Introduction
Endothelial dysfunction is one of the most important
mechanisms underlying the development and progression of
salt-induced hypertension (1).
Reduced bioavailability of nitric oxide (NO) in the vasculature is
a major feature of endothelial dysfunction, which leads to
increased endothelial permeability, platelet aggregation, leukocyte
adhesion, and cytokine generation (2,3).
Endothelial NO synthase (eNOS) is the predominant NOS isoform,
which is responsible for generating the majority of NO in the
vasculature. Reductions in the expression or activity of eNOS, and
its uncoupling, have a crucial role in the process of endothelial
dysfunction (4). Asymmetric
dimethylarginine (ADMA) is an endogenous inhibitor of NOS, the
dysregulation of which is associated with endothelial dysfunction
and salt-sensitive hypertension (5–7).
The small GTPase Ras homolog gene family, member A
(RhoA) and its downstream target Rho-associated protein kinase
(ROCK) are important in the regulation of endothelial barrier
function. The RhoA/ROCK pathway regulates cell motility, migration,
proliferation and angiogenesis via the control of
actin-cytoskeletal assembly and cell contraction (8). Furthermore, it has previously been
demonstrated that the RhoA/ROCK pathway has a central role in
impaired production of NO, due to its numerous actions on eNOS
(9–11).
ADMA is synthesized endogenously during the
methylation of protein arginine residues by protein arginine
methyltransferases (PRMTs). PRMTs are a protein family that
monomethylate or dimethylate the guanidino nitrogen atoms on
arginine side chains. PRMT-1 is the predominant type I PRMT in
mammalian cells, which catalyzes arginine residues facilitating the
formation of ADMA residues (12).
In the cardiovascular system, ADMA is generated by PRMT-1, which is
expressed in the heart, and smooth muscle and endothelial cells. It
has previously been reported that altering ADMA metabolism in
vivo and in vitro may affect the activity of Rho
GTPases, which are key regulators of actin dynamics, endothelial
cell motility and angiogenesis (13). The plasma levels of ADMA are
elevated concomitantly with increased ROCK activity and ROCK1
expression in the pulmonary arteries of Sprague-Dawley rats
(14). In addition, in pulmonary
endothelial cells, exogenous and endogenous ADMA increases RhoA
activity (15,16).
Animal and population studies have demonstrated that
high salt intake can induce a small increase (2–4 mmol/l) in plasma
sodium levels, which may influence blood pressure (17,18).
High salt intake may result in hypertension and cardiovascular
complications via its effects on signals triggered by augmented
extracellular NaCl concentration and/or osmolality of extracellular
fluids (19). Endothelial cells,
as vascular salt sensors, are highly sensitive to extracellular
sodium (20). The present study
tested the hypothesis that high salt medium could increase the
secretion of ADMA, further upregulate RhoA/ROCK expression and
activity, and that downregulation of ADMA by RNA interference
(RNAi) could significantly reverse these processes.
Materials and methods
Cell culture
The EA.hy926 cells (Cell Bank of the Chinese Academy
of Sciences, Shanghai, China) were maintained in high glucose
Dulbecco's modified Eagle's medium (DMEM; Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine
serum (Hyclone; GE Healthcare Life Sciences). The cells were
cultured at 37°C in an atmosphere containing 5% CO2 and
95% air, and were subcultured by trypsinization every 36–48 h.
Cells from the 5th–10th passage were used in
subsequent experiments.
Cell viability assay
To determine the optimum concentration of high salt
medium on EA.hy926 cells, the cells were treated with serum-free
DMEM (109 mmol/l, which contained the standard concentration of
NaCl) as a control; with high salt medium at various concentrations
of salinity (115, 130, 137, 147 and 160 mmol/l); or with isotonic
mannitol (solid mannitol diluted in DMEM) at 12 or 56 mmol/l as a
hyperosmotic control, for 48 h. Cell viability was determined using
WST-8 dye (Beyotime Institute of Biotechnology, Haimen, China)
according to manufacturer's protocol. A total of 5×103
cells/well (100 µl) were seeded into a 96-well plate, and
were incubated at 37°C for 48 h. Subsequently, the cells were
treated with the aforementioned media for 24 h, each group
consisted of four wells. WST-8 dye solution (10 µl) was then
added to each well, and the cells were incubated at 37°C for a
further 2 h. Absorbance (A) was finally determined by
spectrophotometry at 560 nm using a microplate reader (BC Biotech
Engineering Ltd., London, UK). Cell viability was calculated using
the following equation: Cell viability = (A450 of
treatment wells - A450 of control wells) /
A450 of control wells × 100%. All data were expressed as
the mean ± standard error of the mean of three experiments.
PRMT-1 knockdown with RNAi
Three PRMT-1-specific small interfering RNAs
(siRNAs), negative and positive control siRNAs were designed and
synthesized by Shanghai GeneChem Co., Ltd. (Shanghai, China). To
optimize transfection, various concentrations of siRNA (25, 50, 100
and 150 nM) in serum-free DMEM were transfected into the cells in
six-well plates using TurboFect siRNA Transfection Reagent
(Fermentas; Thermo Fisher Scientific, Inc., Pittsburgh, PA, USA)
according to the manufacturer's protocol. Cells were collected 48 h
post-transfection to assess gene silencing using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). The
siRNA for PRMT-1 (siPRMT1-1#) at the final concentration
of 50 nM was found to be the most efficient at knocking down gene
expression, with minimal toxicity. The sequence is as follows:
Sense, 5′-CCAUCGACCUGGACUUCAATT-3′ and antisense,
5′-UUGAAGUCCAGGUCGAUGGTT-3′.
Cell treatment
Once the cell monolayers reached 80% confluence, the
cells were incubated with serum-free medium for 16 h, and were then
treated with various concentrations of high salt medium. The cells
were treated with DMEM; high salt medium (115 or 137 mmol/l); high
salt medium (115 mmol/l) and siRNA-PRMT-1 (50 nM); high salt medium
(137 mmol/l) and siRNA-PRMT-1 (50 nM); or with isotonic mannitol
(12 or 56 mmol/l) as an osmotic control, for 48 h, after which they
were harvested for further analysis.
Measurement of ADMA in culture
supernatant
Following a 48 h incubation, the medium was
transferred to microcentrifuge tubes and was centrifuged at 10,000
× g for 10 min at 4°C. The concentration of ADMA was determined by
high-performance liquid chromatography. To prepare samples, the
supernatant (100 µl) was diluted with 0.1 mol/l hydrochloric
acid to 1.5 ml. The samples (20 µl) and standards (20
µl) were incubated for 3 min with o-phthaldialdehyde
reagent (10 mg/mL OPA in borate buffer, pH 9.0, containing 0.4%
mercaptoethanol. ADMA and o-phthaldialdehyde were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Briefly, a Shimadzu LC-10A
liquid chromatograph equipped with a Model 7125i injection valve
and a Shimadzu RF-10AXL fluorescence detector was used (Shimadzu
Corporation, Kyoto, Japan). N2010 (Zhejiang University, Hangzhou,
China) was used as a data processor. A 5 µM Waters Symmetry
C18 (5 µm; 150×3.9 mm) coupled to a Waters Sentry Symmetry
C18 guard column (5 µm; 3.9×20 mm) (Waters UK, Elstree, UK)
was operated at room temperature. The mobile phase consisted of
77:23 (v/v) potassium phosphate-buffer (pH 3.5):acetonitrile. The
flow rate was 1.0 ml/min. The injection volume was 20 µl.
o-Phthaldialdehyde adducts of methylated amino acids and
internal standard ADMA produced by pre-column mixing were monitored
at 338 and 447 nm, respectively.
RNA isolation and RT-qPCR
Total RNA was isolated from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). cDNA was synthesized from RNA using the
RevertAid™ First strand cDNA Synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc.). Briefly, the RNA/primer mixture was
placed into a sterile, nuclease free tube on ice; 4 µl 5X
Reaction Buffer, 1 µl Ribolock RNase inhibitor, 2 µl
10 mM dNTP mix and Reverse Transcriptase were mixed and centrifuged
at 10,00 × g, and incubated for 60 min at 42°C. The reaction was
terminated by heating at 70°C for 5 min. The mRNA expression levels
of PRMT-1, eNOS and RhoA were detected by qPCR, which was performed
on an iQ5 Real-Time PCR Detection system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) using SYBR® Premix Ex
Taq™ II (Takara Bio, Inc., Shiga, Japan). The reaction volume
was 50 µl and included 37.5 µl ddH2O, 1
µl 10 mM dNTP, 5 µl 10X PCR buffer, 3 µl 25 mM
MgCl2, 1 µl forward primer, 1 µl reverse
primer and 1 µl cDNA. The thermocycling conditions were as
follows: 3 min at 95°C, followed by 40 cycles at 95°C for 10 sec,
58°C for 30 sec and 72°C for 30 sec. The data were collected at the
annealing step (72°C) of each cycle. The primer sequences (Sangon,
Shanghai, China) are presented in Table I. Relative gene expression levels
were normalized to glyceraldehyde 3-phosphate dehydrogenase
(GAPDH). Data were analyzed on the basis of the relative expression
method, using the 2−ΔΔCq relative expression formula,
where ΔΔCq = ΔCq (experimental group) - ΔCq (control group), ΔCq =
Cq (target gene) - Cq (GAPDH), and Cq is the cycle at which the
threshold is crossed (21).
 | Table IPrimer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Sequence |
|---|
| GAPDH | F:
ATCGTGCGTGACATTAAGGAGAAG |
| R:
AGGAAGGAAGGCTGGAAGAGAG |
| PRMT-1 | F:
GCCTCCAGCCGCCCTCTTG |
| R:
CACCTCGTCCTTCAGCATCTCC |
| eNOS | F:
CACCGCTACAACATCCAG |
| R:
GCCTTCTGCTCATTCTCC |
| RhoA | F:
TGCTTGCTCATAGTCTTCAG |
|
R:CACATCAGTATAACATCGGTATC |
Western blot analysis
The cells were homogenized and lysed with
radioimmunoprecipitation assay lysis buffer (GSE Bio, Shaanxi,
China). The supernatant was collected following centrifugation at
10,000 × g for 15 min at 4°C. Proteins in the supernatant were
quantified by the Bradford assay using a protein assay kit. Equal
amounts of protein (45 µg) were separated by 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis, and were
transferred to nitrocellulose membranes (EMD Millipore, Billerica,
MA, USA) at 250 mA for 2 h. Non-specific binding was blocked with
5% skim milk for 2 h at room temperature. Immunoblotting was then
performed using PRMT-1 (mouse monoclonal; cat. no. ab12189; Abcam,
Cambridge, UK); eNOS (mouse monoclonal; cat. no. 9572), RhoA
(rabbit monoclonal; cat. no. 2117), phosphorylated (p)-myosin
phosphatase target subunit 1 (MYPT-1; rabbit monoclonal; cat. no.
5163) and total MYPT-1 (rabbit monoclonal; cat. no. 2634) (all
1:1,000; Cell Signaling Technology, Inc., Beverly, MA, USA); and
GAPDH (rabbit monoclonal; 1:5,000; cat. no. AP0063; Bioworld
Technology, Inc., St. Louis Park, MN, USA) antibodies at 4°C
overnight. The membranes were then washed and probed with
horseradish peroxidase-conjugated goat anti-rabbit (cat. no.
31460), goat anti-rat (cat. no. 31430) (both 1:5,000; Thermo Fisher
Scientific, Inc., Waltham, MA, USA); and mouse anti-goat (1:1,000;
cat. no. bse-0294M; Beijing Biosynthesis Biotechnology Co., Ltd.,
Beijing, China) antibodies for 1 h at room temperature.
Subsequently, the membranes were washed with Tris-buffered saline
containing 0.1% Tween. Densitometric analysis was performed using
Bio-Rad iQ5 image software (Bio-Rad Laboratories, Inc.), and the
ratio relative to GAPDH expression was calculated for each sample.
Blots were visualized using chemiluminescent solution (GSE Bio).
ROCK activity was assessed as the relative ratio of p-MYPT-1/total
MYPT-1.
Statistical analysis
All experiments were performed in triplicate. Data
are presented as the mean ± standard error of the mean. Differences
between the treatment groups were compared by unpaired t-test or
one-way analysis of variance, followed by the Student-Newman-Keuls
post-hoc test for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
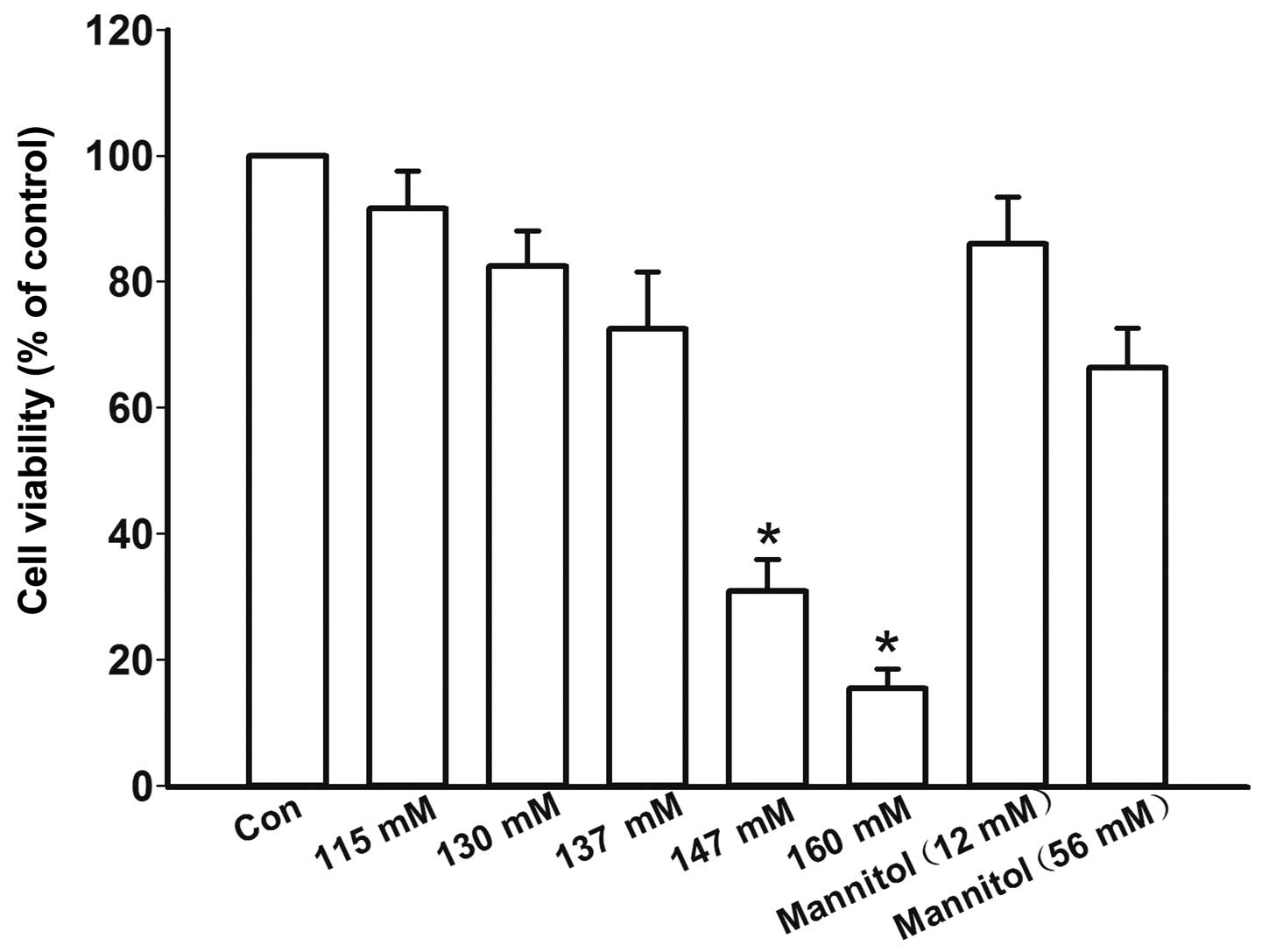
Effects of high salt medium on cell
viability
In order to determine the optimal high salt
concentration without affecting cell viability, the cells were
initially treated with high salt medium containing various
concentration of NaCl, or with mannitol as an osmotic control. High
salt medium at a concentration of 147 mM (30.1±5.0%) and 160 mM
(15.5±3.1%) significantly decreased cell viability compared with in
the control group (P=0.01 and P<0.001, respectively; Fig. 1). High salt medium at a
concentration of 115, 130 and 137 mM caused a slight decrease in
cell viability; however, the difference was statistically
insignificant among the groups (P=0.07, P=0.06 and P=0.06,
respectively, Fig. 1). Therefore,
115 and 137 mmol/l concentrations were chosen to treat the cells,
in order to observe the effects of high salt medium.
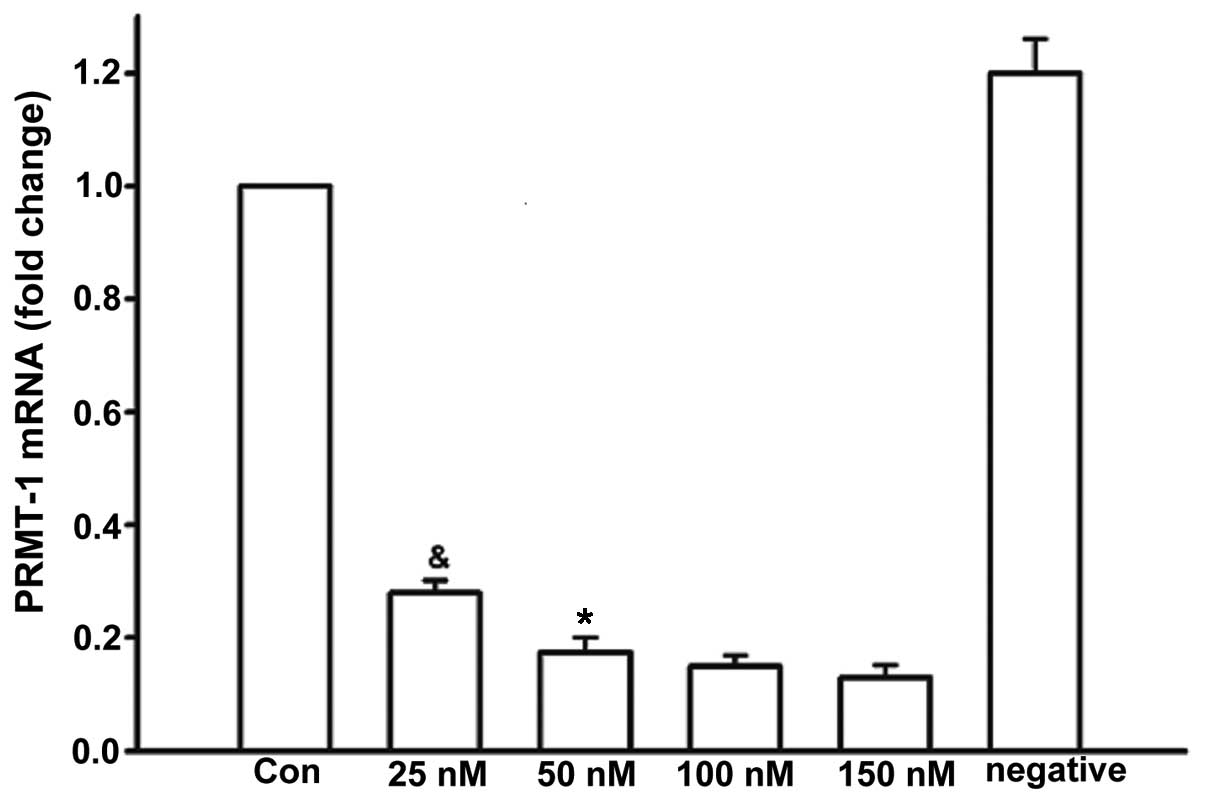
Optimization of siRNA-PRMT-1 cell
transfection
RNAi was used to knockdown the expression of PRMT-1,
which synthesizes ADMA. After 48 h, the gene silencing efficiency
of siRNA-PRMT-1 was detected using RT-qPCR. The mRNA expression
levels of PRMT-1 in the 25 nM group (0.28±0.02) were significantly
higher than in the 50 nM (0.18±0.03; P=0.03), 100 nM (0.15±0.02;
P=0.03) and 150 nM groups (0.13±0.02; P=0.02) (Fig. 2). Transfection with 50 nM
siRNA-PRMT-1 induced a five-fold reduction in PRMT-1 mRNA
expression compared with the control group (1.2±0.06; P=0.01;
Fig. 2). Since high concentrations
of siRNA may exhibit cytotoxicity, 50 nM was selected as the
concentration of siRNA used in subsequent experiments.
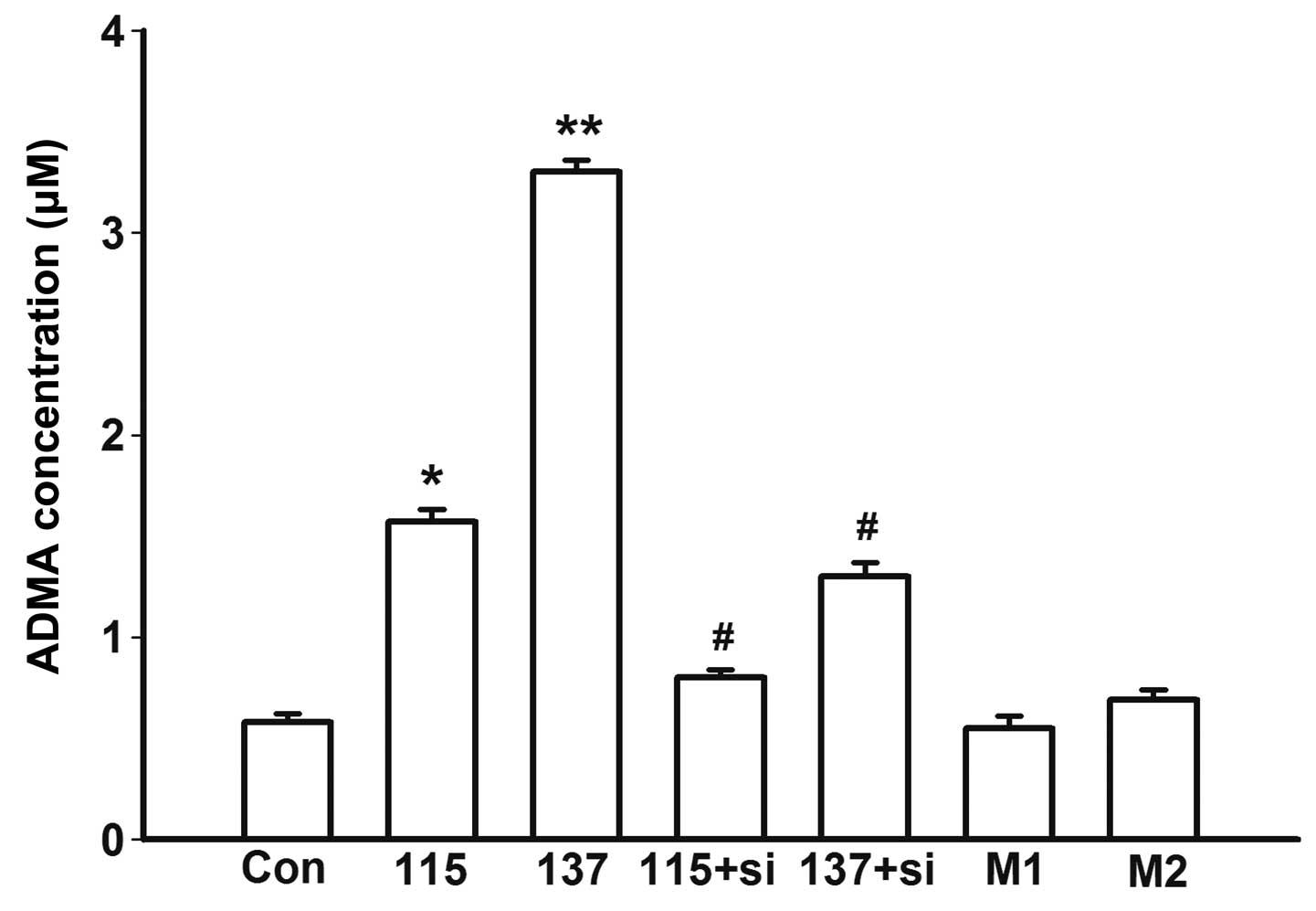
Effects of various treatments on ADMA
concentration
The cells were treated with high salt medium for 48
h, and the concentration of ADMA was measured in the culture
supernatants. Treatment with high salt medium induced a marked
increase in ADMA in a concentration-dependent manner (115 vs. Con,
P=0.01; 137 vs. Con, P<0.001; Fig.
3). The increased concentration of ADMA was verified to be
induced by high salt since there was no change in ADMA
concentration in the mannitol osmotic control groups (M1 vs. Con,
P=0.17; M2 vs. Con, P=0.25; Fig.
3).
Subsequently, RNAi was used to knockdown PRMT-1
expression, and the cells were treated with high salt medium for 48
h, prior to measurement of ADMA concentration in the supernatants.
ADMA concentration in the siRNA-transfected (50 nM) groups was
significantly decreased compared with in the untransfected groups
(115+si vs. 115, P=0.03; 137+si vs. 137, P=0.01; Fig. 3). Knockdown of PRMT-1 almost
abrogated the effects of high salt medium on ADMA secretion. These
results indicate that salt levels, but not osmotic pressure, in
high salt medium have an effect on ADMA biosynthesis (Fig. 3).
Effects of various treatments on the mRNA
and protein expression levels of PRMT-1, eNOS and RhoA, and ROCK
activity
In order to determine whether high salt treatment
was able to alter the expression of PRMT-1, eNOS, RhoA and ROCK,
the cells were cultured in high salt medium for 48 h and RNA and
protein were extracted (Figs. 4
and 5). mRNA expression levels
were quantified using RT-qPCR. Treatment with high salt medium
significantly increased the mRNA expression levels of PRMT-1 (137
vs. Con, P=0.02; Fig. 4A) and RhoA
(115 vs. Con, P=0.01; 137 vs. Con, P=0.01; Fig. 4B), and partially inhibited the
expression of eNOS (137 vs. Con, P=0.01; Fig. 4C). The same trend was detected at
the protein level (Fig. 5A–C and
E). The ratio of p-MYPT-1/total MYPT-1 (Thr 696) was used to
determine RhoA/ROCK activity (22). ROCK activity was significantly
elevated in response to high salt medium compared with the control
group (115 vs. Con, P=0.02; 137 vs. Con, P=0.01; Fig. 5D). Treatment with mannitol (56
mmol/l) significantly enhanced the protein expression of RhoA but
to a lower extent than high salt medium treatment (M2 vs. Con,
P=0.04; M2 vs. 137, P=0.03; Fig.
5C). Furthermore, high salt medium, but not osmotic pressure,
significantly increased MYPT-1 phosphorylation (115 vs. Con,
P=0.02; 137 vs. Con, P=0.01; Fig.
5D).
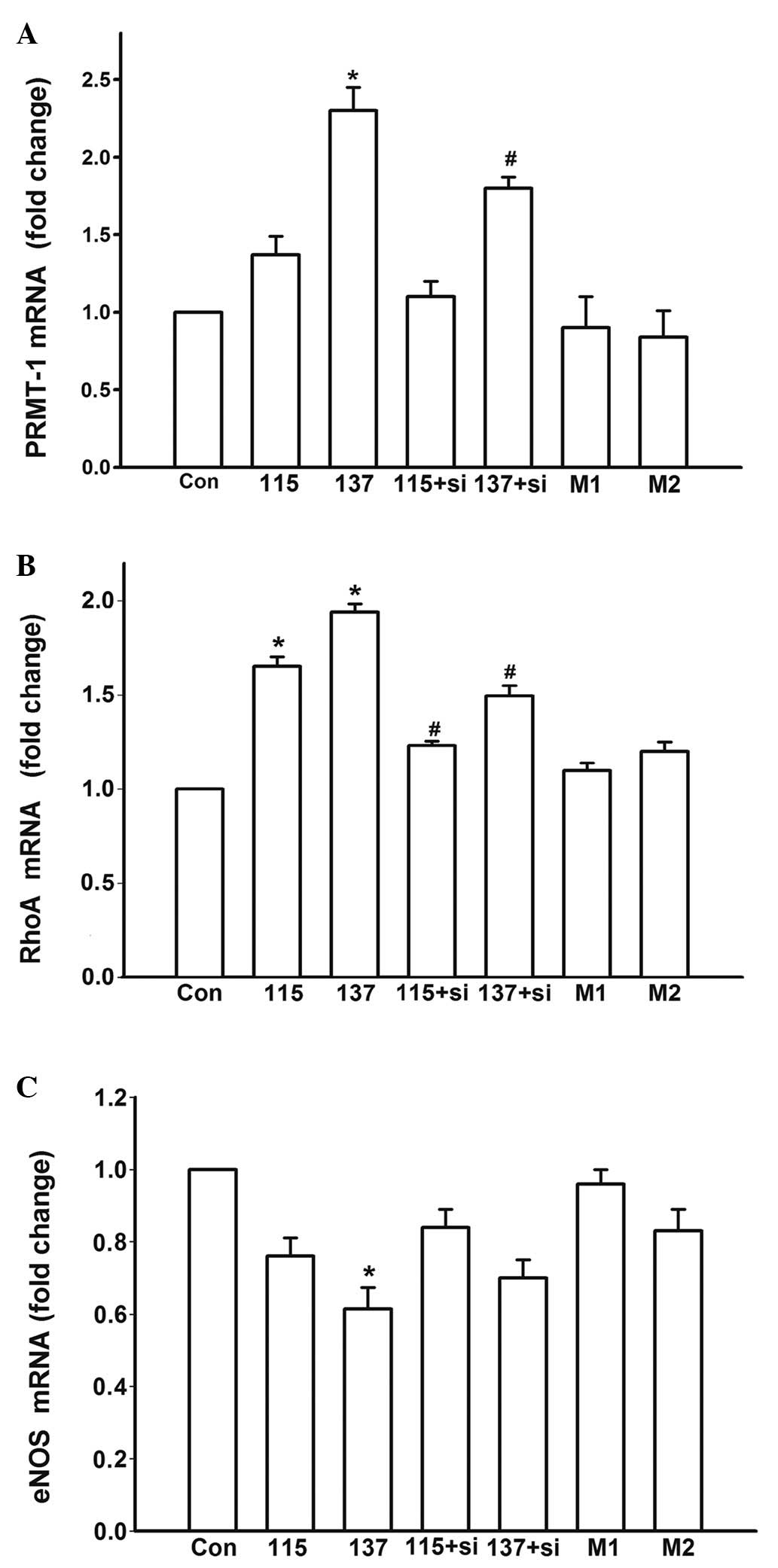 | Figure 4Effects of various treatments on the
mRNA expression levels of PRMT-1, RhoA and eNOS after a 48 h
incubation. Relative (A) PRMT-1, (B) RhoA and (C) eNOS mRNA
expression. Con, control group; 115+si, 115 mmol/l high salt medium
treatment and 50 nM siRNA-PRMT-1 transfection; 137+si, 137 mmol/l
high salt medium treatment and 50 nM siRNA-PRMT-1 transfection; M1,
12 mmol/l mannitol; M2, 56 mmol/l mannitol; si, small interfering
RNA; PRMT-1, protein arginine methyltransferase 1; eNOS,
endothelial nitric oxide synthase; RhoA, Ras homolog gene family,
member A. *P<0.05 compared with the Con group;
#P<0.05 compared with the high salt medium-treated
untransfected groups. |
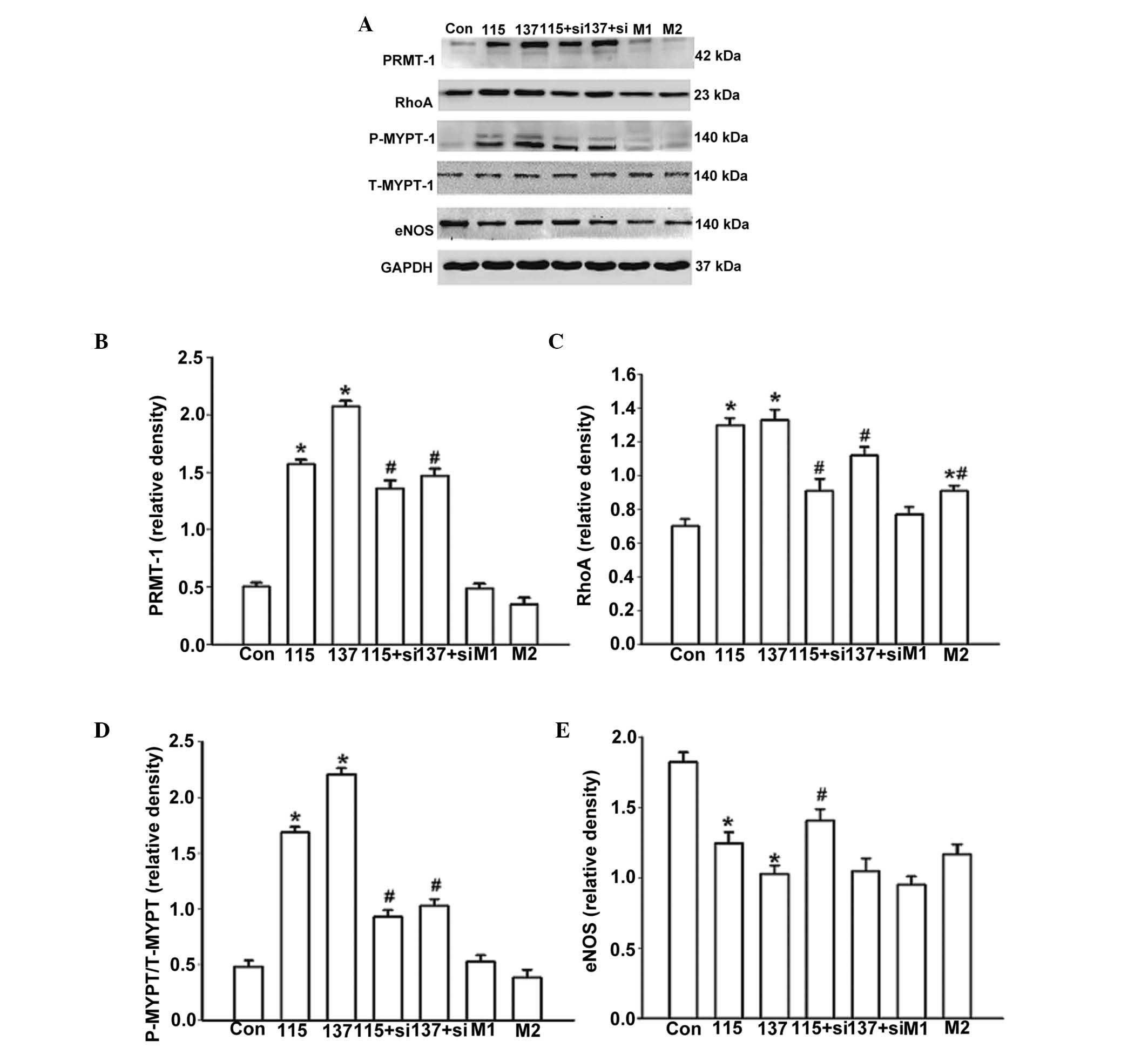 | Figure 5Effects of various treatments on the
protein expression of PRMT-1, eNOS, RhoA and ROCK after 48 h. (A)
Western blot analysis was conducted to detect the protein
expression levels of PRMT-1, RhoA, p-MYPT-1, total MYPT-1, eNOS and
GAPDH. Relative quantification of (B) PRMT-1 protein expression;
(C) RhoA protein expression; (D) ROCK activity and (E) eNOS protein
expression. Con, control group; 115+si, 115 mmol/l high salt medium
treatment and 50 nM siRNA-PRMT-1 transfection; 137+si, 137 mmol/l
high salt medium treatment and 50 nM siRNA-PRMT-1 transfection; M1,
12 mmol/l mannitol; M2, 56 mmol/l mannitol; si, small interfering
RNA;PRMT-1, protein arginine methyltransferase 1; eNOS, endothelial
nitric oxide synthase; RhoA, Ras homolog gene family, member A;
ROCK Rho-associated protein kinase. *P<0.05 compared
with the Con group; #P<0.05 compared with the high
salt medium-treated untransfected groups. |
To explore the potential signaling pathways, cells
were treated with siRNA-PRMT-1 to inhibit ADMA production. The
aforementioned high salt-induced alterations of gene expression
were significantly inhibited following PRMT-1 knockdown (137+si vs.
137, P=0.02, Fig. 4A; 115+si vs.
115, P=0.03; 137+si vs. 137, P=0.02, Fig. 4B; 115+si vs. 115, P=0.03; 137+si
vs. 137, P=0.02, Fig. 4B; 115+si
vs. 115, P=0.04; 137+si vs. 137, P=0.02, Fig. 5B; 115+si vs. 115, P=0.02, Fig. 5E). Specific PRMT-1 RNAi inhibited
RhoA/ROCK activity (115+si vs. 115, P=0.02; 137+si vs. 137, P=0.03,
Fig. 5C; 115+si vs. 115, P=0.01;
137+si vs. 137, P=0.01, Fig. 5D).
These results strongly indicate that to some extent, high salt
intake may regulate the expression of RhoA and eNOS via
downregulation of ADMA.
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate that high salt medium significantly
upregulates ADMA concentration, PRMT-1 and RhoA expression, and
ROCK activity, and downregulates eNOS expression in EA.hy926 cells.
In addition, these effects could be partially attenuated when
PRMT-1 expression was silenced by RNAi.
Previous studies have reported that plasma ADMA
levels are increased after salt loading and decreased after salt
restriction in salt-sensitive subjects (7,23,24).
Animal experiments have also demonstrated that salt loading induces
an acute increase in oxidative stress, which contributes to
salt-sensitive hypertension (25,26).
Furthermore, elevation of NaCl induces oxidative stress and ROS
elevation in vitro (27–29).
The activities of PRMTs and eNOS have previously been revealed to
be regulated in a redox-sensitive fashion (30). ROS and ADMA are able to form a
tightly coupled amplification system, thus aggravating the
pathological progression of endothelial dysfunction. The present
study demonstrated that the expression levels of PRMT-1 and ADMA
were elevated in response to high salt medium in a dose-dependent
manner. Therefore, it may be hypothesized that high salt medium
induces oxidative stress and stimulates ADMA production via the
upregulation of PRMT-1.
Hypertonic stress induces activation of the
RhoA/ROCK signaling pathway in cells (31,32).
These previous findings are consistent with the findings of the
present study, which demonstrated that high salt medium and
mannitol (56 mmol/l) were able to induce RhoA upregulation;
however, the extent of the increase was lower in the mannitol
groups than in the high salt medium groups (115 and 137 mmol/l).
These results indicated that, in addition to osmotic pressure, salt
is the main driving force associated with the upregulation of RhoA
expression and activity. PRMT-1 expression was unaffected by an
increase in mannitol, thus excluding the possibility that the gene
alterations were induced by osmolality.
The present study knocked down PRMT-1 expression
using siRNA, in order to explore the role of ADMA in high
salt-mediated RhoA/ROCK activation and eNOS inhibition. PRMTs are a
family of proteins that either monomethylate or dimethylate the
guanidino nitrogen atoms on arginine side chains. PRMT-1 is the
predominant type I PRMT in mammalian cells, which catalyzes
arginine residues and facilitates the formation of ADMA residues
(12). Aberrant PRMT expression
has been reported to contribute to the pathogenesis of pulmonary
diseases (33). The present study
demonstrated that PRMT-1-specific siRNA attenuated high
salt-induced increases in ADMA secretion, confirming that PRMT-1 is
one of the key upstream regulators of ADMA secretion, and that the
knockdown of PRMT-1 is sufficient to decrease ADMA secretion. Via
knockdown of PRMT-1, the levels of ADMA could be manipulated, and
the hypotheses that ADMA has an important role in high salt-induced
RhoA pathway activation and eNOS inhibition could be verified. The
results of the present study indicated that downregulation of ADMA
was able to inhibit the upregulated expression and activity of RhoA
and the expression of eNOS. Therefore, ADMA secretion and RhoA/ROCK
activation may exert synergistic effects on eNOS expression in
response to high salt medium.
In conclusion, the results of the present study
indicated that high salt medium can increase ADMA secretion,
activate RhoA activity and inhibit eNOS expression. Furthermore,
ADMA was revealed to have a crucial role in the mediation of these
effects. The present study was the first, to the best of our
knowledge, to identify a mechanism and provide information
regarding how high salt intake induces endothelial dysfunction
in vitro. PRMT-siRNA may be considered as a novel gene
therapy used to reduce ADMA production. The present study provided
a novel theoretical basis for the prevention and treatment of
salt-sensitive hypertension.
Acknowledgments
The present study was supported by funds from the
Nature Science Foundation of China (grant no. 30900616); the
National Program on Key Basic Research Project of China (973
Program; grant no. 2012CB517804); the Department of Cardiovascular
Medicine, First Affiliated Hospital of Medical College of Xi'an
Jiaotong University; the Institute of Cardiovascular Channelopathy,
Key Laboratory of Environment and Genes Related to Diseases (Xi'an
Jiaotong University), Ministry of Education; and the Key Laboratory
of Molecular Cardiology, Shannxi Province.
References
|
1
|
Miyoshi A, Suzuki H, Fujiwara M, Masai M
and Iwasaki T: Impairment of endothelial function in salt-sensitive
hypertension in humans. Am J Hypertens. 10:1083–1090. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Félétou M and Vanhoutte PM: Endothelial
dysfunction: A multifaceted disorder (The Wiggers Award Lecture).
Am J Physiol Heart Circ Physiol. 291:H985–H1002. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(23 Suppl 1):
III27–III32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Förstermann U and Münzel T: Endothelial
nitric oxide synthase in vascular disease: From marvel to menace.
Circulation. 113:1708–1714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bragulat E and de la Sierra A: Salt
intake, endothelial dysfunction, and salt-sensitive hypertension. J
Clin Hypertens (Greenwich). 4:41–46. 2002. View Article : Google Scholar
|
|
6
|
Toda N and Arakawa K: Salt-induced
hemodynamic regulation mediated by nitric oxide. J Hypertens.
29:415–424. 2011. View Article : Google Scholar
|
|
7
|
Schmidlin O, Forman A, Leone A, Sebastian
A and Morris RC Jr: Salt sensitivity in blacks: Evidence that the
initial pressor effect of NaCl involves inhibition of
vasodilatation by asymmetrical dimethylarginine. Hypertension.
58:380–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Riento K and Ridley AJ: Rocks:
Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol.
4:446–456. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kobayashi N, Takeshima H, Fukushima H,
Koguchi W, Mamada Y, Hirata H, Machida Y, Shinoda M, Suzuki N,
Yokotsuka F, et al: Cardioprotective effects of pitavastatin on
cardiac performance and remodeling in failing rat hearts. Am J
Hypertens. 22:176–182. 2009. View Article : Google Scholar
|
|
10
|
Loirand G and Pacaud P: The role of Rho
protein signaling in hypertension. Nat Rev Cardiol. 7:637–647.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laufs U and Liao JK: Post-transcriptional
regulation of endothelial nitric oxide synthase mRNA stability by
Rho GTPase. J Biol Chem. 273:24266–24271. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang J, Frankel A, Cook RJ, Kim S, Paik
WK, Williams KR, Clarke S and Herschman HR: PRMT1 is the
predominant type I protein arginine methyltransferase in mammalian
cells. J Biol Chem. 275:7723–7730. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fiedler L and Wojciak-Stothard B: The
DDAH/ADMA pathway in the control of endothelial cell migration and
angiogenesis. Biochem Soc Trans. 37:1243–1247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li XH, Peng J, Tan N, Wu WH, Li TT, Shi RZ
and Li YJ: Involvement of asymmetric dimethylarginine and Rho
kinase in the vascular remodeling in monocrotaline-induced
pulmonary hypertension. Vascul Pharmacol. 53:223–229. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fiedler L: The DDAH/ADMA pathway is a
critical regulator of NO signalling in vascular homeostasis. Cell
Adh Migr. 2:149–150. 2008. View Article : Google Scholar
|
|
16
|
Wojciak-Stothard B, Torondel B, Zhao L,
Renné T and Leiper JM: Modulation of Rac1 activity by ADMA/DDAH
regulates pulmonary endothelial barrier function. Mol Biol Cell.
20:33–42. 2009. View Article : Google Scholar :
|
|
17
|
de Wardener HE, He FJ and MacGregor GA:
Plasma sodium and hypertension. Kidney Int. 66:2454–2466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He FJ, Markandu ND, Sagnella GA, de
Wardener HE and MacGregor GA: Plasma sodium: Ignored and
underestimated. Hypertension. 45:98–102. 2005. View Article : Google Scholar
|
|
19
|
Orlov SN and Mongin AA: Salt-sensing
mechanisms in blood pressure regulation and hypertension. Am J
Physiol Heart Circ Physiol. 293:H2039–H2053. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oberleithner H, Kusche-Vihrog K and
Schillers H: Endothelial cells as vascular salt sensors. Kidney
Int. 77:490–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
van Nieuw Amerongen GP and van Hinsbergh
VW: Cytoskeletal effects of Rho-like small guanine
nucleotide-binding proteins in the vascular system. Arterioscler
Thromb Vasc Biol. 21:300–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang Y, Mu JJ, He LC, Wang SC and Liu ZQ:
Salt loading on plasma asymmetrical dimethylarginine and the
protective role of potassium supplement in normotensive
salt-sensitive Asians. Hypertension. 48:724–729. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujiwara N, Osanai T, Kamada T, Katoh T,
Takahashi K and Okumura K: Study on the relationship between plasma
nitrite and nitrate level and salt sensitivity in human
hypertension: Modulation of nitric oxide synthesis by salt intake.
Circulation. 101:856–861. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou MS, Adam AG, Jaimes EA and Raij L: In
salt-sensitive hypertension, increased superoxide production is
linked to functional upregulation of angiotensin II. Hypertension.
42:945–951. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taylor NE, Maier KG, Roman RJ and Cowley
AW Jr: NO synthase uncoupling in the kidney of Dahl S rats: Role of
dihydrobiopterin. Hypertension. 48:1066–1071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang T, Zhang A, Honeggar M, Kohan DE,
Mizel D, Sanders K, Hoidal JR, Briggs JP and Schnermann JB:
Hypertonic induction of COX-2 in collecting duct cells by reactive
oxygen species of mitochondrial origin. J Biol Chem.
280:34966–34973. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Z, Dmitrieva NI, Park JH, Levine RL
and Burg MB: High urea and NaCl carbonylate proteins in renal cells
in culture and in vivo, and high urea causes 8-oxoguanine lesions
in their DNA. Proc Natl Acad Sci USA. 101:9491–9496. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou X, Ferraris JD and Burg MB:
Mitochondrial reactive oxygen species contribute to high
NaCl-induced activation of the transcription factor TonEBP/OREBP.
Am J Physiol Renal Physiol. 290:F1169–F1176. 2006. View Article : Google Scholar
|
|
30
|
Sydow K and Münzel T: ADMA and oxidative
stress. Atheroscler Suppl. 4:41–51. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Di Ciano-Oliveira C, Sirokmány G, Szászi
K, Arthur WT, Masszi A, Peterson M, Rotstein OD and Kapus A:
Hyperosmotic stress activates Rho: Differential involvement in Rho
kinase-dependent MLC phosphorylation and NKCC activation. Am J
Physiol Cell Physiol. 285:C555–C566. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Malek AM, Xu C, Kim ES and Alper SL:
Hypertonicity triggers RhoA-dependent assembly of myosin-containing
striated polygonal actin networks in endothelial cells. Am J
Physiol Cell Physiol. 292:C1645–C1659. 2007. View Article : Google Scholar
|
|
33
|
Zakrzewicz D, Zakrzewicz A, Preissner KT,
Markart P and Wygrecka M: Protein arginine methyltransferases
(PRMTs): Promising targets for the treatment of pulmonary
disorders. Int J Mol Sci. 13:12383–12400. 2012. View Article : Google Scholar : PubMed/NCBI
|