Introduction
Optic atrophy 1 gene (OPA1) is a
multifunctional protein located within the mitochondrial inner
membrane, which regulates a number of critical cellular functions,
including mitochondrial fusion, mitochondrial DNA maintenance and
apoptosis (1). OPA1 belongs
to the dynamin family with which it shares three conserved regions,
a GTPase domain, a middle domain, and a GTPase effector domain
(2). Mutations in the OPA1
gene (3q28-q29, OMIM 165500) are known to result in autosomal
dominant optic atrophy (DOA, OMIM 165500), which is the most
commonly diagnosed inherited optic neuropathy in clinical practice
(2). Up to 20% of OPA1
mutation carriers develop phenotypes that have been designated as
DOA+, where the optic atrophy is complicated by a wide range of
neuromuscular features, including ataxia, myopathy, peripheral
neuropathy, sensorineural deafness, and notably, chronic
progressive external ophthalmoplegia (3). With the exception of these autosomal
dominant forms, only a few syndromic cases have so far been
reported with compound heterozygous OPA1 mutations
suggestive of either recessive or semi-dominant patterns of
inheritance (3–6). However, the clinical spectrum of
these emerging double-mutant OPA1-associated disorders
remains to be elucidated.
The majority of patients with OPA1 mutations
possess a simple heterozygous mutation. However, only seven
patients have been genetically confirmed with compound heterozygous
mutations and, of these, six were diagnosed with Behr syndrome
(OMIM 210000), which is characterized by early-onset optic atrophy,
ataxia, pyramidal signs, peripheral neuropathy, mental retardation
and developmental delay (7–8).
Although it has been demonstrated to be inherited in an autosomal
recessive manner, simple heterozygous OPA1 mutations have
also been reported to result in Behr syndrome (9). Although it is understood to be
inherited in an autosomal recessive manner, it had been reported
autosomal dominant inheritance.
To date, ~35 OPA1 mutations have been
identified in multiple domains (3), and it is hypothesized that the final
common pathway of OPA1 results in retinal ganglion cell loss
and optic nerve degeneration, in addition to more widespread
neuronal loss with multi-systemic manifestations. In the present
study, the detailed clinical manifestations of autosomal recessive
optic atrophy, sensorimotor neuropathy and cataracts associated
with the novel compound heterozygous mutations in the OPA1
gene was reported, with the aims of expand the clinical spectrum of
OPA1 mutations.
Patients and methods
Patients
The present study included a total of three
individuals of Korean origin; one patient with optic atrophy and
his parents who exhibited the normal phenotype. Clinical
characteristics are presented in Table
I. Paternity was determined by genotyping 15 microsatellites
using a PowerPlex 16 system (Promega Corporation, Madison, WI,
USA). In addition to this family, 300 healthy individuals were
examined as controls. Written informed consent was obtained from
all participants according to a protocol approved by the
Institutional Review Board for Sungkyunkwan University, Samsung
Medical Center (Seoul, South Korea), and Kongju National University
(Chungnam, South Korea; Industry-Cooperation Foundation).
 | Table IClinical characteristics of patients
with compound heterozygous optic atrophy 1 gene mutations. |
Table I
Clinical characteristics of patients
with compound heterozygous optic atrophy 1 gene mutations.
| Parameter | Ethnicity
|
|---|
| Korean | French | French | French | French | American | American | German |
|---|
| Reference | The present
study | Bonneau et al
(6) | Bonneau et al
(6) | Bonneau et al
(6) | Bonneau et al
(6) | Schaaf et al
(5) | Schaaf et al
(5) | Pesch et al
(4) |
| Gender | Male | Female | Female | Male | Male | Male | Female | Female |
| Age at exam
(years) | 10 | 6 | 11 | 14 | 16 | 8 | 3 | 30 |
| Age at onset | 12 months | 42 months | 12 months | 18 months | 3 years | 12 months | 6 months | Childhood |
| Mutation |
| Amino acid
change | Arg905Gln | Ile382Met | Val402Met | Ile382Met | Ile382Met | Ile382Met | Ile382Met | Glu270Lys |
| Leu620fs | Arg557a | Val903Glyfsa | Arg824a | Glu487Lys | Val903Glyfsa | Val903Glyfsa | Arg290Trp |
| Domain | GED | GTPase | GTPase | GTPase | GTPase | GTPase | GTPase | GTPase |
| MID | MID | GED | MID | GTPase | GED | GED | GTPase |
| Clinical
manifestation |
| Optic atrophy | Global pallor | Yes | Yes | Yes | Yes | Diffuse severe | Diffuse severe | Global pallor |
| Visual acuity | Blind | 0.01/0.01 | Unknown | 0.01/0.01 | Blind | Severely
reduced | Unknown | 0.2/0.2 |
| Cataracts | Yes | Unknown | Unknown | No | Unknown | No | No | No |
| Peripheral
neuropathy | Yes | Yes | Yes | Yes | Yes | Unknown | Unknown | Unknown |
| Ataxia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Unknown |
| Laboratory
findings |
| VEP | Absent | Prolonged | Prolonged | Prolonged | Prolonged | Unknown | Unknown | Prolonged |
| NCS | Axonal
sensorimotor | Axonal sensory | Axonal
sensorimotor | Axonal sensory | Axonal
sensorimotor | Unknown | Unknown | Unknown |
| Brain MRI | Normal | Normal | Cerebellar
atrophy | Cerebellar
atrophy | Cerebellar
atrophy | Normal | Unknown | Unknown |
| Lower leg MRI | Fatty
infiltration | NA | NA | NA | NA | NA | NA | NA |
| Nerve biopsy | Axonal
neuropathy | NA | NA | NA | NA | NA | NA | NA |
Whole exome sequencing (WES) and
determination of causative mutations
Peripheral blood DNA was isolated using a QIAamp
blood DNA purification kit (Qiagen GmbH, Hilden, Germany). Exome
sequencing has been recently introduced to inherited peripheral
neuropathy as an efficient molecular diagnostic tool (10). WES was performed using the SeqCap
EZ Human Exome Library version 3.0 (Roche NimbleGen, Inc., Madison,
WI, USA), and the HiSeq 2000 Genome Analyzer (Illumina, Inc., San
Diego, CA, USA) for the three individuals (designated I-1, I-2, and
II-1 in the FC394 family) using a previously described method
(10). The UCSC assembly hg19
(NCBI build 37.1) was used as the reference sequence. In the
present study variants with >20 single nucleotide polymorphism
(SNP) quality were used. Functionally significant variants
(missense, nonsense, exonic indel and splicing site variants) were
selected from hereditary peripheral neuropathy- and optic
atrophy-associated genes, and these were then compared with the
dbSNP142 database (http://www.ncbi.nlm.nih.gov), the 1,000 Genomes
Project database (http://www.1000genomes.org/) and the Exome Variant
Project (http://evs.gs.washington.edu/EVS/).
Candidate causative variants were confirmed by
Sanger sequencing for the three individuals using an ABI3130XL
Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Mutations were considered causative when
they: (i) Were inherited in a recessive pattern or occurred by
de novo mutation; (ii) co-segregated with affected members;
(iii) were not found in any of the 300 healthy controls; and (iv)
were not reported in the dbSNP142 database, the 1000 Genomes
Project database or the Exome Variant Project. Genotype-phenotype
correlation was also considered for candidate variants. Genomic
evolutionary rate profiling (GERP) scores were determined by the
GERP++ program (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html).
In silico analyses were performed using the prediction
algorithms of SIFT (http://sift.jcvi.org), MUpro (http://www.ics.uci.edu/~baldig/mutation), and
PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/).
Clinical and electrophysiological
assessments
All subjects were comprehensively assessed by a team
of experienced neurologists and ophthalmologists in order to
determine the extent and severity of their ophthalmic, auditory and
neurological deficits. Ophthalmic examination included fundoscopic
examination, color vision discrimination and automated visual field
assessment. Patients were examined for motor and sensory
impairment, deep tendon reflexes and muscle atrophy by two
independent neurologists, and pure-tone audiometric evaluation was
performed.
Nerve conduction studies were performed on all
extremities of the patient (Medtronic Keypoint 4; Medtronic Inc.,
Minneapolis, MN, USA). Motor nerve conduction velocities (MNCVs) of
the median and ulnar nerves were determined by stimulation at the
elbow or wrist, while recording compound muscle action potentials
(CMAPs) over the abductor pollicis brevis and adductor digiti
quinti, respectively. Similarly, the MNCVs of the peroneal and
tibial nerves were determined by stimulation at the knee and ankle,
while recording CMAPs over the extensor digitorum brevis and
adductor hallucis, respectively. CMAP amplitudes were measured from
baseline to negative peak values. Sensory nerve conduction
velocities were obtained over the finger-wrist segment from the
median and ulnar nerves by orthodromic scoring, and were also
recorded for sural nerves. Sensory nerve action potential (SNAP)
amplitudes were measured from positive peaks to negative peaks.
Magnetic resonance imaging (MRI) scans of
lower limbs and brain
On a follow-up visit at 8 years old, the patient
(II-1) was evaluated using a 3.0-T system (Siemens AG, Munich,
Germany). Lower limb imaging was obtained in the axial [field of
view (FOV), 24–32 cm; slice thickness, 6 mm; and slice gap, 0.5–1.0
mm] and coronal planes (FOV, 38–40 cm; slice thickness, 4–5 mm;
slice gap, 0.5–1.0 mm). The following protocol was used:
T1-weighted spin-echo (SE) [repetition time (TR)/echo time (TE)
570-650/14-20, 512 matrices], T2-weighted SE (TR/TE
2800-4000/96-99, 512 matrices), and fat-suppressed T2-weighted SE
(TR/TE 3090-4900/85-99, 512 matrices).
Sural nerve biopsy
Histopathological analysis of the distal sural nerve
biopsy was performed on the patient (II-1) at 6 years of age. In
addition to light microscopic examination (BX51; Olympus
Corporation, Tokyo, Japan) of formalin-fixed sections, electron
microscopic observations were made using specimens fixed in 2%
glutaraldehyde in 0.025 M cacodylate buffer (pH 7.4; both
Sigma-Aldrich, St. Louis, MA, USA) and processed for semi-thin and
ultra-thin studies. Semi-thin sections were stained with toluidine
blue (Sigma-Aldrich) for evaluation under light microscopy.
Ultra-thin sections (60–65 nm) were contrasted with uranyl acetate
and lead citrate (both Sigma-Aldrich) for ultrastructural studies
(H-7650; Hitachi, Ltd., Tokyo, Japan). The density of myelinated
fibers (MFs), axonal diameter, myelin thickness, and the g-ratio of
MFs were determined from semi-thin transverse sections using a
computer-assisted image analyzer (AnalySIS; Olympus Soft Imaging
Solutions, GmbH, Münster, Germany).
Results
Identification of compound heterozygous
mutations in OPA1
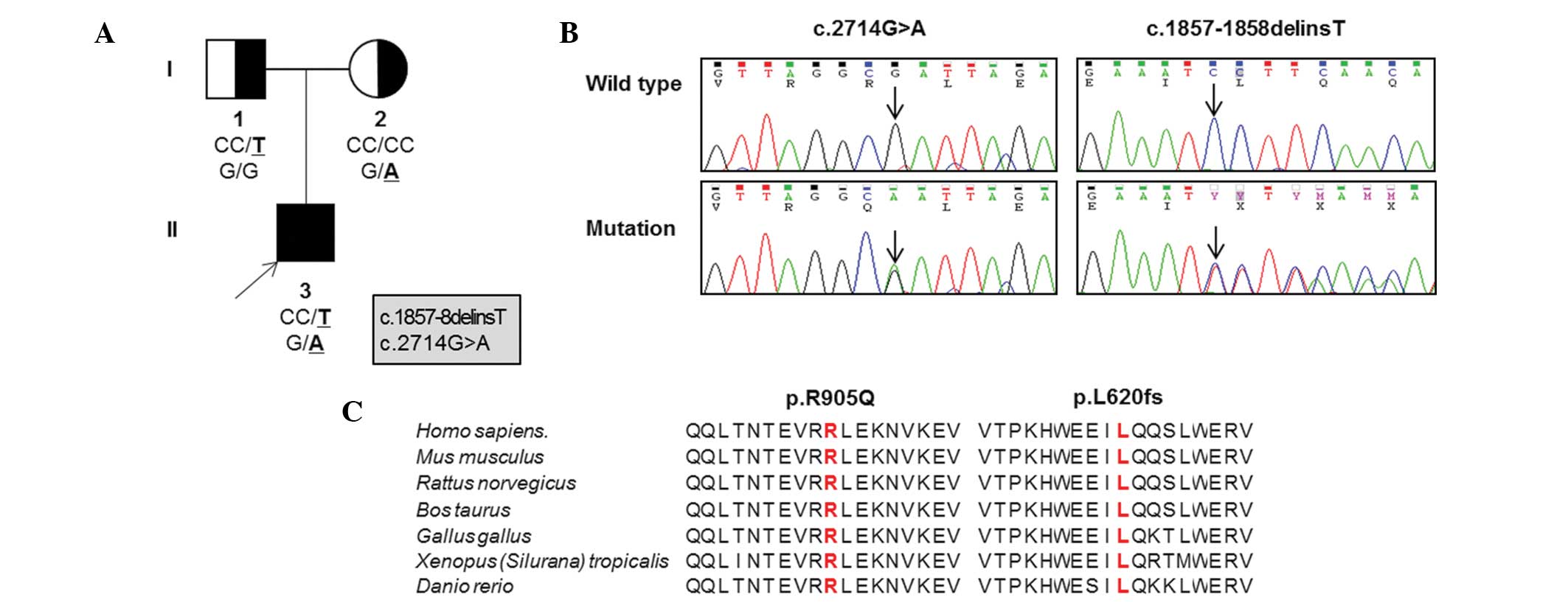
Based on WES in the father-mother-patient trio
(Table II; Fig. 1A), the patient possessed a pair of
novel compound heterozygous mutations in OPA1,
p.L620fs*13 (c.1857–1858delinsT) in exon 20 and p.R905Q
(c.G2714A) in exon 27 (Fig. 1B).
These mutations were located in the middle domain and the guanosine
triphosphatase (GTPase) effector domain, respectively. Parental
analysis indicated that the mother was heterozygous for the p.R905Q
missense mutation and the father was heterozygous for the p.L620fs
frameshift mutation. Neither mutation was detected in the 300
healthy Korean controls, dbSNP138 (http://www.ncbi.nlm.nih.gov), the 1,000 Genomes
Database (http://www.1000genomes.org/) or the
Exome Variant Server (http://evs.gs.washington.edu/EVS/). Amino acid
sequences surrounding the mutation sites were highly conserved
throughout different vertebrate species (Fig. 1C). Three in silico analyses
(SIFT, PolyPhen2 and MUpro) indicated pathogenic prediction
(Table III). The GERP scores of
the mutations were high (5.650 at c.1858C and 5.890 at c.2714G).
Thus, these novel compound heterozygous mutations in the
OPA1 gene were likely the underlying cause of the patient's
phenotype.
| Table IIWhole exome sequencing analysis of
the proband and his parents. |
Table II
Whole exome sequencing analysis of
the proband and his parents.
| Items/samples | I-1 (father) | I-2 (mother) | II-1 (patient) |
|---|
| Total yield
(Gbp) | 14,27 | 9,48 | 5,30 |
| Mappable reads
(%) | 99.2 | 99.4 | 92.1 |
| Coverage of the
target region (≥1X, %) | 95.7 | 95.2 | 97.9 |
| Coverage of the
target region (≥10X, %) | 90.5 | 88.2 | 92.9 |
| Mean read depth of
the target region (X) | 58.7 | 40.6 | 57.9 |
| Total number of
SNPs | 86,313 | 84,206 | 49,686 |
| Number of coding
SNPs | 20,265 | 20,498 | 19,472 |
| Total number of
indels | 7,902 | 7,472 | 6,898 |
| Number of coding
indels | 390 | 398 | 508 |
| Table IIISummary of optic atrophy 1 gene
mutations and in silico scores. |
Table III
Summary of optic atrophy 1 gene
mutations and in silico scores.
| Domain | Nucleotide
change | Amino acid
changea | in silico
analysisb
| GERP score |
|---|
| SIFT | PolyPhen2 | MUpro |
|---|
| MID |
c.1857–1858delinsT | L620fs*13 | – | – | – | 5.650 |
| GED | c.2714G>A | R905Q | 0.000c | 1.000c | −0.630c | 5.890 |
In addition to the OPA1 causative mutations,
multiple functionally notable variants were detected in optic
atrophy and hereditary peripheral neuropathy associated genes.
However, these were considered polymorphic or rare private
variants, as they were observed in controls, including various
human genome variants databases. Multiple variants were not
observed in the controls, but they did not co-segregate with the
affected individual in the present family.
Clinical manifestations
Clinical manifestations are summarized in Table I. A 10-year-old boy (II-1; Fig. 1A), was born healthy at full term by
spontaneous vaginal delivery to healthy non-consanguineous Korean
parents. He visited the Department of Neurology at Samsung Medical
Center (Seoul, Korea) aged 10 years due to gait disturbance. He was
diagnosed with congenital cataracts at the age of one and was
treated with a corrective operation; at that point, optic atrophy
and nystagmus were noted in his eyes. The optic atrophy progressed
to the point that he was declared legally blind at six years of
age. By age two, he was not able to walk without support, and by
the age of 10, he harbored severe ataxia and postural instability.
Neurological examination at 10 years of age indicated muscle
weakness and atrophy of the bilateral distal muscles, predominantly
in the lower limbs. Bilateral pes cavus, steppage gait, and
atrophic changes of the intrinsic foot and calf muscles were noted.
Sensitivity to pinprick, touch, position, and vibration was
decreased. Knee and ankle jerks were absent and pyramidal and
cerebellar signs were not observed. At 10 years of age, the patient
was not able to count fingers at ~15 cm with either eye but did
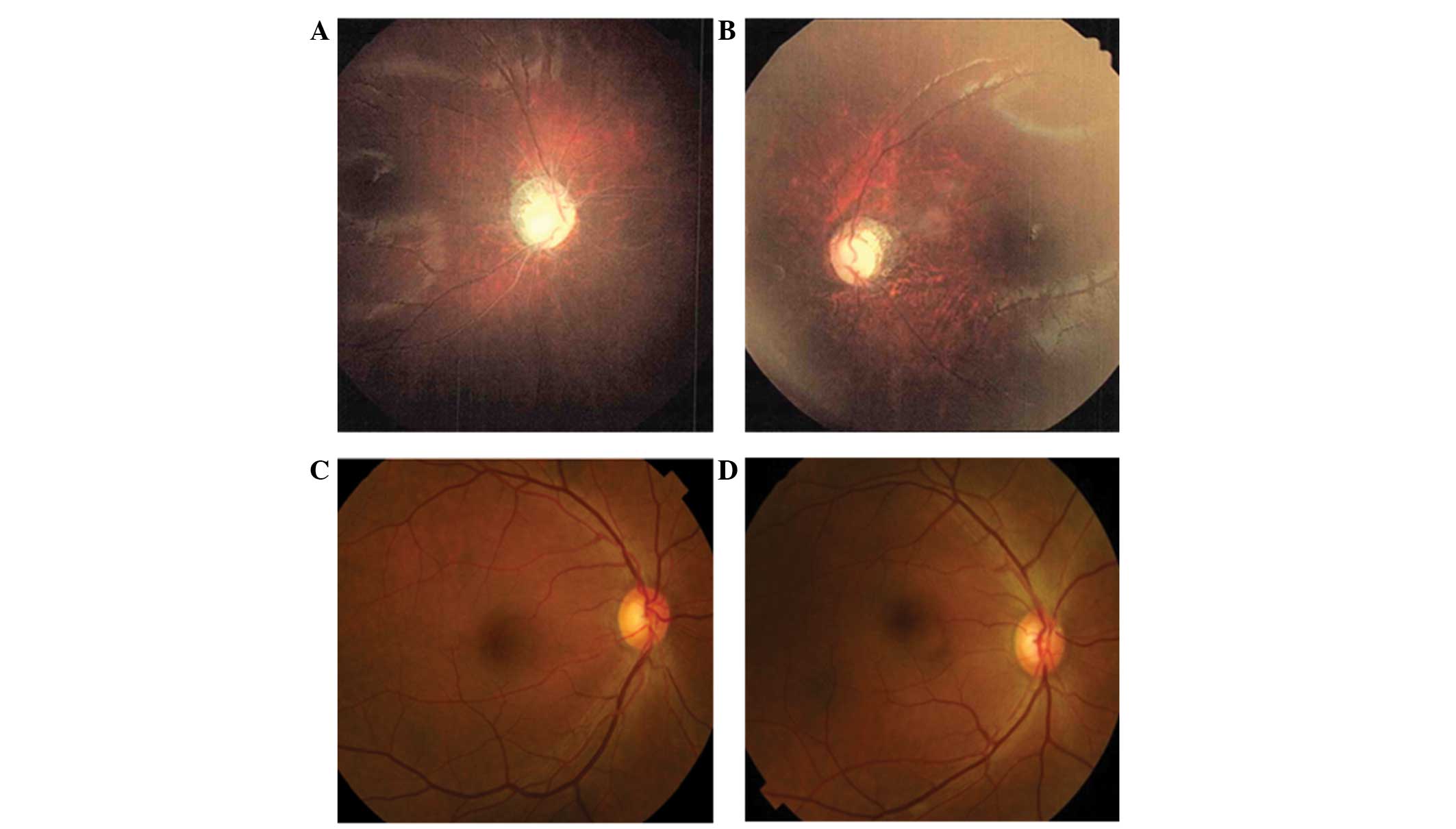
perceive light. A dilated fundus examination demonstrated diffuse
severe optic atrophy in his eyes (Fig.
2A and B). Audiological evaluation indicated normal hearing
sensitivity. Laboratory tests, such as glucose level, liver
enzymes, and renal function were normal, and echocardiography
indicated a normal sinus rhythm. Although his father and mother had
an OPA1 heterozygous mutation, no abnormalities following
ophthalmological and electrophysiological evaluation were observed
(I-1, and I-2; Fig. 1A). They did
not complain of any visual problems and dilated fundus examination
results, including assessment of each optic nerve and formal color
vision test, were normal (Fig. 2C and
D).
Electrophysiological features are summarized in
Table IV. SNAPs were absent in of
the patient at all extremities at 6, 8 and 10 years of age. CMAPs
were decreased in the peroneal and tibial nerves, compatible with
axonal polyneuropathy. When visual evoked potentials (VEP) were
performed at 6 years, no potential was demonstrated. However,
brainstem auditory evoked potentials (BAEP) were normal. In the
parents, VEP and BAEP were normal.
| Table IVSerial electrophysiological studies
in a patient (II-1) with novel compound mutations in the optic
atrophy 1 gene. |
Table IV
Serial electrophysiological studies
in a patient (II-1) with novel compound mutations in the optic
atrophy 1 gene.
| Parameter | Sidea
| Sideb
| Sidec
| Normal value |
|---|
| Right | Left | Right | Left | Right | Left |
|---|
| Median nerve |
| TL (ms) | 3.0 | 2.8 | 3.4 | 2.9 | 3.3 | 3.0 | <3.9 |
| CMAP (mV) | 7.9 | 8.8 | 7.3 | 6.9 | 6.2 | 7.6 | >6.0 |
| MNCV (m/sec) | 54.2 | 54.2 | 51.8 | 53.7 | 48.0 | 55.0 | >50.5 |
| Ulnar nerve |
| TL (ms) | 2.4 | 2.4 | 2.4 | 2.8 | 2.8 | 2.6 | <3.0 |
| CMAP (mV) | 8.0 | 6.1 | 7.4 | 6.7 | 8.1 | 7.4 | >8.0 |
| MNCV (m/sec) | 53.8 | 54.0 | 53.6 | 53.4 | 55.0 | 53.0 | >51.1 |
| Peroneal nerve |
| TL (ms) | 2.3 | 2.9 | 2.7 | 3.0 | 3.8 | 3.6 | <5.3 |
| CMAP (mV) | 1.1 | 1.3 | 1.1 | 1.0 | 0.6 | 1.7 | >1.6 |
| MNCV (m/sec) | 46.3 | 48.6 | 42.2 | 43.5 | 45.0 | 50.0 | >41.2 |
| Tibial nerve |
| TL (ms) | 2.3 | 2.6 | 2.9 | 3.0 | 4.1 | 3.9 | <5.4 |
| CMAP (mV) | 4.7 | 5.1 | 2.6 | 2.8 | 2.9 | 2.2 | >6.0 |
| MNCV (m/sec) | 45.5 | 46.3 | 42.0 | 43.8 | 41.0 | 45.0 | >41.1 |
| Median sensory
nerve |
| SNAP
(μV) | A | A | A | A | A | A | >8.8 |
| SNCV (m/sec) | A | A | A | A | A | A | >39.3 |
| Ulnar sensory
nerve |
| SNAP
(μV) | A | A | A | A | A | A | >7.9 |
| SNCV (m/sec) | A | A | A | A | A | A | >37.5 |
| Sural nerve |
| SNAP
(μV) | A | A | A | A | A | A | >6.0 |
| SNCV (m/sec) | A | A | A | A | A | A | >32.1 |
Length-dependent patterns in the lower
extremity MRI
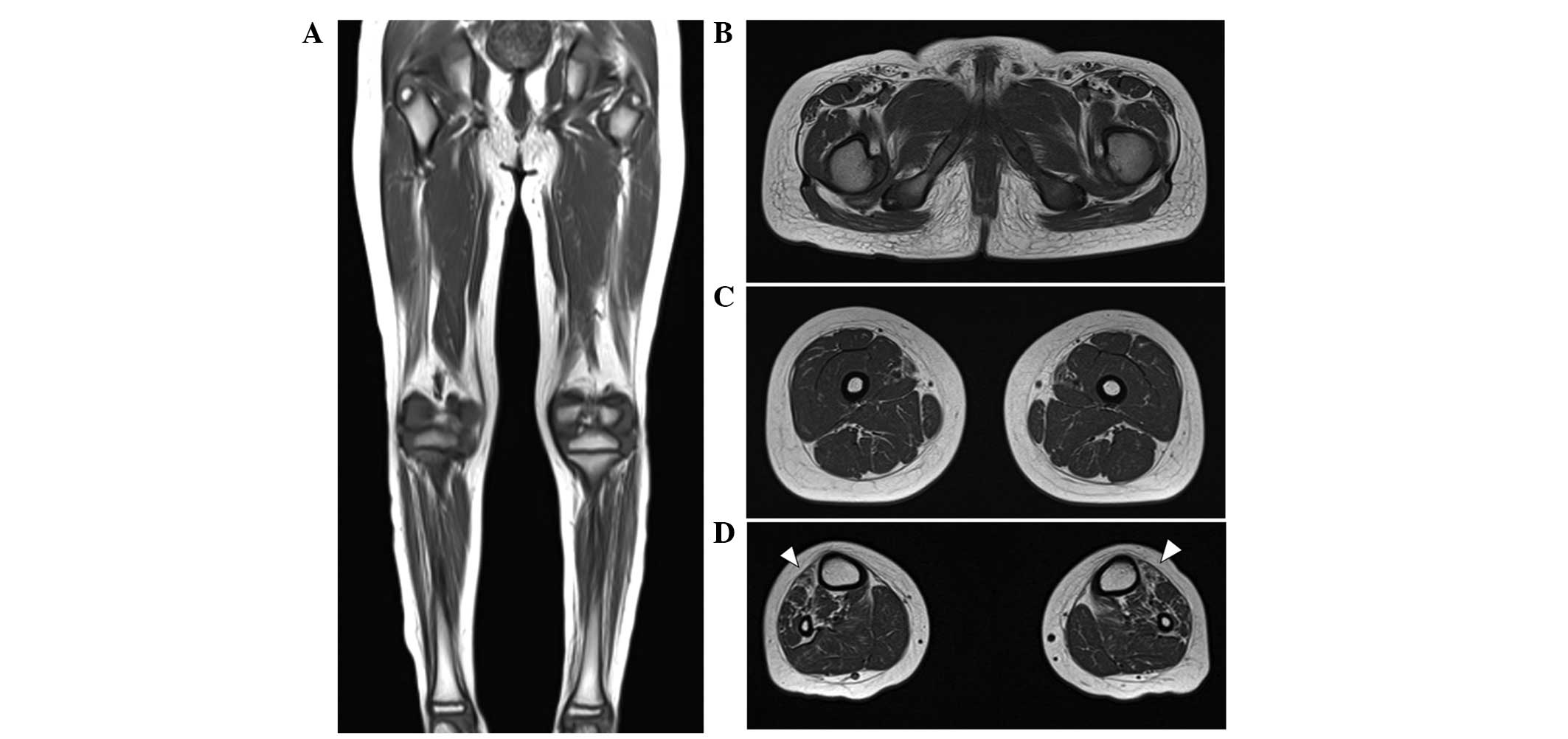
At the age of 10 years, T1-weighted MRIs indicated
muscle atrophy and fatty replacement in the lower calf muscles but
close to normal hip and thigh muscles. These findings are
compatible with length-dependent axonal degeneration. In addition,
fatty replacement of the calf muscles was predominant in the
anterior compartment, including the tibialis anterior muscle,
consistent with his foot drop and steppage gait (Fig. 3A–D). A brain MRI performed when he
was 8 years old indicated the cerebral cortex and cerebellum were
normal in structure and size.
Histopathological findings
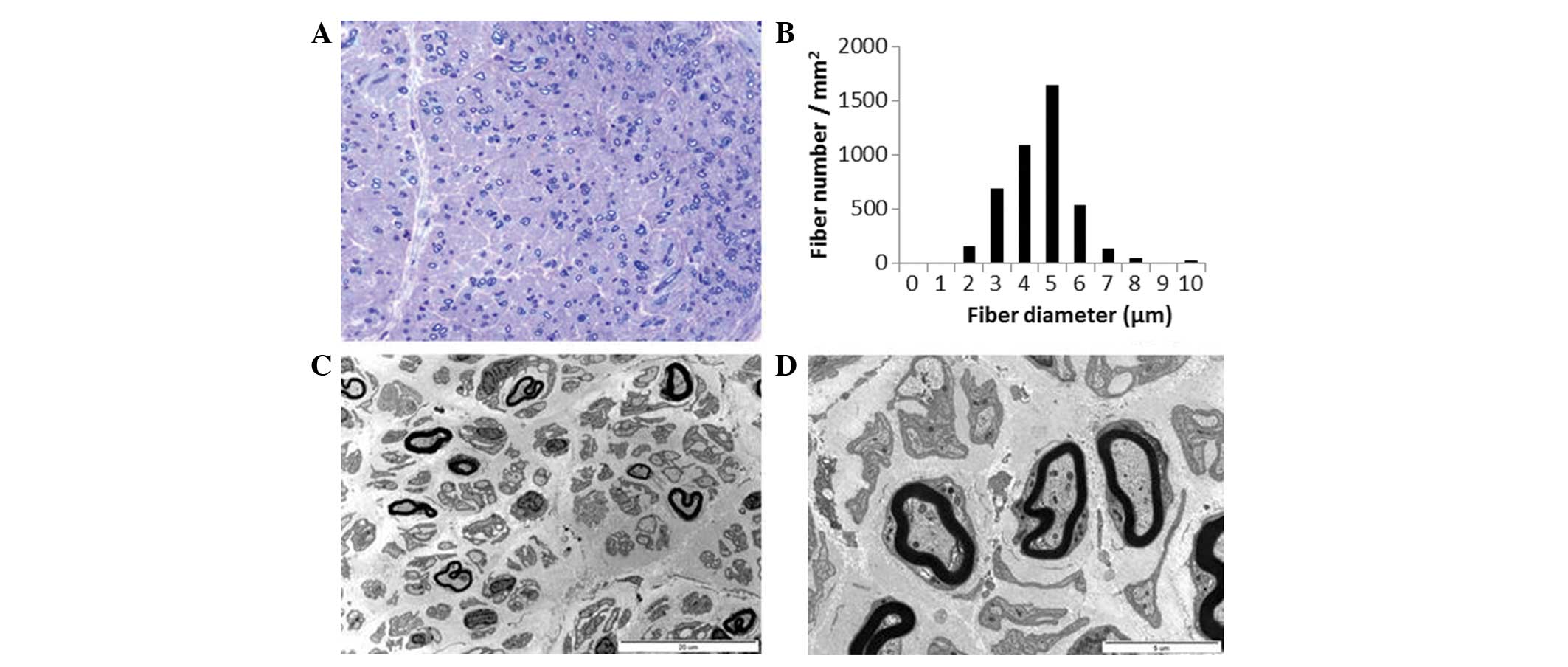
Semi-thin transverse sections of the distal sural
nerve demonstrated medium and small MFs with complete loss of large
MFs. Compared with a healthy distal sural nerve MF count of
14,170±2,250/mm2 in a normal 2-year-old female, 4,245
MFs/mm2 were counted (Fig.
4A). The range and average MF diameter were 2.5–10.3 μm
and 5.0 μm, respectively. A histogram indicated a unimodal
distribution pattern; MFs with a diameter <3 μm comprised
3.6% of the total population (Fig.
4B), the MFs with a diameter >8 μm comprised 0.9% of
the total population. The MF% area in this case was 8.8%. The
remaining MFs demonstrated occasional excessive folding of myelin,
with very rare evidence of regeneration. There was no evidence of
demyelination, remyelination, or onion bulb formation. Electron
microscopic examination indicated that the remaining MFs had focal
abnormalities of the myelin, including irregular myelin sheaths,
distorted shapes or focal noncompaction (Fig. 4C). Unmyelinated axons were
relatively well preserved with occasional rarefaction (Fig. 4D). Endoneurial fibroblast
proliferation and large quantities of collagen deposition were
documented.
Discussion
Novel compound heterozygous OPA1 mutations
were identified in a patient with recessive optic atrophy,
sensorimotor neuropathy and congenital cataracts. OPA1 is a
multifunctional protein located within the mitochondrial inner
membrane (1) and mutations in the
OPA1 gene are known to cause DOA (11). The majority of patients with DOA
have isolated optic atrophy and ~20% of patients present with
extra-ocular manifestations, termed DOA+ (3,12).
Excluding these autosomal dominant forms, only seven cases have so
far been genetically confirmed as compound heterozygous OPA1
mutations. The majority of patients with compound heterozygous
mutations presented with an earlier onset and more severe disease
manifestation, suggestive of either recessive or semi-dominant
patterns of inheritance (4–6). In
the present family, the proband demonstrated early onset severe
optic atrophy whereas neither parents had ophthalmic or neurologic
symptoms, and the results of electrophysiologic testing, such as
nerve conduction studies, VEP and BAEP were normal. These results
may support either recessive or semi-dominant inheritance with
incomplete penetrance.
Clinical and electrophysiological features of seven
cases with confirmed compound heterozygous OPA1 mutations
were summarized in Table I. Six of
them were diagnosed with Behr syndrome except one case in which
extra-ocular manifestations were not described. Behr syndrome is
characterized by the association of early-onset optic atrophy,
ataxia, pyramidal signs, peripheral neuropathy, mental retardation
and developmental delay (7,8).
However, six patients did not have pyramidal signs, mental
retardation or marked developmental delay. All the cases presented
with early-onset severe optic atrophy and ataxia. Four patients had
peripheral neuropathies and three showed signs of cerebellar
atrophy on brain MRIs. Genetically, six of the patients had either
one or two OPA1 mutations in the GTPase domain. The present
case presented with early-onset severe atrophy, sensorimotor
neuropathy, and ataxia, and he did not have pyramidal signs,
developmental delay or mental retardation, which was similar to
previously reported cases (4–6). In
addition, he had congenital cataracts, which have not, to the best
of our knowledge, been previously described in patients with
OPA1 mutations. In addition, the patient did not have any
mutation in the GTPase domain.
The present case had bilateral severe congenital
cataracts, congenital cataracts occur in infants due to various
known causes that include inherited conditions, infection,
metabolic disorders and ocular diseases (13,14).
Inherited conditions are the likely cause of congenital cataracts
in this case as there were no specific causes demonstrated by the
medical records or laboratory findings, and symptoms were severe
and symmetric. Aijaz et al (15) reported that expression of
OPA1 was observed in the lens and these results may support
this assertion. The present case may expand the clinical spectrum
of Behr syndrome with compound heterozygous OPA1
mutations.
In conclusion, the present study identified novel
compound heterozygous OPA1 mutations in a patient with
recessive optic atrophy, sensorimotor neuropathy and congenital
cataracts. Congenital cataracts that were described in the present
case may expand the clinical spectrum in patients with OPA1
mutations. Thus, OPA1 mutations should be considered when
screening patients with recessive optic atrophy, particularly those
with peripheral neuropathy, and cataracts.
Acknowledgments
The present study was supported by the Korean Health
Technology R&D Project, Ministry of Health & Welfare (grant
nos. HI12C0135 and HI14C3484) and by a National Research Foundation
of Korea grant funded by the Korean government (MSIP; grant no.
2014R1A2A2A01004240).
References
|
1
|
Lenaers G, Hamel C, Delettre C,
Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P and Milea D:
Dominant optic atrophy. Orphanet J Rare Dis. 7:462012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eiberg H, Kjer B, Kjer P and Rosenberg T:
Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I.
Linkage analysis. Hum Mol Genet. 3:977–980. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu-Wai-Man P, Griffiths PG, Gorman GS,
Lourenco CM, Wright AF, Auer-Grumbach M, Toscano A, Musumeci O,
Valentino ML, Caporali L, et al: Multi-system neurological disease
is common in patients with OPA1 mutations. Brain. 133:771–786.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pesch UE, Leo-Kottler B, Mayer S, Jurklies
B, Kellner U, Apfelstedt-Sylla E, Zrenner E, Alexander C and
Wissinger B: OPA1 mutations in patients with autosomal dominant
optic atrophy and evidence for semi-dominant inheritance. Hum Mol
Genet. 10:1359–1368. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schaaf CP, Blazo M, Lewis RA, Tonini RE,
Takei H, Wang J, Wong LJ and Scaglia F: Early-onset severe
neuromuscular phenotype associated with compound heterozygosity for
OPA1 mutations. Mol Genet Metab. 103:383–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonneau D, Colin E, Oca F, Ferré M,
Chevrollier A, Guéguen N, Desquiret-Dumas V, N'Guyen S, Barth M,
Zanlonghi X, et al: Early-onset Behr syndrome due to compound
heterozygous mutations in OPA1. Brain. 137:e3012014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beh r C: Die komplizierte,
hereditäre-familiäre Optikusatrophie des Kindesalters-ein bisher
nicht beschriebener Symptomkomplex. Klin Mbl Augenheilkd.
39:138–60. 1909.In German.
|
|
8
|
Thomas PK, Workman JM and Thage O: Behr's
syndrome. A family exhibiting pseudodominant inheritance. J Neurol
Sci. 64:137–148. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marelli C, Amati-Bonneau P, Reynier P,
Layet V, Layet A, Stevanin G, Brissaud E, Bonneau D, Durr A and
Brice A: Heterozygous OPA1 mutations in Behr syndrome. Brain.
134:e1692011.author reply e170. View Article : Google Scholar
|
|
10
|
Choi BO, Koo SK, Park MH, Rhee H, Yang SJ,
Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, et al: Exome
sequencing is an efficient tool for genetic screening of
Charcot-Marie-Tooth disease. Hum Mutat. 33:1610–1615. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delettre C, Lenaers G, Griffoin JM,
Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J,
Turc-Carel C, Perret E, et al: Nuclear gene OPA1, encoding a
mitochondrial dynamin-related protein, is mutated in dominant optic
atrophy. Nat Genet. 26:207–210. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Amati-Bonneau P, Milea D, Bonneau D,
Chevrollier A, Ferré M, Guillet V, Gueguen N, Loiseau D, de
Crescenzo MA, Verny C, et al: OPA1-associated disorders: Phenotypes
and pathophysiology. Int J Biochem Cell Biol. 41:1855–1865. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lloyd IC, Goss-Sampson M, Jeffrey BG,
Kriss A, Russell-Eggitt I and Taylor D: Neonatal cataract:
Aetiology, pathogenesis and management. Eye (Lond). 6:184–196.
1992. View Article : Google Scholar
|
|
14
|
Chan WH, Biswas S, Ashworth JL and Lloyd
IC: Congenital and infantile cataract: Aetiology and management.
Eur J Pediatr. 171:625–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aijaz S, Erskine L, Jeffery G,
Bhattacharya SS and Votruba M: Developmental expression profile of
the optic atrophy gene product: OPA1 is not localized exclusively
in the mammalian retinal ganglion cell layer. Invest Ophthalmol Vis
Sci. 45:1667–1673. 2004. View Article : Google Scholar : PubMed/NCBI
|