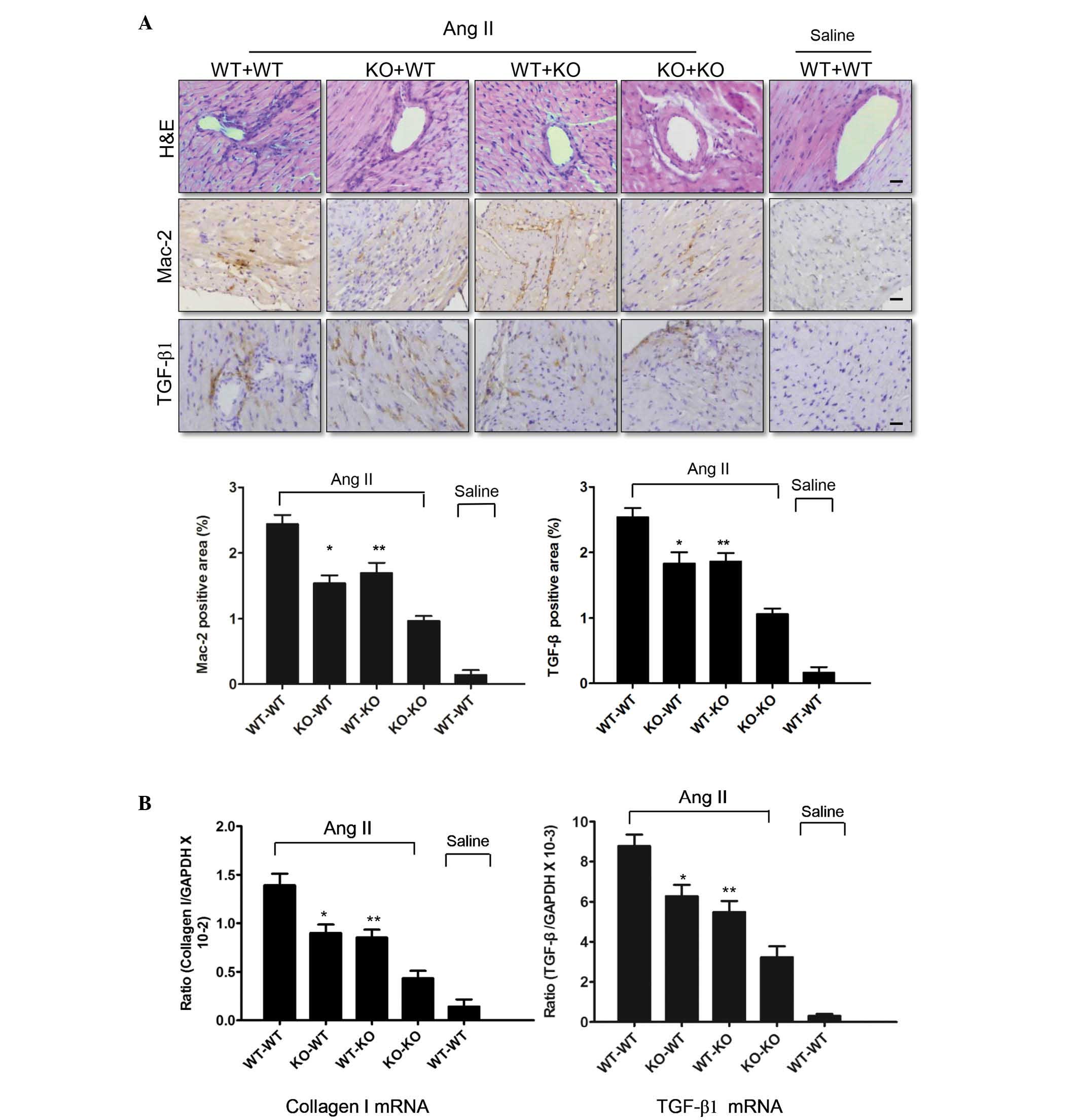
Platelet-secreted P-selectin is important
in the response to Ang II stimulation
A platelet cross-transplantation system was used to
determine whether P-selectin predominantly originates from
platelets in hypertension-associated cardiac inflammation and
fibrosis. Platelets [1.5 ml (108/ml)] were injected into
the tail vein of the recipient mice, which were treated with
busulfan for 5 days. A marked difference in cardiac fibrosis was
observed between the KO-WT and KO-KO groups, in addition to between
the WT-KO and KO-KO groups. As presented in Fig. 3A, more inflammatory cells
infiltrated the hearts of Ang II-infused KO-WT and WT-KO mice than
those of Ang II-infused KO-KO mice. Immunohistochemistry
demonstrated that the levels of TGF-β1 and the number of
Mac-2-positive macrophages in the KO-WT and WT-KO mice were
significantly (P<0.05) lower compared with the WT-WT mice
infused with Ang II (Fig. 3A), but
significantly greater (P<0.05) compared with the KO-KO mice.
qPCR analysis demonstrated that P-selectin deficiency decreased
TGF-β1 and collagen I expression (Fig.
3B). Thus, platelet-derived P-selectin increased Ang II-induced
cardiac inflammation and fibrosis. However, endothelial
cell-derived P-selectin is also involved in hypertension-associated
cardiac inflammation and fibrosis.
Discussion
There is accumulating evidence that P-selectin is a
key cytokine in cardiovascular disease (16). A clinical trial demonstrated that
P-selectin levels were markedly higher in the plasma of
hypertensive patients (17). The
results in the current study are consistent with this clinical
observation. The plasma levels of P-selectin in WT mice infused
with Ang II were significantly higher than those in the control
group (P<0.01). In addition, myocardial P-selectin expression
was increased in the Ang II-infused group. Thus, Ang II may
stimulate the secretion of P-selectin, which exists in a soluble
form in the plasma and heart. Following Ang II infusion, P-selectin
expression was induced by the type 1 angiotensin II (AT1) receptor,
and this upregulation may be inhibited by the AT1 receptor blocker,
valsartan, in hypertensive patients (18). However, the source of soluble
P-selectin remains to be elucidated. Plasma soluble P-selectin
levels were associated with plasma platelet count and
platelet-associated protein expression. However, in a previous
study, no association was observed between the expression levels of
P-selectin and von Willebrand factor (vWF), which is a marker of
activated endothelial cells (19).
It was identified that vWF was stored in Weibl-Palade bodies in the
endothelial cells, and was secreted into the plasma once the
endothelial cells were activated (19). Thus, the majority of the soluble
P-selectin was likely secreted by platelets.
Larsson et al (20) observed elevated blood pressure and
platelet activation following injection of Ang II into healthy
volunteers. Reciprocal activation between platelets and leukocytes
promotes the secondary capture of leukocytes (21). Selectin is the predominant
leukocyte adhesion molecule. Wang et al (22) demonstrated that impaired leukocyte
adhesion in P-sel KO mice was ameliorated by the binding of soluble
P-selectin and its ligand, PSGL-1. Myocardial P-selectin expression
may be mediated by adhesive leukocytes in the blood. Activated
leukocytes express PSGL-1, which binds to P-selectin expressed on
endothelial cells or platelet surfaces. These leukocytes then
invade cardiac tissue through gaps between endothelial cells to
exert pro-inflammatory effects.
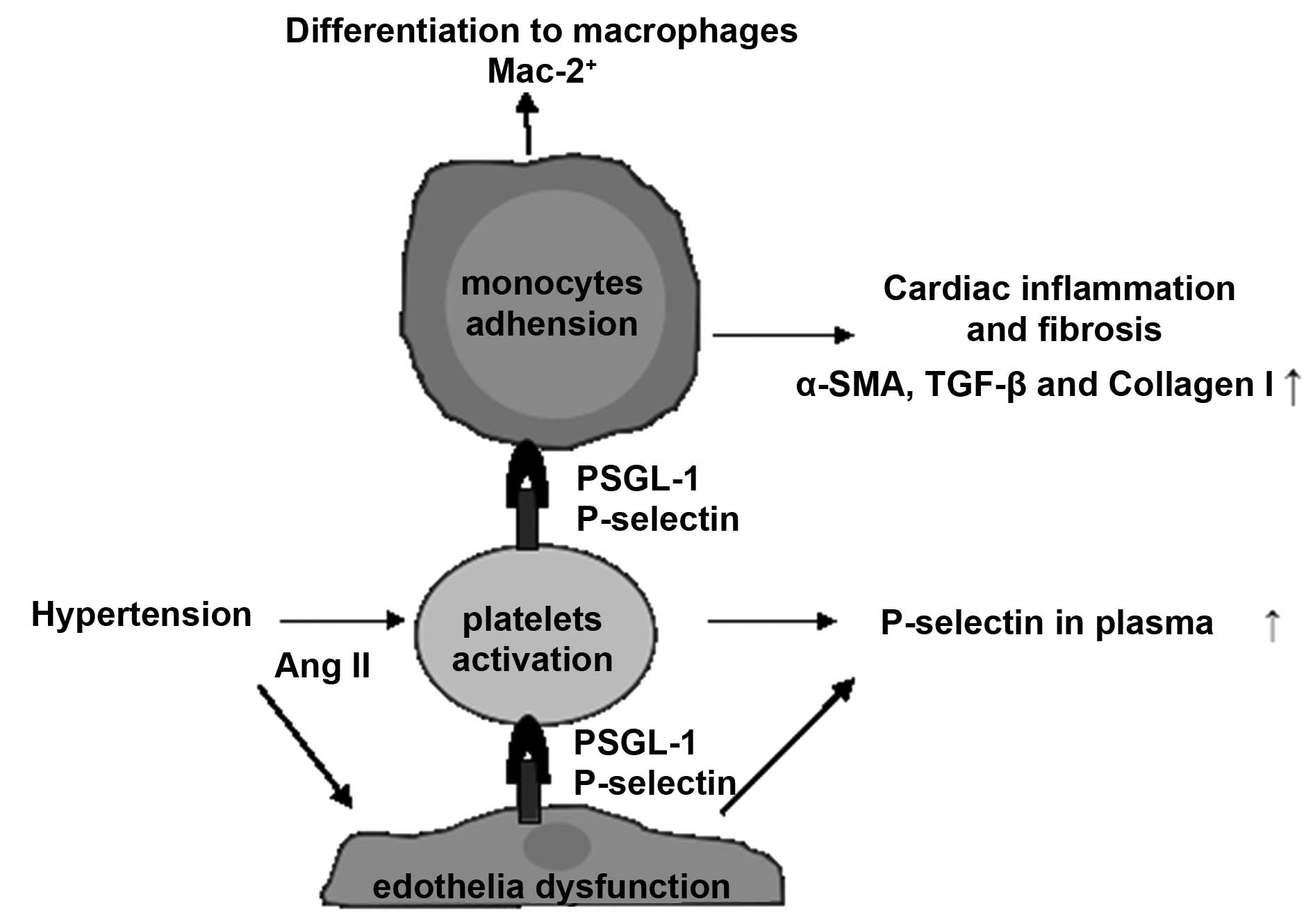
Fig. 4 summarizes
the signaling pathways involved in the present study. Platelet
activation in response to Ang II was recently reported to be an
early event that stimulates platelet-leukocyte conjugation and
inflammatory cell recruitment into the heart, resulting in cardiac
fibrosis (2). However, the
mechanism by which platelets mediate inflammation remained to be
elucidated. Results from the present study demonstrate direct
evidence that P-selectin, a marker of platelet activation, is
involved in Ang II-induced hypertension-associated cardiac
inflammation and fibrosis. Western blotting and
immunohistochemistry demonstrated that the expression levels of the
profibrotic cytokine, TGF-β1, and the fibroblast differentiation
marker, α-SMA, was decreased in the P-sel KO mice, suggesting that
P-selectin is key in cardiac fibrosis following Ang II infusion.
Consistently, the number of Mac-2-positive inflammatory cells,
which are upregulated under inflammatory conditions, were markedly
reduced in the P-sel KO mice (KO-KO vs. WT-WT), indicating that
P-selectin deficiency decreased Ang II-induced cardiac
inflammation. However, cardiac function was not affected after 7
days of Ang II infusion in the P-sel KO mice, which was consistent
with previous studies demonstrating that cathepsin S, intercellular
adhesion molecule 1 and IL-6 deficiencies did not alter the Ang
II-induced elevated blood pressure and cardiac dysfunction
(11,23). A 4-week-long Ang II infusion period
induced cardiac hypertrophy, remodeling and dysfunction in mice
(24,25), whereas short-term Ang II infusion
(7 days) may induce cardiac inflammation but is insufficient to
result in cardiac dysfunction. The focus of the current study was
to investigate the role of platelet P-selectin in the early stage
of hypertensive cardiac inflammation and fibrosis, thus, a
short-term Ang II infusion animal model was selected.
Clinical and animal studies have reported platelet
activation and inflammation in hypertensive mice (26) and patients (27–29).
Injection of Ang II may induce platelet activation in humans and
mice (16,26). The binding of P-selectin to PSGL-1
mediates platelet-leukocyte conjugation in early-stage myocardial
infarction, resulting in cardiac inflammation and remodeling
(30). In a previous study on the
murine cutaneous Arthus reaction, platelets regulated leukocyte
recruitment in a P-selectin/PSGL-1 interaction-dependent manner
(31). Numerous studies have
illustrated that circulating platelet activation and
platelet-leukocyte conjugation are key inflammatory mediators.
P-selectin-deficient conditions may be rescued with WT platelets.
In the present study, using platelet cross-transplantation
demonstrated that the P-selectin involved in Ang II-induced
hypertension-associated cardiac inflammation and fibrosis
predominantly originated from platelets. The KO-WT group (injection
of activated WT platelets) exhibited increased inflammatory cells
and fibrotic tissue compared with the KO-KO group (injection of
P-sel KO platelets) but the fibrotic tissue was reduced compared
with the WT-WT group.
Monocyte recruitment is regulated by activated
platelets and platelet-derived P-selectin in response to Ang
II-induced hypertension. Monocytes are recruited into the cardiac
tissue, where they transform into macrophages, which secrete
numerous cytokines and elicit fibroblast differentiation into
myofibroblasts within the myocardial infarction area (32–34).
The activated myofibroblasts release an abundance of extracellular
matrix proteins, including collagen I/III and fibronectin, thus
inducing cardiac fibrosis (35–37).
The WT-KO group, which predominantly demonstrated
the effects of endothelial cell-derived P-selectin, exhibited a
similar trend to the KO-WT group. This suggests that in addition to
platelets, endothelial cells and endothelial cell-derived
P-selectin affects the hypertension-associated cardiac fibrosis.
The Ang II-induced elevated blood pressure resulted in platelet
activation and endothelial dysfunction, leading to P-selectin
secretion into the plasma. Similarly, in atherosclerosis, platelet-
and endothelial cell-derived P-selectin promote the progression of
atherosclerotic lesion development (8).
In conclusion, P-selectin expression levels were
increased in myocardium and plasma samples from Ang II-infused
mice, and P-selectin deficiency reduced cardiac inflammation and
fibrosis. P-selectin produced by activated platelets and by
endothelial cells during Ang II stimulation resulted in the
development of hypertension-associated cardiac inflammation and
fibrosis.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant no.
81070090).
References
|
1
|
Marvar PJ, Thabet SR, Guzik TJ, Lob HE,
McCann LA, Weyand C, Gordon FJ and Harrison DG: Central and
peripheral mechanisms of T-lymphocyte activation and vascular
inflammation produced by angiotensin II-induced hypertension. Circ
Res. 107:263–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jia LX, Qi GM, Liu O, Li TT, Yang M, Cui
W, Zhang WM, Qi YF and Du J: Inhibition of platelet activation by
clopidogrel prevents hypertension-induced cardiac inflammation and
fibrosis. Cardiovasc Drugs Ther. 27:521–530. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mantovani A and Garlanda C:
Platelet-macrophage partnership in innate immunity and
inflammation. Nat Immunol. 14:768–770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Gils JM, Zwaginga JJ and Hordijk PL:
Molecular and functional interactions among monocytes, platelets,
and endothelial cells and their relevance for cardiovascular
diseases. J Leukoc Biol. 85:195–204. 2009. View Article : Google Scholar
|
|
5
|
von Hundelshausen P and Weber C: Platelets
as immune cells: Bridging inflammation and cardiovascular disease.
Circ Res. 100:27–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
National Research Council (US) Institute
for Laboratory Animal Research: Guide for the Care and Use of
Laboratory Animals. National Academies Press; Washington, DC: pp.
85–23. 1996
|
|
7
|
Gresele P, Momi S, Berrettini M, Nenci GG,
Schwarz HP, Semeraro N and Colucci M: Activated human protein C
prevents thrombin-induced thromboembolism in mice. Evidence that
activated protein c reduces intravascular fibrin accumulation
through the inhibition of additional thrombin generation. J Clin
Invest. 101:667–676. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burger PC and Wagner DD: Platelet
P-selectin facilitates atherosclerotic lesion development. Blood.
101:2661–2666. 2003. View Article : Google Scholar
|
|
9
|
Huang XR, Chung AC, Yang F, Yue W, Deng C,
Lau CP, Tse HF and Lan HY: Smad3 mediates cardiac inflammation and
fibrosis in angiotensin II-induced hypertensive cardiac remodeling.
Hypertension. 55:1165–1171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
11
|
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H,
Qi Y and Du J: Macrophage-stimulated cardiac fibroblast production
of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis
induced by angiotensin II. PLoS One. 7:e351442012. View Article : Google Scholar
|
|
12
|
Usher MG, Duan SZ, Ivaschenko CY, Frieler
RA, Berger S, Schütz G, Lumeng CN and Mortensen RM: Myeloid
mineralocorticoid receptor controls macrophage polarization and
cardiovascular hypertrophy and remodeling in mice. J Clin Invest.
120:3350–3364. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Totani L and Evangelista V:
Platelet-leukocyte interactions in cardiovascular disease and
beyond. Arterioscler Thromb Vasc Biol. 30:2357–2361. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Honsho S, Nishikawa S, Amano K, Zen K,
Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K,
et al: Pressure-mediated hypertrophy and mechanical stretch induces
IL-1 release and subsequent IGF-1 generation to maintain
compensative hypertrophy by affecting Akt and JNK pathways. Circ
Res. 105:1149–1158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bujak M, Dobaczewski M, Chatila K, Mendoza
LH, Li N, Reddy A and Frangogiannis NG: Interleukin-1 receptor type
I signaling critically regulates infarct healing and cardiac
remodeling. Am J Pathol. 173:57–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo L, Sun G, Wang G, Ning W and Zhao K:
Soluble P-selectin promotes acute myocardial infarction onset but
not severity. Mol Med Rep. 11:2027–2033. 2015.
|
|
17
|
Spencer CG, Gurney D, Blann AD, Beevers DG
and Lip GY; ASCOT Steering Committee Anglo-Scandinavian Cardiac
Outcomes Trial: Von willebrand factor, soluble P-selectin, and
target organ damage in hypertension: A substudy of the
angloscandinavian cardiac outcomes trial (ASCOT). Hypertension.
40:61–66. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Piqueras L, Kubes P, Alvarez A, O'Connor
E, Issekutz AC, Esplugues JV and Sanz MJ: Angiotensin II induces
leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2)
receptor-mediated P-selectin upregulation. Circulation.
102:2118–2123. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kawai Y and Montgomery RR: Endothelial
cell processing of von Willebrand proteins. Ann N Y Acad Sci.
509:60–70. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Larsson PT, Schwieler JH and Wallén NH:
Platelet activation during angiotensin II infusion in healthy
volunteers. Blood Coagul Fibrinolysis. 11:61–69. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuebler WM: Selectins revisited: The
emerging role of platelets in inflammatory lung disease. J Clin
Invest. 116:3106–3108. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL,
Xu T, Huo Y, Zhu X, Plow EF, Chen M and Geng JG: P-selectin primes
leukocyte integrin activation during inflammation. Nat Immunol.
8:882–892. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan L, Li Y, Jia L, Qin Y, Qi G, Cheng J,
Qi Y, Li H and Du J: Cathepsin S deficiency results in abnormal
accumulation of autophagosomes in macrophages and enhances Ang
II-induced cardiac inflammation. PLoS One. 7:e353152012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dai DF, Johnson SC, Villarin JJ, Chin MT,
Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla
TA, et al: Mitochondrial oxidative stress mediates angiotensin
II-induced cardiac hypertrophy and Galphaq overexpression-induced
heart failure. Circ Res. 108:837–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Izumiya Y, Kim S, Izumi Y, Yoshida K,
Yoshiyama M, Matsuzawa A, Ichijo H and Iwao H: Apoptosis
signal-regulating kinase 1 plays a pivotal role in angiotensin
II-induced cardiac hypertrophy and remodeling. Circ Res.
93:874–883. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bujak M, Dobaczewski M, Chatila K, Mendoza
LH, Li N, Reddy A and Frangogiannis NG: Interleukin-1 receptor type
I signaling critically regulates infarct healing and cardiac
remodeling. Am J Pathol. 173:57–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferroni P, Guagnano MT, Falco A, Paoletti
V, Manigrasso MR, Michetti N, Santilli F, Guadagni F, Basili S and
Davì G: Association of low-grade inflammation and platelet
activation in patients with hypertension with microalbuminuria.
Clin Sci (Lond). 114:449–455. 2008. View Article : Google Scholar
|
|
28
|
Davì G, Gresele P, Violi F, Basili S,
Catalano M, Giammarresi C, Volpato R, Nenci GG, Ciabattoni G and
Patrono C: Diabetes mellitus, hypercholesterolemia, and
hypertension but not vascular disease per se are associated with
persistent platelet activation in vivo. Evidence derived from the
study of peripheral arterial disease. Circulation. 96:69–75. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nadar SK, Blann AD, Kamath S, Beevers DG
and Lip GY: Platelet indexes in relation to target organ damage in
high-risk hypertensive patients: A substudy of the
anglo-scandinavian cardiac outcomes trial (ASCOT). J Am Coll
Cardiol. 44:415–422. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y, Gao XM, Fang L, Jennings NL, Su Y,
Q X, Samson AL, Kiriazis H, Wang XF, Shan L, et al: Novel role of
platelets in mediating inflammatory responses and ventricular
rupture or remodeling following myocardial infarction. Arterioscler
Thromb Vasc Biol. 31:834–841. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hara T, Shimizu K, Ogawa F, Yanaba K,
Iwata Y, Muroi E, Takenaka M, Komura K, Hasegawa M, Fujimoto M and
Sato S: Platelets control leukocyte recruitment in a murine model
of cutaneous arthus reaction. Am J Pathol. 176:259–269. 2010.
View Article : Google Scholar :
|
|
32
|
Larsen E, Celi A, Gilbert GE, Furie BC,
Erban JK, Bonfanti R, Wagner DD and Furie B: PADGEM protein: A
receptor that mediates the interaction of activated platelets with
neutrophils and monocytes. Cell. 59:305–312. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Theilmeier G, Lenaerts T, Remacle C,
Collen D, Vermylen J and Hoylaerts MF: Circulating activated
platelets assist THP-1 monocytoid/endothelial cell interaction
under shear stress. Blood. 94:2725–2734. 1999.PubMed/NCBI
|
|
34
|
Mollmann H, Nef HM and Troidl C: Turning
the right screw: Targeting the interleukin-6 receptor to reduce
unfavourable tissue remodelling after myocardial infarction.
Cardiovasc Res. 87:395–396. 2010. View Article : Google Scholar
|
|
35
|
Staufenberger S, Jacobs M, Brandstatter K,
Hafner M, Regitz-Zagrosek V, Ertl G and Schorb W: Angiotensin II
type 1 receptor regulation and differential trophic effects on rat
cardiac myofibroblasts after acute myocardial infarction. J Cell
Physiol. 187:326–335. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lijnen PJ, Petrov VV and Fagard RH:
Angiotensin II-induced stimulation of collagen secretion and
production in cardiac fibroblasts is mediated via angiotensin II
subtype 1 receptors. J Renin Angiotensin Aldosterone Syst.
2:117–122. 2001.
|
|
37
|
Hafizi S, Wharton J, Morgan K, Allen SP,
Chester AH, Catravas JD, Polak JM and Yacoub MH: Expression of
functional angiotensin-converting enzyme and AT1 receptors in
cultured human cardiac fibroblasts. Circulation. 98:2553–2559.
1998. View Article : Google Scholar : PubMed/NCBI
|