Introduction
Coronary artery disease (CAD) is a leading cause of
mortality (1). Similar to other
common complex diseases, the pathogenesis of CAD and associated
myocardial infarction (MI) is multifactorial, and is influenced by
complex interactions between environmental and genetic factors
(1). Several risk factors,
including hypertension, dyslipidemia, obesity and smoking, have
been established for CAD (2).
Genome-wide association studies have uncovered several
susceptibility loci and candidate genes that are associated with
CAD, either by directly participating in the pathogenesis of CAD or
by indirectly regulating the contributing risk factors (3–10).
As a multifactorial complex disease, common sequence variants or
mutations in numerous genes are commonly associated with CAD
(4). In these cases, the genetic
contribution of each gene is relatively small; however, it has
previously been suggested that CAD/MI may manifest via autosomal
dominant inheritance in some families (4).
A mutation in the human myocyte enhancer factor 2A
(MEF2A) gene, which is a member of the myocyte enhancer
family of transcription factors, has previously been detected in an
autosomal dominant form of CAD (1). Genetic linkage analysis of a large
Caucasian family exhibiting an autosomal dominant inheritance
pattern of premature CAD indicated a positive linkage to a single
locus on chromosome 15q26, which includes ~90 annotated genes.
Resequencing of the MEF2A gene, which is a prime candidate
gene in the linked locus, revealed a 21-base pair (bp) coding
sequence deletion at exon 11 in all affected family members
(1). Although this initial study
suggested the involvement of MEF2A variants in the risk of
CAD/MI, they have not been supported by more recent reports. Weng
et al identified these variants in elderly Caucasian control
subjects without CAD (11),
whereas other studies found no evidence of any linkage or
association between MEF2A and CAD in 1,700 patients with
sporadic MI and multiple families with apparent Mendelian
inheritance of the disease (12,13).
These findings suggested that these mutations may be rare and
isolated only to families exhibiting premature CAD, or that the
MEF2A gene is unrelated to CAD. The present study aimed to
identify the genetic defect responsible for familial premature
CAD/MI in an extended Chinese Han pedigree of 34 members exhibiting
an autosomal dominant pattern.
Materials and methods
Participants and clinical evaluation
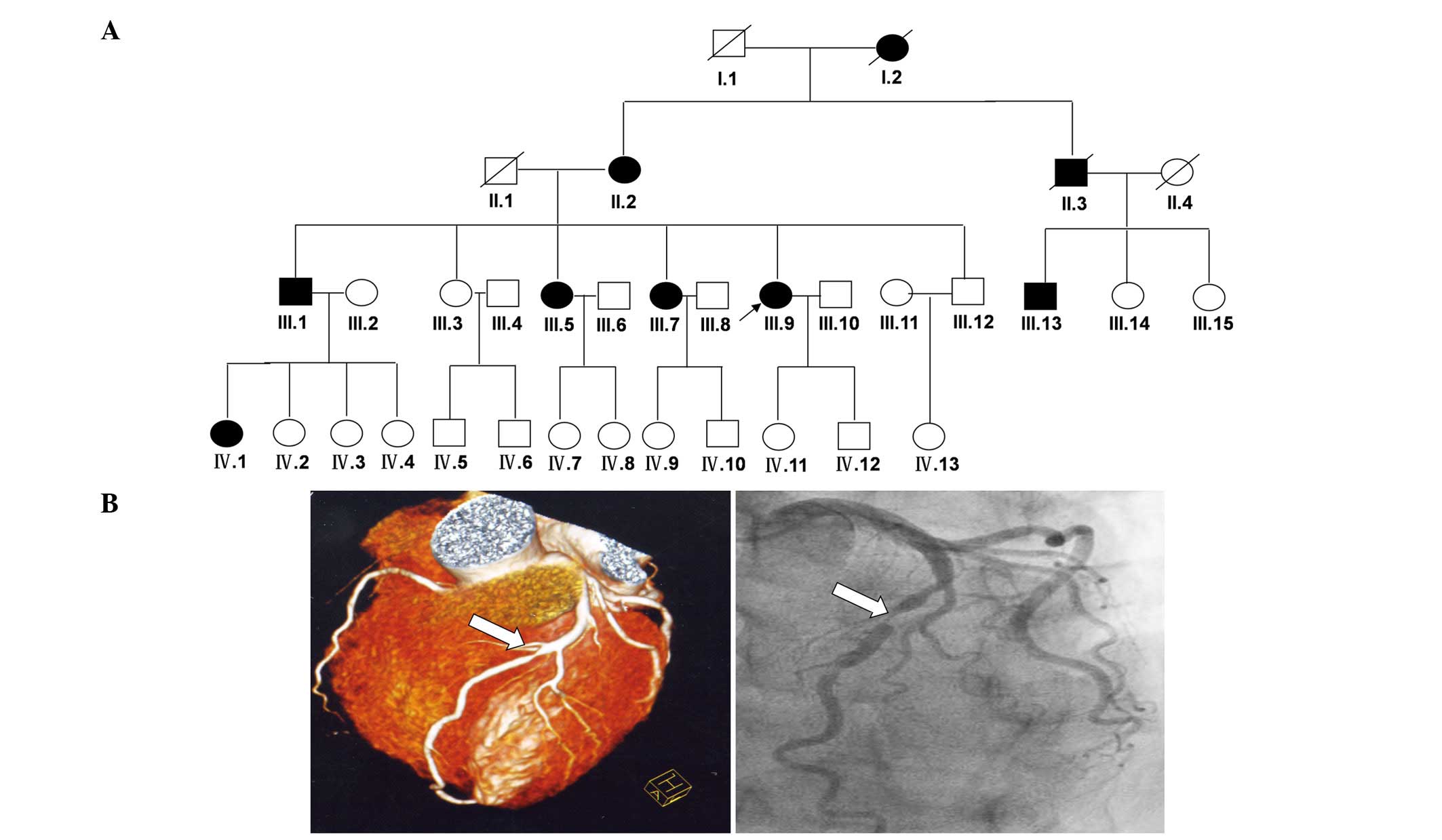
A four-generation, 34-member Chinese Han family with
familial CAD was recruited from the Qilu Hospital, Shandong
University (Jinan, China) through reviewing the records of patients
displaying the clinical features of CAD/MI. CAD/MI in this family
followed an autosomal dominant pattern of inheritance (Fig. 1A). Upon basic clinical examination,
members of the family with preceding or existing indications of
CAD/MI (based on the existence of at least two of the following
criteria: Prolonged chest pain, electrocardiography patterns
consistent with acute MI, or significant elevation of cardiac
enzymes) underwent coronary computed tomography analysis. Coronary
angiograms were subsequently carried out on all subjects to confirm
a diagnosis of CAD (Fig. 1B).
According to angiographic appearance, a vessel was regarded as
diseased if it contained at least one stenosis involving >50%
loss of lumen diameter. Seven living patients in this family were
identified as having CAD (II-2, III-1, III-5, III-7, III-9, III-13
and IV-1) (Fig. 1A). In addition,
all family members were subjected to a physical examination, blood
testing, and a standardized interview that included questions
related to medical history, physical activity, medication and
personal habits (Table I). In
addition, blood pressure was taken according to the MONICA
guidelines (3), using the
random-zero method and using standard mercury sphygmomanometers
after the subjects had been resting in a seated position (Table I). The control group consisted of
311 patients (mean age, 46.3±10.12; male/female, 151/160) who had
attended the Cardiology Departments of the Qilu Hospital between
2005 and 2008, and had suffered a first episode of MI, as defined
according to World Health Organization criteria (14). The healthy control group consisted
of 323 Chinese individuals (mean age: 46 years), including both
obese and normal weight subjects, without a history of premature
CAD, who were recruited separately from the Jinan region. All
subjects provided written informed consent for the present study,
which was approved by the ethics committee of Qilu Hospital,
Shandong University.
 | Table IClinical characteristics of family
members. |
Table I
Clinical characteristics of family
members.
| ID no. | Current age
(years) | Gender | Premature CAD | TC | TG | LDL-C | HDL-C | TC/HDL-C | HTN | Smoker | BMI | FBG |
|---|
| I-1 | – | M | – | – | – | – | – | – | – | – | – | – |
| I-2 | – | F | – | – | – | – | – | – | – | – | – | – |
| II-1 | | M | – | – | – | – | – | – | – | – | – | – |
| II-2 | 84 | F | Yes | 183.6 | 83.7 | 115.6 | 65.73 | 2.79 | No | No | 22.2 | 5.8 |
| II-3 | – | M | – | – | – | – | – | – | – | – | – | – |
| II-4 | – | F | – | – | – | – | – | – | – | – | – | – |
| III-1 | 59 | M | Yes | 179.1 | 71.5 | 131.2 | 69.4 | 2.58 | No | No | 24.1 | 4.7 |
| III-2 | 60 | F | No | 221.8 | 88.5 | 148.2 | 79.7 | 2.78 | No | No | 24.7 | 4.9 |
| III-3 | 58 | F | No | 200.4 | 76.1 | 124.2 | 83.9 | 2.38 | No | No | 22.3 | 5.1 |
| III-4 | 59 | M | No | 213.5 | 89.3 | 138.9 | 83.9 | 2.54 | No | No | 21.1 | 5.5 |
| III-5 | 55 | F | Yes | 121.9 | 77.9 | 65.4 | 43.9 | 2.77 | No | No | 23.4 | 4.2 |
| III-6 | 56 | M | No | 235.7 | 101.7 | 143.5 | 99.8 | 2.36 | No | No | 21.6 | 3.9 |
| III-7 | 53 | F | Yes | 143.6 | 46.9 | 79.3 | 91.6 | 1.57 | No | No | 22.7 | 5.3 |
| III-8 | 51 | M | No | 223.5 | 101.2 | 131.5 | 86.4 | 2.59 | No | No | 23 | 5.6 |
| III-9 | 49 | F | Yes | 145.9 | 86.7 | 90.8 | 53.5 | 2.73 | No | No | 26.8 | 5.1 |
| III-10 | 49 | M | No | 201.6 | 90.4 | 126.3 | 81.4 | 2.47 | No | No | 22.8 | 3.8 |
| III-11 | 46 | F | UK | 173.8 | 61.9 | 115.3 | 59.3 | 2.93 | No | No | 24.1 | 4.6 |
| III-12 | 41 | M | No | 207.2 | 74.8 | 121.3 | 81.7 | 2.54 | No | No | 22.6 | 5.3 |
| III-13 | 57 | M | Yes | 167.7 | 88.5 | 101.2 | 71.7 | 2.34 | No | No | 24.5 | 4.3 |
| III-14 | 49 | F | No | 146.2 | 94.8 | 111.1 | 61.5 | 2.38 | No | No | 23.9 | 5.8 |
| III-15 | 51 | F | No | 168.6 | 90.6 | 131.1 | 73.4 | 2.3 | No | No | 22.8 | 5.2 |
| IV-1 | 41 | F | Yes | 143.9 | 47.5 | 76.9 | 93.9 | 1.53 | No | No | 20.6 | 4.4 |
| IV-2 | 38 | F | No | 220.9 | 124.8 | 137.4 | 78.5 | 2.81 | No | No | 23.8 | 3.7 |
| IV-3 | 35 | F | UK | 216.7 | 36.3 | 144.7 | 97.8 | 2.21 | No | No | 22.4 | 4.8 |
| IV-4 | 32 | F | No | 142.4 | 56.6 | 89.4 | 65.5 | 2.18 | No | No | 26.6 | 5.5 |
| IV-5 | 36 | M | No | 226 | 312.4 | 127.3 | 44.7 | 5.06 | No | No | 22.8 | 3.9 |
| IV-6 | 33 | M | No | 153.6 | 109.7 | 93.7 | 48.5 | 3.17 | No | No | 19.9 | 4.2 |
| IV-7 | 30 | F | UK | 193.5 | 84.9 | 118.8 | 66.9 | 2.89 | No | No | 23.1 | 4.7 |
| IV-8 | 28 | F | UK | 179.7 | 67.4 | 113.5 | 61.5 | 2.92 | No | No | 25.9 | 5.1 |
| IV-9 | 28 | F | No | 172.6 | 46.2 | 92.9 | 102 | 1.69 | No | No | 19.8 | 4.7 |
| IV-10 | 10 | M | UK | 177.3 | 65.9 | 87.6 | 103.7 | 1.71 | No | No | 21.4 | 5 |
| IV-11 | 27 | F | UK | 178.8 | 90.3 | 77.8 | 106 | 1.68 | No | No | 22.3 | 4.1 |
| IV-12 | 26 | M | No | 159.8 | 37.2 | 93.7 | 78.93 | 2.02 | No | No | 24.3 | 5.3 |
| IV-13 | 22 | F | No | 166.8 | 62.8 | 99.1 | 81.62 | 2.04 | No | No | 22.1 | 4.7 |
Exome capture
Genomic DNA was extracted from peripheral blood
using the standard phenol-chloroform extraction method (15). The genomic DNA of three patients in
the Qilu hospital (III-1, III-5 and III-7) was sheared by
sonication and was then hybridized to the Nimblegen SeqCap EZ
Library (Roche Diagnostics, Basel, Switzerland), in order to enrich
exonic DNA in each library, according to the manufacturer's
protocol. Sequencing of the enriched library was performed using
the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA) to
generate 90-bp paired-end reads (16). A mean exome coverage of 78.87x was
obtained, allowing each selected region of the genome to be
checked. Such coverage provided sufficient depth to accurately call
variants at 99.34% of the targeted exome (17).
Read mapping and variant analysis
The human reference genome was obtained from the
online University of California, Santa Cruz database (http://genome.ucsc.edu/), version hg19 (build 37.1).
Alignment of patient sequences was performed using the Short
Oligonucleotide Analysis Package (SOAP) aligner (soap2.21;
http://soap.genomics.org.cn/soapsnp.html) and single
nucleotide polymorphisms (SNPs) were called using the SOAP snp set
with the default parameters, after the duplicated reads [obtained
mainly in the polymerase chain reaction (PCR) step] were deleted.
PCR was performed accoreding to the protocol of Illumina Paired-End
Sample Prep Kit (Illumina). A reaction volume of 50 µl was
used, containing 100 ng gDNA and 10 pmol of each primer, and PCR
was performed in a 9700 Thermal Cycler system (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The reaction was
performed for 35 cycles of dena turation at 95°C for 30 sec,
annealing at 58°C for 35 sec and extension at 72°C for 50 sec, and
a final extension step at 72°C for 5 min. Small insertions or
deletions (indels) affecting coding sequence or splicing sites were
detected (16,17). The thresholds for calling SNPs and
short indels included the number of unique mapped reads supporting
a SNP ≥4 and a consensus quality score ≥20. The quality score
represents a Phred score, generated by the program SOAP snp 1.05,
where quality = −10log (error rate). It is unlikely that causative
variants are present in the general population. All candidate
variations identified in the patient sequences were filtered
against the SNP database (dbSNP build 137; http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi),
1000 Genomes Project (1000genomes release_20100804; http://www.1000genomes.org/), HapMap project
(2010-08_phase II+III; http://hapmap.ncbi.nlm.nih.gov/) and YanHuang project
(http://yh.genomics.org.cn/). Sorting
intolerant from tolerant prediction (http://sift.jcvi.org/) was performed to evaluate
whether amino acid substitutions, amino acid indels and
frameshifting indels could affect protein function (16).
Mutation validation
Locus-specific PCR and detection primers were
designed (Boshang Biotechnology Company, Jinan, China). Sanger
sequencing was performed to determine the presence and identity of
potential disease-causing variants using the ABI3500 sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). PCR
amplification and Sanger sequencing were conducted as described
previously (18). The primer
sequences used to identify MEF2A disease-linked variants
were as follows: forward, 5′-GCATCAAGTCCGAACCGATT-3′, and reverse,
5′-GGAGCGACCCATTTCCTGTC-3′.
Results
Clinical characteristics of the
family
The present study identified an extended Chinese
family containing 34 members (five members deceased) with a history
of CAD/MI (Fig. 1A). The family
consisted of 20 females and 14 males distributed in four
generations; nine members were diagnosed with CAD/MI (two of which
were deceased). The proband (III-9) with CAD was identified at the
Department of Cardiology, Qilu Hospital, Shandong University at 36
years of age, with right coronary artery stenosis with >80%
severity (Table II).
Subsequently, the patient's two elder sisters, one elder brother,
and male cousin developed symptoms of CAD. Subjects III-1 [left
anterior descending coronary artery (LAD) angiogram >75%
stenosis] and III-13 (RCA angiogram >80% stenosis) were
diagnosed at the ages of 49 and 46, respectively; subjects III-5
(LAD angiogram >90% stenosis) and III-7 (LAD angiogram >90%
stenosis) were diagnosed at the ages of 43 and 45, respectively.
Subject III-1 was diagnosed 10 years ago, at the age of 49 years
old. Subjects III-1 and III-5 suffered MI and stroke at the ages of
49 and 51 years old, respectively. The severity of the disease in
female patients was greater than that in males (Table II). As shown in Table I, a few members of the family
exhibited mildly elevated serum levels of total cholesterol and
triglycerides, but all had normal serum levels of low-density and
high-density lipoproteins, and none of the family members were
cigarette smokers or had hypertension, diabetes or obesity. These
clinical manifestations strongly suggested heritable CAD in this
family. Pedigree analysis of the family suggested autosomal
dominant inheritance of CAD (Fig.
1A).
| Table IICharacteristics of family members
with coronary artery disease and myocardial infarction (MI). |
Table II
Characteristics of family members
with coronary artery disease and myocardial infarction (MI).
| Individual ID
No. | Current age
(years) | Age at time of
diagnosis (years) | Clinical
diagnosis |
|---|
| II-2 | 84 | 51 | MI, stroke |
| III-1 | 59 | 49 | LAD angiogram
>75% stenosis |
| III-5 | 55 | 43 | LAD angiogram
>90% stenosis |
| III-7 | 53 | 45 | LAD angiogram
>90% stenosis |
| III-9 | 49 | 36 | RCA angiogram
>80% stenosis |
| III-13 | 57 | 46 | RCA angiogram
>80% stenosis |
| IV-1 | 41 | 40 | LAD angiogram
>50% stenosis |
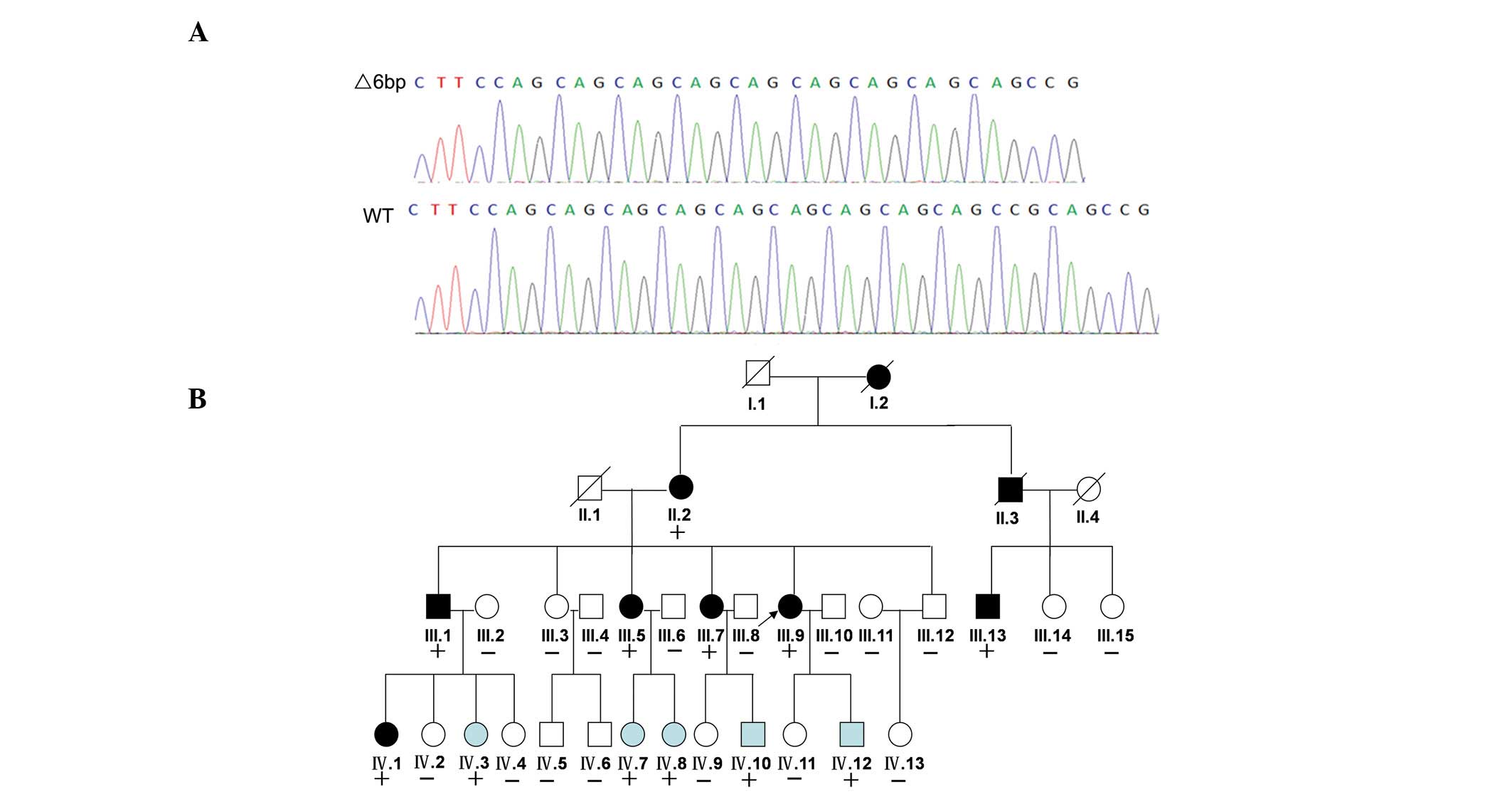
Identification of a 6-bp deletion in the
MEF2A gene
Using the filtering criteria as described previously
(19), MEF2A was identified
as a CAD-causing candidate gene after exome sequencing of genomic
DNA. Following validation by Sanger sequencing, a 6-bp deletion
(CAGCCG) was identified in exon 11 in all seven family members with
CAD and in five non-CAD members (IV-3, IV-7, IV-8, IV-10 and
IV-12). The 6-bp deletion was located at position 1671 to 1677 in
the cDNA sequence of the MEF2A gene (Fig. 2A). The CAGCCG deletion also
contained the first CAG repeat of the (CAG)n repeats polymorphism
in exon 11, which resulted in variable expression and was
associated with CAD (5). The 6-bp
deletion was identified in family members with a normal phenotype
(IV-3, IV-7, IV-8, IV-10 and IV-12) and segregated with CAD in the
family, thus suggesting that this variant is the pathogenic
mutation (Fig. 2B). The five
family members (IV-3, IV-7, IV-8, IV-10 and IV-12) with the
MEF2A deletion but without CAD likely have not yet developed
the phenotype due to their younger ages (all <40 years old). In
order to deter-mine whether the mutation is present in sporadic CAD
cases, the exon 11 coding sequence of MEF2A was sequenced in
311 unrelated subjects with an established diagnosis of CAD and in
323 healthy subjects. The 6-bp deletion was not detected in these
subjects. The entire coding region of MEF2A and the
intron-exon boundaries were also screened for mutations in all
members of the family. No mutations were identified in any other
exons (data not shown).
Discussion
The present study identified a novel mutation in
MEF2A in a Chinese family with inherited CAD. To the best of
our knowledge, the present study is the first to report a causative
association between a MEF2A mutation and CAD in the Chinese
population, and a novel mutation of MEF2A due to a 6-bp
deletion in exon 11.
Wang et al reported a possible role for
MEF2A variants in the pathogenesis of CAD, describing a
21-bp deletion as the disease-causing genetic mutation for
Caucasian familial CAD/MI without common risk factors (1). A subsequent functional study revealed
that the 21-bp MEF2A gene mutation resulted in the deletion
of seven amino acids in exon 11 of MEF2A, thus disrupting
the transcriptional activity and blocking nuclear localization of
the MEF2A protein (1). In a
previous study, three genetic variants of the MEF2A gene
(N263S, P279L and G283D) were detected in four out of 207 unrelated
Caucasian patients (1.9%) with CAD (7). Furthermore, a (CAG)n repeat in exon
11 has been reported to be associated with CAD in a small Chinese
case-control study (5). These data
suggested that MEF2A may have a significant role in the
pathogenesis of CAD in non-familial (sporadic) cases. However, this
hypothesis was not supported by subsequent studies. Weng et
al did not detect an MI causative MEF2A mutation in 300
cases of sporadic CAD in Caucasian patients (11). Furthermore, Lieb et al
failed to detect the 21-bp deletion in the MEF2A gene in
1,481 individuals with a positive family history of CAD (3).
The present study identified a MEF2A gene
mutation in a family with CAD. To the best of our knowledge, this
is only the second report to identify a MEF2A mutation in a
family with CAD (4). These results
strongly supported a causative role of MEF2A gene mutations
in the pathogenesis of CAD. High MEF2A expression in the
endothelium of coronary arteries suggests that an early step, or
triggering event, in the development of CAD may involve the
dysregulation of specific MEF2A transcriptional pathways in
the endothelium, which is expected to result in endothelial
dysfunction (5,10). Endothelial dysfunction is
associated with atherosclerotic plaque formation and rupture, and
subsequent thrombosis, which are common causes of MI and sudden
cardiac death (6,7).
The present study demonstrated that the effects of
MEF2A and its mutation on the pathogenesis of CAD are not
confined to a single ethnic group, since it was originally reported
in a Caucasian family (1). It has
been well established that CAD is a multifactorial disease that is
associated with an array of genes and their variants (8). Previous studies have clearly
demonstrated that CAD is a multifactorial disease that is affected
by multiple genes. It is therefore uncommon to observe a
multifactorial common disease manifesting in a dominant Mendelian
inheritance unless the mutation has a predominant effect on the
pathogenesis of CAD/MI (9). In
these situations, the functional effects of the mutation on the
target gene are significant and dominant, and therefore may
override other risk factors and induce pathological outcomes in
subjects with the mutation. The lack of MEF2A mutations in
sporadic CAD cases may be attributed to two factors. Firstly, the
mutation may be too rare to be detectable in 311 subjects with CAD;
therefore, a much larger number of subjects may be needed in order
to detect the mutation. Secondly, the mutation may be confined to
autosomal dominant CAD cases and may not contribute in a
significant way to common, sporadic CAD cases. The mutational
effect observed in the present study was so large that, when it
occurred, it affected family members in a Mendelian fashion. This
hypothesis is not contradictory to the established relationship
between common sporadic CAD cases and multiple genes with small
effects. Additional functional studies of the MEF2A gene and
mutation in both in vitro and in vivo models are
required to further elucidate the functional implications.
Considered alongside a growing body of evidence, the findings of
the present study strongly indicated that the MEF2A gene,
and its possible causal relationship with the pathogenesis of CAD,
is too important to ignore.
In conclusion, the discovery of a novel mutation in
the MEF2A gene in a Chinese family with autosomal dominant
CAD suggests that MEF2A may have a significant role in the
pathogenesis of CAD.
Acknowledgments
The present study was supported by the National 973
Basic Research Program of China (grant no. 2015CB553604), the
National Natural Science Foundation of China (grant no. 91439201,
81170275, 81370412), and the State Program of National Natural
Science Foundation of China for Innovative Research Group (grant
no. 81321061).
References
|
1
|
Wang L, Fan C, Topol ES, Topol EJ and Wang
Q: Mutation of MEF2A in an inherited disorder with features of
coronary artery disease. Science. 302:1578–1581. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
González P, García-Castro M, Reguero JR,
Batalla A, Ordóñez AG, Palop RL, Lozano I, Montes M, Alvarez V and
Coto E: The Pro279Leu variant in the transcription factor MEF2A is
associated with myocardial infarction. J Med Genet. 43:167–169.
2006. View Article : Google Scholar
|
|
3
|
Lieb W, Mayer B, König IR, Borwitzky I,
Götz A, Kain S, Hengstenberg C, Linsel-Nitschke P, Fischer M,
Döring A, et al: Lack of association between the MEF2A gene and
myocardial infarction. Circulation. 117:185–191. 2008. View Article : Google Scholar
|
|
4
|
Wang Q: Advances in the genetic basis of
coronary artery disease. Curr Atheroscler Rep. 7:235–241. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han Y, Yang Y, Zhang X, Yan C, Xi S and
Kang J: Relationship of the CAG repeat polymorphism of the MEF2A
gene and coronary artery disease in a Chinese population. Clin Chem
Lab Med. 45:987–992. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mayer B, Erdmann J and Schunkert H:
Genetics and heritability of coronary artery disease and myocardial
infarction. Clin Res Cardiol. 96:1–7. 2007. View Article : Google Scholar
|
|
7
|
Topol EJ, Smith J, Plow EF and Wang QK:
Genetic susceptibility to myocardial infarction and coronary artery
disease. Hum Mol Genet. 15:R117–R123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Incalcaterra E, Hoffmann E, Averna MR and
Caimi G: Genetic risk factors in myocardial infarction at young
age. Minerva Cardioangiol. 52:287–312. 2004.PubMed/NCBI
|
|
9
|
Broeckel U, Hengstenberg C, Mayer B,
Holmer S, Martin LJ, Comuzzie AG, Blangero J, Nürnberg P, Reis A,
Riegger GA, et al: A comprehensive linkage analysis for myocardial
infarction and its related risk factors. Nat Genet. 30:210–214.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ikewaki K, Matsunaga A, Han H, Watanabe H,
Endo A, Tohyama J, Kuno M, Mogi J, Sugimoto K, Tada N, et al: A
novel two nucleotide deletion in the apolipoprotein A-I gene,
apoA-I Shinbashi, associated with high density lipoprotein
deficiency, corneal opacities, planar xanthomas, and premature
coronary artery disease. Atherosclerosis. 172:39–45. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weng L, Kavaslar N, Ustaszewska A, Doelle
H, Schackwitz W, Hébert S, Cohen JC, McPherson R and Pennacchio LA:
Lack of MEF2A mutations in coronary artery disease. J Clin Invest.
115:1016–1020. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Altshuler D and Hirschhorn JN: MEF2A
sequence variants and coronary artery disease: A change of heart? J
Clin Invest. 115:831–833. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bhagavatula MR, Fan C, Shen GQ, Cassano J,
Plow EF, Topol EJ and Wang Q: Transcription factor MEF2A mutations
in patients with coronary artery disease. Hum Mol Genet.
13:3181–3188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
World Health Organization: Nomenclature
and criteria for diagnosis of ischemic heart disease: Report of the
Joint International Society and Federation of Cardiology/World
Health Organization task force on standardization of clinical
nomenclature. Circulation. 59:607–609. 1979. View Article : Google Scholar
|
|
15
|
Guo Y, Yuan L, Yi J, Xiao J, Xu H, Lv H,
Xiong W, Zheng W, Guan L, Zhang J, et al: Identification of a GJA3
mutation in a Chinese family with congenital nuclear cataract using
exome sequencing. Indian. J Biochem Biophys. 50:253–258. 2013.
|
|
16
|
Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang
JG, Liang Y, San A, Li N, Chen SQ, et al: Identification of PRRT2
as the causative gene of paroxysmal kinesigenic dyskinesias. Brain.
134:3493–3501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi Y, Li Y, Zhang D, Zhang H, Li Y, Lu F,
Liu X, He F, Gong B, Cai L, et al: Exome sequencing identifies
ZNF644 mutations in high myopia. PLoS Genet. 7:e10020842011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan L, Song Z, Xu H, Gu S, Zhu A, Gong L,
Zhao Y and Deng H: EIF4G1 Ala502Val and Arg1205His variants in
Chinese patients with Parkinson disease. Neurosci Lett. 543:69–71.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan L, Wu S, Xu H, Xiao J, Yang Z, Xia H,
Liu A, Hu P, Lu A, Chen Y, et al: Identification of a novel PHEX
mutation in a Chinese family with X-linked hypophosphatemic rickets
using exome sequencing. Biol Chem. 396:27–33. 2015. View Article : Google Scholar
|