Introduction
Podocytes are among the structures of the glomerulus
filtration barrier and are located around the glomerular basement
membrane (GBM). As they have a similar morphology and structure to
that of neurocytes, they are classified as a terminally
differentiated cell type, in which the cell structures contain a
cell body, preliminary processes and secondary processes (foot
process). The intercellular junction of podocytes is formed in a
staggered manner, based on the foot process (1). Differing from endothelial cells and
the GBM, podocytes have an essential role in the selective
glomerular filtration function. The abnormal cellular structure and
function of podocytes can directly result in proteinuria, which may
further develop into glomerulonephritis. Podocyte injury
constitutes a common pathology in most glomerular diseases
(2). In recent years, a large
number of studies have focused on the mechanisms underlying
podocyte injury and have led to a marked progression in this field
(3,4). In a clinical study, podocyte injury
was targeted for the treatment of recurrent and refractory focal
segmental glomerular sclerosis, ultimately resulting in partial or
even complete remission (5). This
study indicated great prospects for treatments targeting podocytes
and suggested that further elucidation of podocyte injury
mechanisms may be beneficial to the specific therapies for focal
segmental glomerular sclerosis, membranous nephropathy, diabetic
nephropathy, and so forth.
Lipopolysaccharide (LPS)-mediated podocyte injury is
a commonly used model in studies on podocyte injury (6,7).
Previous studies have illustrated possible mechanisms of the
LPS-induced injury in podocytes. For instance, LPS-induced
activation of B7-1 results in the reorganization of vital slit
diaphragm proteins (6),
LPS-induced downregulation of the cell survival factor contributes
to podocyte apoptosis (8) and
LPS-induced activation of PTP1B leads to an increase in cell
migration (9). A recent study by
our group showed that LPS increased the activity of urokinase
receptor-mediated beta 3 integrin signals to promote podocyte
activity, thus resulting in cell injury (10,11).
Apart from these effects, the mechanisms of LPS-induced podocyte
injury have remained elusive.
Autophagy, a lysosome-dependent bioactivity that
results in the degradation of the cell's own biomacromolecules and
organelles, has a significant role in the maintenance of cellular
functions and resistance to exogenous stress (12). In contrast to other types of
glomerular cells, normal podocytes demonstrate high levels of
autophagic activity (13).
Knockout of the autophagy-associated gene Atg5 was shown to
decrease the high levels of autophagic activity in podocytes,
resulting in the accumulation of oxidized and ubiquitinated
proteins as well as endoplasmic reticulum (ER) stress. Furthermore,
proteinuria, loss of podocytes and glomerular sclerosis were
demonstrated in aged mice with Atg5 deficiencies (13). The essential role of autophagy in
the maintenance of the bioactivities of podocytes is well known.
However, whether changes in autophagic activity have a role during
podocyte injury has remained elusive.
The present study generated an in vitro model
of podocyte injury using LPS to assess the association between
autophagic activity and podocyte injury.
Materials and methods
Reagents and equipment
Chloroquine, rapamycin and 3-methyladenine (3-MA)
were all purchased from Sigma-Aldrich (St. Louis, MO, USA). LPS, as
well as rabbit anti-podocin (cat. no. P0372) and rabbit anti-light
chain (LC)3B (cat. no. L7543) antibodies were purchased from
Sigma-Aldrich. Rabbit anti-Beclin-I (cat. no. 3738s), rabbit
anti-P62 (cat. no. 5114s) and mouse-anti-CHOP (cat. no. 2895s)
antibodies were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Mouse anti-glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) antibody was obtained from Bioworld
Technology (St. Louis Park, MN, USA). Polyclonal horseradish
peroxidase (HRP)-conjugated goat anti-mouse IgG (cat. no.
115-035-003) and goat anti-rabbit IgG (cat. no. 111-035-003) were
purchased from Jackson ImmunoResearch Laboratories, Inc., West
Grove, PA, USA). Red fluorescence protein - green fluorescence
protein - LC3 double-tagged adenovirus (mRFP-GFP-LC3) was provided
by Hanbio (Shanghai, China).
Podocyte culture
Differentiated podocytes are unable to replicate,
and primary podocytes would result in rapid growth arrest when
culturing. Conditionally immortalized mouse podocyte clones (MPCs)
have overcome these difficulties, and conditionally immortalized
MPC cells retain a differentiation potential similar to that of
podocytes in vivo (14).
With these characteristics, MPCs are widely used as a cell model in
research focusing on human podocyte injury. Conditionally
immortalized mouse podocytes were a gift from Professor J. Reiser
of Rush University (Chicago, IL, USA). The recovered podocytes were
cultured and passaged with Corning® cellgro®
RPMI-1640 (Cellgro; Mediatech, Manassas, VA, USA) containing
interferon (IFN)-γ (10–100 U/ml; ProSpec, Tany Technogene Ltd.,
Rehovot, Israel), penicillin (100 U/ml), streptomycin (100
µg/ml) and 10% fetal bovine serum (FBS; Gibco®;
Thermo Fisher Scientific, Waltham, MA, USA) at 33°C in a humidified
atmosphere containing 5% CO2 for 3–4 days. After
proliferation was completed in the above medium, the podocytes were
transferred into IFN-γ-free RPMI-1640 medium containing 5% FBS and
cultured at 37°C in a humidified atmosphere containing 5%
CO2 for 10–14 days for differentiation and maturation
prior to use in the experiments of the present study.
Podocyte grouping and treatment
The matured podocytes were divided into seven groups
as follows: i) CQ0h group/Control group (podocytes were cultured
with normal medium); ii) CQ2h group [podocytes were cultured with
chloroquine (10 µM) for 2 h]; iii) CQ4h group [podocytes
were cultured with chloroquine (10 µM) for 4 h]; iv) CQ6h
group [podocytes were cultured with chloroquine (10 µM) for
6 h]; v) LPS group [podocytes were cultured with LPS (100
µg/ml) for 24 h]; vi) LPS + rapamycin group [podocytes were
cultured with LPS (100 µg/ml) and rapamycin (5 nM) for 24
h]; and vii) LPS + 3-MA group [podocytes were cultured with LPS
(100 µg/ml) and 3-MA (10 mM) for 24 h]. The starting cell
number for differentiating podocytes was ~40,000 per well on a
6-well plate with coverslips (120,000 per dish).
Western blot analysis
Following treatment with LPS and optionally with
rapamycin or 3-MA, the podocytes were washed there times with cold
phosphate-buffered saline (PBS) and then lysed on ice for 10 min
using cell lysis buffer (Beyotime Institute of Biotechnology,
Shanghai, China) containing proteinase inhibitor
(phenylmethylsulfonyl fluoride; Guangzhou Genebase Bioscience Co.,
Ltd, Guangzhou, China) with scraping. The total protein was
quantified using a bicinchoninic acid protein quantification kit
(Thermo Fisher Scientific) After denaturation at 98°C for 10 min,
the protein (20 µg per lane) was subjected to 12% sodium
dodecyl sulfate polyacrylamide gel electrophoresis (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and then transferred onto a
nitrocellulose membrane (Millipore Corp., Boston, MA, USA) at 200
mA. The blots were incubated with skimmed milk powder (5%; YiLi,
Inner Mongolia, China) for 1 h and then probed with the specific
antibody at 4°C overnight. The blots were then incubated with the
secondary antibody for 1 h and treated with an enhanced
chemiluminescence (ECL) reagent (ECL Western Blotting substrate;
Thermo Fisher Scientific). Images of the blots were captured on
Kodak X-ray film (Eastman Kodak, Rochester, NY, USA). OPTIMAX X-ray
film processor (Protec GmbH & Co., Oberstenfeld, Germany) was
used to capture the images, and scans of the images were subjected
to quantitative analysis of the protein bands by densitometric
analysis using ImageJ software (National Institutes of Health,
Bethesda, MD, USA). The ratios of protein band intensities to GAPDH
were determined and subsequently normalized to the control group or
the CQ 0 h group.
Detection of autophagosomes and
autolysosomes
Podocytes proliferating at 33°C were seeded into
six-well plates containing coverslips coated with collagen type I
(BD Biosciences, Franklin Lakes, NJ, USA) for 10–14 days (40,000
per well) and cultured until differentiation and maturation were
achieved. The podocytes were inoculated with the mRFP-GFP-LC3
adenovirus at a concentration of 1×107 PFU/well. The
culture medium was replaced with fresh medium at 6 h
post-inoculation and treatment [with LPS (100 µg/ml)
intervention for 24 h] was performed at 24 h post-inoculation. The
cells were washed with PBS three times, fixed with 4%
paraformaldehyde (Guangzhou Whiga Technology Co., Ltd., Guangzhou,
China) for 10 min and permeabilized with 0.5% Triton X-100
(Sigma-Aldrich) for 10 min. The cells were stained with
4′,6-diamidino-2-phenylindole (DAPI; Guangzhou Whiga Technology
Co., Ltd.) for 6 min, mounted for anti-fluorescence quenching
[using Antifade Mounting medium (Beyotime Institute of
Biotechnology), a reagent that is able to achieve the effect of
anti-fluorescence quenching], and observed using a Fluoview FV1000
laser scanning confocal microscope (Olympus, Tokyo, Japan). The
mRFP-GFP-LC3 adenovirus-infected cells showed dual green and red
fluorescence. LC3 II was recruited to the autophagosomal membranes,
which was indicated by fluorescent spots. Red fluorescent spots
indicated the autolysosome due to hydrolysis of GFP, whereas the
autophagosome featured dual fluorescence, indicated by yellow
spots. The numbers of autophagosomes and autolysosomes were
recorded using Image J software (National Institutes of Health,
Bethesda, MD, USA).
Observation of ER and autophagosomes by
transmission electron microscopy (TEM)
Following the indicated treatments, the cells were
harvested in Eppendorf tubes (1.5 ml), fixed with 2% glutaraldehyde
(Sigma-Aldrich) at 4°C for 1 h and washed with cacodylate (0.15 mM;
Sigma-Aldrich) three times for 5 min each. The cells were then
fixed with 1% osmium tetroxide (Sigma-Aldrich) at 4°C for 30 min,
washed with distilled water three times for 5 min each and treated
with 1% uranyl acetate (Sigma-Aldrich) at 4°C for 30 min. This step
was followed by dehydration, embedding into epoxy resin
(Sigma-Aldrich) for 12 h and polymerization at 60°C. Cells prepared
into resin embedded blocks were fixed in ultramicrotome (Leica EM
UC6; Leica, Wetzlar, Germany) and then cut into ultrathin slices.
The ultrathin slices were set into copper mesh. Images were
captured by TEM using a JEM-100CX transmission electron microscope
(Jeol, Tokyo, Japan) at magnifications of ×9,700 or ×37,000.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical analysis was performed using SPSS software,
version 13.0 (SPSS, Inc., Chicago, IL, USA). Statistical
significance was assessed using one-way ANOVA analysis of variance,
followed by a least significant difference test for comparison
between two groups. A two-tailed P<0.05 was considered to
indicate a statistically significant difference between values.
Results
Podocytes have high basal levels of
autophagy
Autophagosomes were identified in normal podocytes
using TEM (Fig. 1A). Chloroquine
(CQ) was used to block the fusion of the autophagosomes and
lysosomes in the podocytes for 2, 4 and 6 h to investigate
autophagosome formation. Autophagy was evaluated based on an
increase in LC3 II expression. The results demonstrated that CQ
significantly increased LC3 II expression in a time-dependent
manner (Fig. 1B), indicating that
matured podocytes have a high basal level of autophagosome
formation.
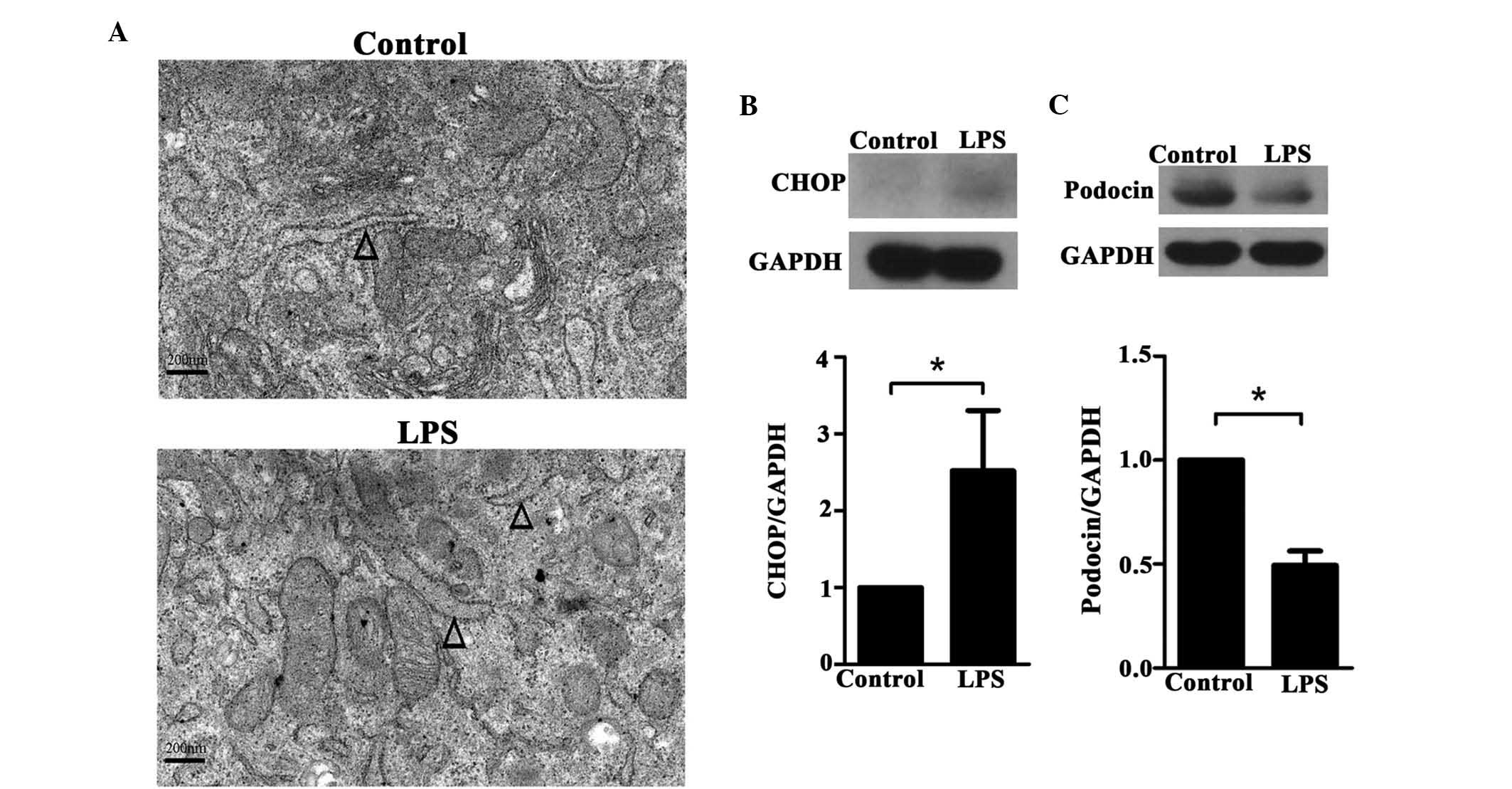
LPS induces podocyte injury
TEM observation indicated dilation of the ER in
LPS-treated podocytes compared with those in the control group
(Fig. 2A). In addition, western
blot analysis revealed elevated protein expression levels of CHOP
in the LPS-treated group compared with the control group (P=0.005)
(Fig. 2B). By contrast, the
expression of podocin was significantly inhibited in the
LPS-treated group (P<0.001) (Fig.
2C).
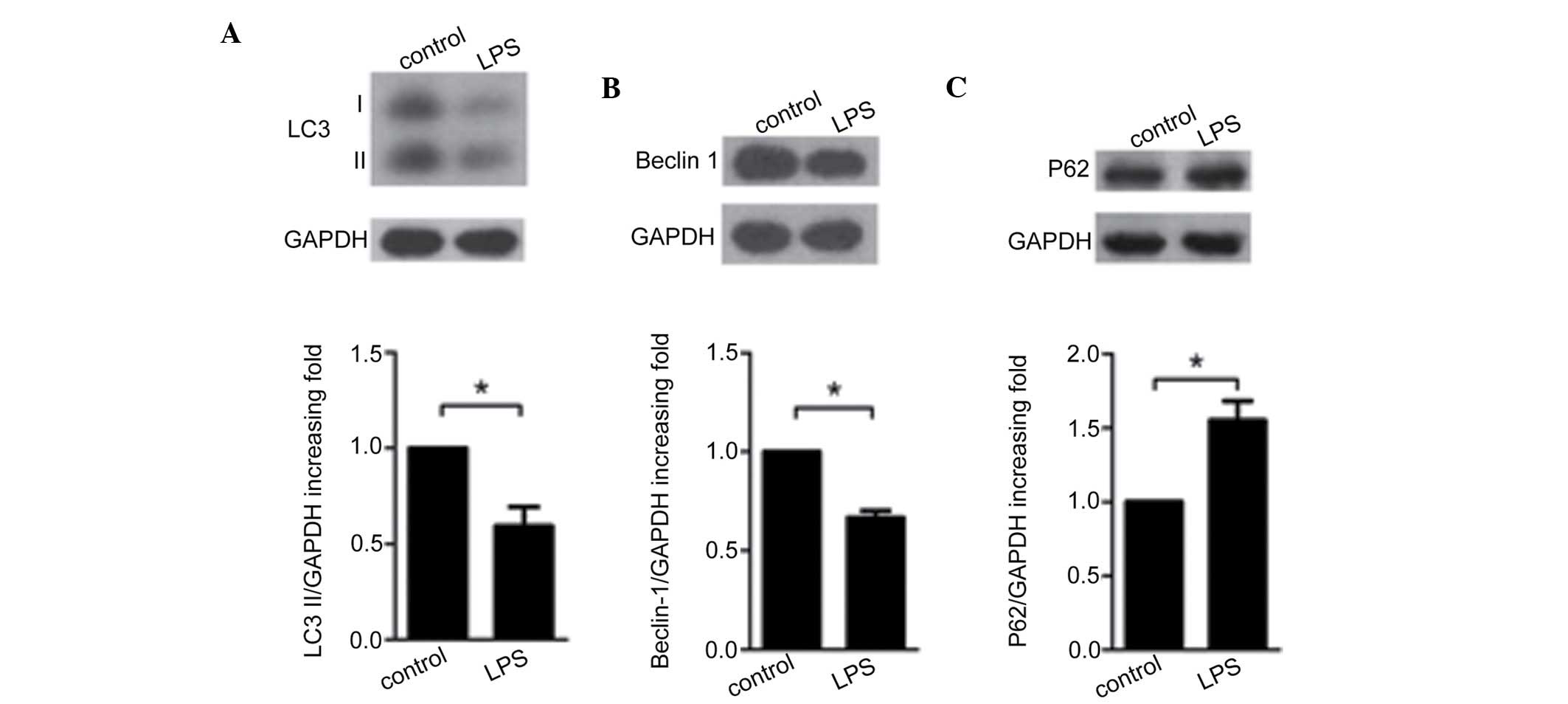
LPS decreases autophagy in podocytes
Decreases in the protein levels of LC3 II (P=0.002)
(Fig. 3A) and beclin-1
(P<0.001) (Fig. 3B), and an
increase in the expression of P62 (P=0.002) (Fig. 3C) were observed in the LPS-treated
group compared with the control group. LC3 I also revealed a
significant difference between the LPS-treated and the control
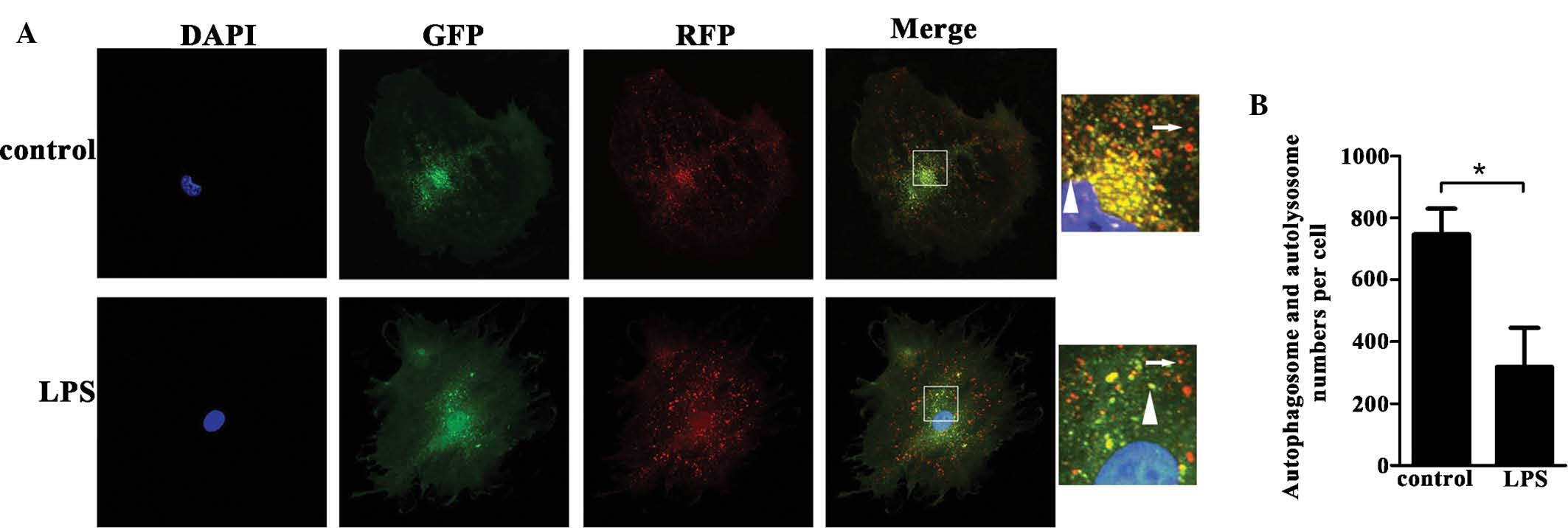
groups, although this is not shown in Figure 3. Furthermore, laser scanning
confocal microscopic observation of podocytes transfected with the
adenovirus mRFP-GFP-LC3, which tagged the autophagosomes in yellow
and the autolysosomes in red, showed a decreased ratio of
autophagosomes to autolysosomes in the LPS-treated podocytes
(P<0.001) (Fig. 4A and B).
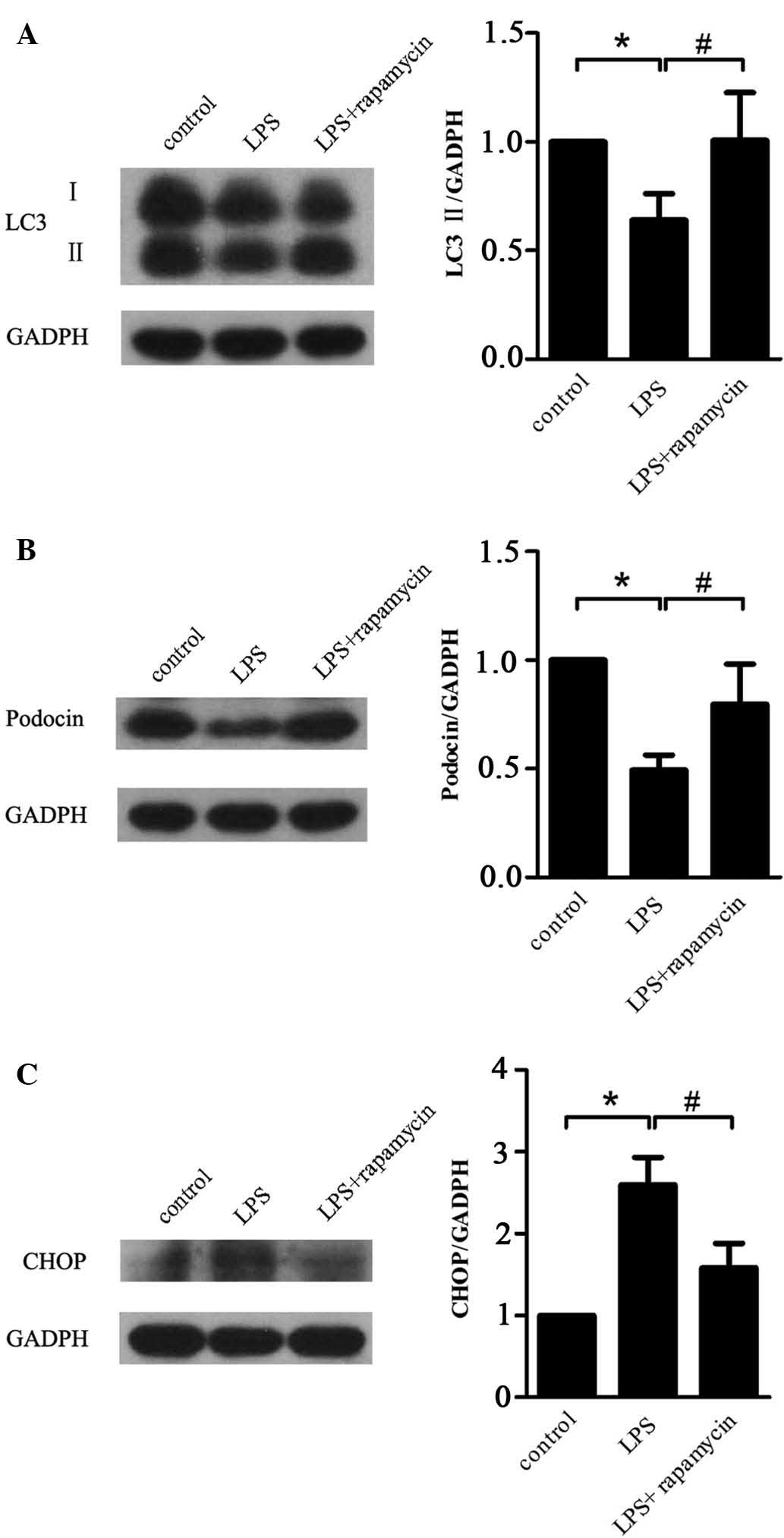
Rapamycin inhibits LPS-induced autophagy
suppression and podocyte injury
Treatment with rapamycin significantly inhibited
LPS-induced decreases in LC3 II (P=0.03) (Fig. 5A). Even though rapamycin did not
reverse the expression level of LC3I (Fig. 5A), this finding indicated that
rapamycin partially restored autophagy in podocytes after LPS
treatment. In addition, rapamycin attenuated LPS-induced decreases
in the expression of podocin (P=0.021) (Fig. 5B) and increases in the expression
of CHOP (P=0.013) (Fig. 5C),
indicating that rapamycin inhibited LPS-induced podocyte
injury.
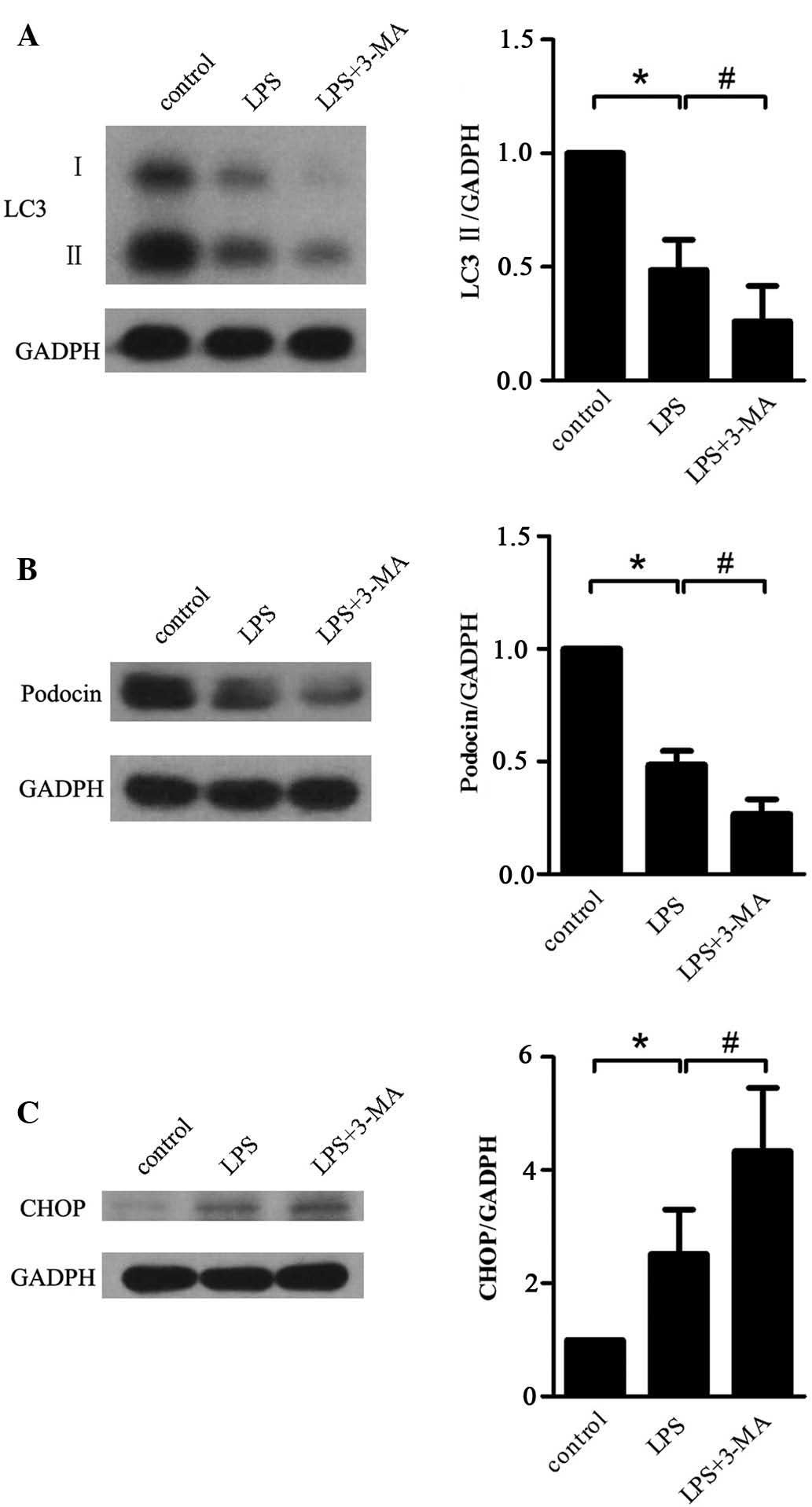
3-MA aggravates LPS-induced autophagy
suppression and cell injury in podocytes
The reduction of LC3 II expression in podocytes
treated with LPS was shown to be aggravated by simultaneous
treatment with 3-MA (P=0.022) (Fig.
6A), indicating that 3-MA contributed to LPS-induced autophagy
suppression. In addition, a similar trend in the variation was
observed in the expression level of LC3 I (Fig. 6A). In line with this, a further
decrease in the levels of podocin (P=0.014) (Fig. 6B) and a further increase in CHOP
expression (P=0.040) (Fig. 6C) by
co-treatment with 3-MA compared with the group treated with LPS
alone was observed.
Discussion
LPS has been used to establish models of renal
injury, such as minimal change disease, focal segmental
glomerulosclerosis (FSGS), acute kidney injury, and so forth
(15–17). As illustrated above, some
mechanisms of podocyte injury have been discovered during LPS
treatment. Recently, some studies have demonstrated that a
renoprotective role of autophagy in kidney tubules during
LPS-inducing acute kidney injury (17,18).
Thus, it is hypothesized that autophagy plays a role in
LPS-inducing podocyte injury. Autophagy is a quality control system
that is involved in the maintenance of intracellular homeostasis.
Its steps include autophagosome formation (engulfment of
cytoplasmic proteins and injured organelles by double-membraned
pre-autophagosomal structures), autolysosome formation (fusion of
the autophagosomes and lysosomes) and autolysosomal degradation
(generation of basal metabolism products) (19). Endogenous as well as exogenous
stress can compel cells to recycle basal nutrition elements via
autophagic degradation of abnormal intracellular components to
maintain cellular structures and biofunctions. Furthermore,
impairment of autophagy results in the accumulation of cell waste
products and abnormal regeneration of long-lived proteins, which
may result in the initiation of ER stress and cell apoptosis
(20,21). Therefore, normal autophagic
activity is a necessity for long-lived, terminally differentiated
cells, particularly neurocytes and podocytes.
In contrast to other cell types in the glomerulus,
podocytes do not proliferate and must therefore attenuate injury
stress by alternative means to cell regeneration or proliferation
(1). Based on the specificity of
their cell structure and biofunctions, podocytes have been found to
be a significant target for glomerular injury (2). Therefore, it is indicated that
podocytes face a particular challenge regarding homeostasis
maintenance. Although the mechanisms of podocyte-associated
pathogenesis of glomerular diseases remain largely elusive, recent
studies have enhanced the current level of knowledge. The
correlation between autophagy levels in podocytes and glomerular
disease susceptibility has been reported by Hartleben et al
(13). This study demonstrated
that aged mice with autophagy-deficient podocytes suffered from
proteinuria, which finally led to glomerular sclerosis (13). This finding emphasized the
significance of high levels of autophagy in the maintenance of
cellular biofunctions. Recent clinicopathological studies analyzing
renal biopsy specimens further supported that autophagic activity
of podocytes has a critical protective role in renal injury
(22–24). Glomeruli and in particular
podocytes from patients with minimal changes in their disease
status had higher levels of autophagic activity than glomeruli from
patients with focal segmental glomerulosclerosis. Repeat renal
biopsies of patients with minimal changes in their disease status
enabled the tracking of podocyte autophagic activity and confirmed
that patients maintaining high autophagic activity in their
podocytes retained minimal change disease status, whereas patients
with decreased autophagic activity progressed to focal segmental
glomerulosclerosis (25).
Consistent with the above studies, the present study detected a
high basal level of autophagy in normal podocytes. In the present
study, CQ treatment was employed to explore autophagic activity.
CQ, an anti-malarial drug, can block the fusion of lysosomes and
autophagosomes by increasing the pH value of the lysosomes. This
process results in the abortive formation of the autolysosomes and
the accumulation of newly generated autophagosomes (26). Microtubule-associated protein LC3,
a mammalian homologue of yeast Atg8, exists in two forms: LC3-I and
LC3-II. LC3-I is an abundant cytoplasmic protein that is cleaved
and lipidated during initiation of autophagy (forming LC3-II),
translocating to and associating with the autophagosome in a
punctate pattern. Thus, LC3-II is considered as an autophagy
biomarker. Based on the characteristics of CQ, the protein levels
of LC3-II were determined at different CQ intervening time points
to indirectly indicate the level of autophagy activity. The results
showed that LC3 II rapidly accumulated following CQ treatment,
indicating a high level of autophagy in the podocytes. Furthermore,
a decrease in the expression level of LC3-I was also observed in
the LPS-treated group. This suggested that the inhibition of
autophagy could involve two aspects: Downregulation of the
expression of LC3-I and the conversion inhibition of LC3-I to
LC3-II.
The results of the present study demonstrated that
LC3 II and beclin-1 were downregulated in LPS-treated podocytes.
P62 has been used in numerous studies as a protein marker to
indicate autophagic activity. It is an adapter protein, which can
bind to LC3 II and ubiquitinated aggregated proteins and facilitate
the transportation of ubiquitinated proteins to the autophagosomes.
Subsequently, P62 is degraded together with the ubiquitinated
proteins in the autolysosomes (27). In the present study, P62 expression
in podocytes was found to be significantly increased following LPS
treatment. The dual-tagged adenovirus mRFP-GFP-LC3 has been
previously used to detect autophagosomes (28), and was used to investigate the
effects of LPS on autophagy in podocytes. In this system, the
stability of mRFP can compensate for the instability of GFP in an
acidic environment, enabling for the simultaneous detection of
autophagosomes and autolysosomes. In the present study, the number
of autophagosomes and autolysosomes was decreased by LPS. All of
these findings indicated that LPS inhibited the high-level
autophagic activity in podocytes.
Previous studies have reported that ER stress and
abnormal slit diaphragm proteins have significant roles in the
injury of podocytes and thus serve as known markers of podocyte
injury (29). In the present
study, LPS treatment resulted in ER dilation, increased expression
levels of the ER stress-associated protein CHOP and a significant
decrease in the a slit diaphragm protein podocin. To investigate
the association between LPS-induced podocyte injury and autophagy,
autophagy enhancer rapamycin and autophagy suppressor 3-MA were
employed (30,31). The results showed that rapamycin
partly restored LPS-mediated decreases of autophagy, and attenuated
LPS-induced ER stress and downregulation of podocin. By contrast,
3-MA aggravated the effects of LPS on autophagy and podocyte
injury.
In spite of the findings of the present and previous
studies, the underlying mechanisms of LPS-induced podocyte injury
and inhibition of autophagy remain to be fully elucidated. The
migration of autophagosomes and lysosomes depends on components of
the cytoskeleton, such as microtubules (32), and it is known that LPS damages the
skeletal systems of podocytes (6).
Whether LPS affects autophagic activity of podocytes by damaging
the skeletal system is worthy of further exploration.
In conclusion, the present study reported that
LPS-induced podocyte injury is, at least in part, mediated via
inhibition of autophagy. 3-MA-induced inhibition of autophagy
significantly enhanced LPS-induced podocyte injury, while
rapamycin-mediated restoration of autophagy attenuated the effects
of LPS. The present study therefore provided a basis for the
development of novel strategies for the treatment of glomerular
diseases, such as the upregulation of autophagy under injury
stress.
Acknowledgments
This study received financial support from the
National Natural Science Foundation of China (81270784; 81470930)
and the National Clinical Key Specialty Construction Projects.
References
|
1
|
Pavenstadt H, Kriz W and Kretzler M: Cell
biology of the glomerular podocyte. Physiol Rev. 83:253–307. 2003.
View Article : Google Scholar
|
|
2
|
Greka A and Mundel P: Cell biology and
pathology of podocytes. Annu Rev Physiol. 74:299–323. 2012.
View Article : Google Scholar
|
|
3
|
Gubler MC: Podocyte differentiation and
hereditary proteinuria/nephrotic syndromes. J Am Soc Nephrol.
14(Suppl 1): S22–S26. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patrakka J and Tryggvason K: New insights
into the role of podocytes in proteinuria. Nat Rev Nephrol.
5:463–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu CC, Fornoni A, Weins A, Hakroush S,
Maiguel D, Sageshima J, Chen L, Ciancio G, Faridi MH, Behr D, et
al: Abatacept in B7-1-positive proteinuric kidney disease. N Engl J
Med. 369:2416–2423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reiser J, von Gersdorff G, Loos M, Oh J,
Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H,
Schwarz K, et al: Induction of B7-1 in podocytes is associated with
nephrotic syndrome. J Clin Invest. 113:1390–1397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei C, Moller CC, Altintas MM, Li J,
Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, et
al: Modification of kidney barrier function by the urokinase
receptor. Nat Med. 14:55–63. 2008. View
Article : Google Scholar
|
|
8
|
Saurus P, Kuusela S, Lehtonen E, Hyvönen
ME, Ristola M, Fogarty CL, Tienari J, Lassenius MI, Forsblom C,
Lehto M, et al: Podocyte apoptosis is prevented by blocking the
Toll-like receptor pathway. Cell Death Dis. 6:e17522015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumagai T, Baldwin C, Aoudjit L, Nezvitsky
L, Robins R, Jiang R and Takano T: Protein tyrosine phosphatase 1B
inhibition protects against podocyte injury and proteinuria. Am J
Pathol. 184:2211–2224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang B, Shi W, Ma J, Sloan A, Faul C, Wei
C, Reiser J, Yang Y, Liu S and Wang W: The calcineurin-NFAT pathway
allows for urokinase receptor-mediated beta3 integrin signaling to
cause podocyte injury. J Mol Med (Berl). 90:1407–1420. 2012.
View Article : Google Scholar
|
|
11
|
Zhang B, Xie S, Shi W and Yang Y:
Amiloride off-target effect inhibits podocyte urokinase receptor
expression and reduces proteinuria. Nephrol Dial Transplant.
27:1746–1755. 2012. View Article : Google Scholar
|
|
12
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hartleben B, Gödel M, Meyer-Schwesinger C,
Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ,
Lindenmeyer MT, et al: Autophagy influences glomerular disease
susceptibility and maintains podocyte homeostasis in aging mice. J
Clin Invest. 120:1084–1096. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mundel P, Reiser J, Zúñiga Mejía Borja A,
Pavenstädt H, Davidson GR, Kriz W and Zeller R: Rearrangements of
the cytoskeleton and cell contacts induce process formation during
differentiation of conditionally immortalized mouse podocyte cell
lines. Exp Cell Res. 236:248–58. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Srivastava T, Sharma M, Yew KH, Sharma R,
Duncan RS, Saleem MA, McCarthy ET, Kats A, Cudmore PA, Alon US, et
al: LPS and PAN-induced podocyte injury in an in vitro model of
minimal change disease: changes in TLR profile. J Cell Commun
Signal. 7:49–60. 2013. View Article : Google Scholar :
|
|
16
|
Reiser J, von Gersdorff G, Loos M, Oh J,
Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H,
Schwarz K, et al: Induction of B7 1 in podocytes is associated with
nephrotic syndrome. J Clin Invest. 113:1390–1397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Howell GM, Gomez H, Collage RD, Loughran
P, Zhang X, Escobar DA, Billiar TR, Zuckerbraun BS and Rosengart
MR: Augmenting autophagy to treat acute kidney injury during
endotoxemia in mice. PLoS One. 8:e695202013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mei S, Livingston M, Hao J, Li L, Mei C
and Dong Z: Autophagy is activated to protect against endotoxic
acute kidney injury. Sci Rep. 6:221712016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y and Kominami E: Loss
of autophagy in the central nervous system causes neurodegeneration
in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen YM, Zhou Y, Go G, Marmerstein JT,
Kikkawa Y and Miner JH: Laminin beta2 gene missense mutation
produces endoplasmic reticulum stress in podocytes. J Am Soc
Nephrol. 24:1223–1233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu CC, Fornoni A, Weins A, Hakroush S,
Maiguel D, Sageshima J, Chen L, Ciancio G, Faridi MH, Behr D, et
al: Abatacept in B7 1 positive proteinuric kidney disease. N Engl J
Med. 369:2416–2423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma J, Zhang B, Liu S, Xie S, Yang Y, Ma J,
Deng Y, Wang W, Xu L, Li R, et al: 1,25-dihydroxyvitamin D(3)
inhibits podocyte uPAR expression and reduces proteinuria. PLoS
One. 8:e649122013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong
Y, Zhang C, Zen K and Liu Z: Podocyte autophagic activity plays a
protective role in renal injury and delays the progression of
podocytopathies. J Pathol. 234:203–213. 2014.PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bjorkoy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou C, Zhong W, Zhou J, Sheng F, Fang Z,
Wei Y, Chen Y, Deng X, Xia B and Lin J: Monitoring autophagic flux
by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3)
reveals that high-dose rapamycin impairs autophagic flux in cancer
cells. Autophagy. 8:1215–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cybulsky AV, Takano T, Papillon J, Kitzler
TM and Bijian K: Endoplasmic reticulum stress in glomerular
epithelial cell injury. Am J Physiol Renal Physiol. 301:F496–F508.
2011. View Article : Google Scholar
|
|
30
|
Wu L, Feng Z, Cui S, Hou K, Tang L, Zhou
J, Cai G, Xie Y, Hong Q, Fu B and Chen X: Rapamycin upregulates
autophagy by inhibiting the mTOR-ULK1 pathway, resulting in reduced
podocyte injury. PLoS One. 8:e637992013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seglen PO and Gordon PB: 3-Methyladenine:
Specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mackeh R, Perdiz D, Lorin S, Codogno P and
Poüs C: Autophagy and microtubules-new story, old players. J Cell
Sci. 126:1071–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|