Introduction
Heatstroke is an illness, which frequently occurs
during the summer. Although substantial progress has been made in
the prevention and treatment of heatstroke, its mortality rate
remains between 20 and 70%. The possible reason for the high
mortality rate of heatstroke is the poor understanding of the
underlying molecular mechanisms, which has resulted in a lack of
targeted and effective treatments (1). Studies have suggested that heatstroke
and its progression to multiple organ dysfunction syndromes are due
to a complex interplay between the acute physiological alterations
associated with direct heat injuries, the inflammatory and
coagulative responses of the host, and systemic inflammatory
response syndrome (SIRS) secondary to immediate heat injury as the
leading cause (2–5). It is currently hypothesized that
intestinal dysfunction is the initiating and stimulating factor
leading to SIRS, and infections caused by intestinal bacteria and
endotoxin translocation have been clinically implicated (2). Studies investigating heatstroke have
also revealed that intestinal lesions are common, and
intestinal-derived endotoxemia has been observed in cases of
heatstroke (6,7). Thus, during heatstroke, tissues and
cells are stimulated by direct heat and subsequent gut-derived
endotoxemia.
Vascular endothelial cells line the entire
circulatory system from the heart to the smallest capillaries.
These cells have distinct and unique functions, and are also
considered to be involved in SIRS (8,9).
Typically, the heat involved in heatstroke is considered to be
directly cytotoxic, and endothelial cell injuries and diffuse
microvascular thrombosis are also prominent features of heatstroke
(2,10,11).
However, as a physical stress, heat stress can also induce cellular
heat shock responses, which are characterized by anti-inflammatory
medium and the expression of protective heat shock proteins (HSPs),
and these protect cells from delayed injury stimulation, including
isch-emia/reperfusion and oxidative injury (12–14).
Therefore, the present study aimed to examine the types of injuries
induced in endothelial cells in the complex condition in which
endothelial cells are stimulated by hyperthermia and gut-derived
endotoxemia.
Cellular apoptosis is typically considered to be the
predominant reason for organ dysfunction, and studies have
suggested that endothelial cell apoptosis appears to be a mechanism
of heatstroke (2,11,15).
However, the detailed molecular changes in endothelial cell
apoptosis, which are induced by heat stress remain to be fully
elucidated. To examine apoptosis in the vascular endothelium during
heatstroke, an in vitro model of human umbilical vascular
endothelial cells (HUVECs) stimulated with heat stress and
lipopolysaccharide (LPS) was used to mimic the in vivo
micro-environment of a direct heat attack and subsequent
gut-derived endotoxemia. Furthermore, heat stress can induce
increases in the expression levels of various HSPs, including
HSP27, HSP90 and small molecular mass HSPs, which may be
responsible for protection against cellular injury and apoptosis
(15–18). B-cell lymphoma 2 (Bcl-2) is
considered to be an important apoptosis-associated protein
(19), and p38 mitogen-activated
protein kinase (MAPK) has been found to affect a multitude of
cellular events, including cell growth and death, differentiation
and inflammation, in response to oxidative stress and LPS (20,21).
Therefore, the HSP27, HSP90, Bcl-2 and p38 MAPK proteins were
selected in the present study as candidates for further
investigation of the possible molecular mechanisms of endothelial
apoptosis in the above-mentioned complex condition of heatstroke,
so as to provide a potential therapeutic method for the prevention
of sepsis-induced endothelial injury.
Materials and methods
Endothelial cells
The HUVECs were harvested from umbilical cords by
collagenase treatment, as previously described (22,23).
Briefly, umbilical cords (length, 20-30 cm) were obtained from
patients at the Victoria Hospital (London, ON, Canada) between
October 2013 and June 2014, with ~1-2 patients per week. All
procedures relevant to HUVEC isolation were approved by the Human
Ethics Committee of the University of Western Ontario (London, ON,
Canada). The umbilical vein was washed and digested with 0.2%
collagenase solution (Roche Applied Science, Mannheim, Germany).
The detached endothelial cells were plated in Medium 199 (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% heat-inactivated fetal calf serum (GE Healthcare Life Sciences,
Logan, TX, USA), thymidine (2.4 mg/l; Sigma-Aldrich, St. Louis, MO,
USA), glutamine (230 mg/l; JRH Biosciences, Lenexa, KS, USA),
heparin sodium (10 U/ml; Sigma-Aldrich), antibiotics (100 IU/ml
penicillin, 100 µg/ml streptomycin and 0.125 µg/ml
amphotericin B) and endothelial cell growth factor (80
µg/ml; Biomedical Technologies Inc., Stoughton, MA, USA).
The cell cultures were incubated at 37°C in a humidified atmosphere
with 5% CO2 and expanded by brief trypsinization using
0.25% trypsin in phosphate-buffered saline (PBS), containing 0.02%
EDTA (Gibco; Thermo Fisher Scientific, Inc.). The first to third
passage HUVECs were used in the experiments. The study was approved
by the ethics committee of the University of Western Ontario.
Drugs and treatment
LPS and SB203580, a specific inhibitor of p38 MAPK,
were purchased from Sigma-Aldrich and Enzo Life Sciences
(Farmingdale, NY, USA), respectively. The cells (1×105)
were plated and incubated for 24 h at 37°C, then treated with LPS
(1 µg/ml) for 24 h at 37°C. Heat stress was induced at 43°C
for 1 h on aluminum plates within a tissue culture incubator to
ensure temperature uniformity. Following the heat stress, the
cultures were transferred to the standard 37°C incubator for
another 23 h. The combination of heat stress and LPS treatment was
administered by applying heat stress at 43°C for 1 h, followed by
exposure to 1 µg/ml LPS for another 23 h. SB203580 was
applied for 1 h prior to the other treatments.
Caspase-3 activity
Cellular caspase-3 activity was measured using a
caspase-3 fluorescent assay kit, according to manufacturer's
protocol (cat. no. C002A20; Thomas Scientific, Swedesboro, NJ,
USA). The cells were harvested and washed with cold Dulbecco's PBS.
The cell pellet was resuspended in ice-cold lysis buffer (50 mM
HEPES, pH 7.4, 0.1% CHAPS, 5 mM DTT, 0.1 mM EDTA; all chemical
reagents were purchased from Sigma-Aldrich). Following incubation
on ice, the lysate was centrifuged at 10,000 × g at 4°C for 10 min.
The resulting supernatant was used for the measurement of caspase-3
activity, and the protein concentrations were quantified using a
micro BCA protein assay kit (cat. no. 23227; Pierce Biotechnology,
Inc., Rockford, IL, USA). The samples (50 µg protein) were
incubated in duplicate with the caspase-3 substrate, Ac-DEVD-AMC,
or Ac-DEVD-AMC and the inhibitor, Ac-DEVD-CHO, at 37°C for 2 h
prior to measurements of caspase-3 activity, which were obtained
with a fluorescent spectrophotometer (Wallac Victor 3 1420
Multi-Label Counter; PerkinElmer, Inc., Waltham, MA, USA), with
excitation at 380 nm and emission at 405 nm. The signals from the
inhibitor-treated samples served as the background.
DNA fragmentation
The HUVECs were pre-labeled with BrdU (Roche Applied
Science) for 24 h at 37°C prior to the other treatments. DNA
fragmentation was measured using a Cellular DNA Fragmentation ELISA
kit (cat. no. 11585045001; Roche Applied Science), according to the
manufacturer's protocol.
Cellviability
Cell viability was evaluated using a 3-(4,
5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
assay kit (cat. no. 11465007001; Roche Applied Science), according
to the manufacturer's protocol. Briefly, 1×104 cells
were plated in 96-well microplates at a final volume of 100
µl culture medium (serum-free Medium 199) per well in a
humidified atmosphere (37°C; 5% CO2). After the
incubation period (24 h) and treatment with LPS and heat stress, 10
µl MTT labeling reagent (final concentration, 0.5 mg/ml) was
added to each well. The microplate was incubated for 4 h in a
humidified atmosphere (37°C; 5% CO2). Solubilization
solution (100 µl) was added to each well. The plate was
allowed to stand overnight in the humidified atmosphere of the
incubator. Upon complete solubilization of purple formazan
crystals, the spectrophoto-metrical absorbance of the samples was
measured using a microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) at a wavelength of 550 nm.
Western blot analysis
Protein samples were extracted from the cultured
HUVECs in extraction buffer [20 mM Tris, pH 0.5, 150 mM NaCl, 1.0
mM EDTA, 1.0 mM EGTA, 0.1% Triton X-100, 2.5 mM sodium
pyrophosphate (Sigma-Aldrich), 1.0 mM β-pyrophosphate glycerol
(Sigma-Aldrich)], which was supplemented with 1.0 mM
Na3Vo4 (Sigma-Aldrich), 1.0 mM
phenylmethanesulfonyl fluoride (Sigma-Aldrich) and a protease
inhibitor cocktail. Following centrifugation at 10,000 × g for 15
min at 4°C, the supernatant was collected and the protein
concentrations were quantified using the micro BCA protein assay
kit. Equal quantities of protein (50 µg) were subjected to
SDS-PAGE (10 or 12%). After separation with SDS-PAGE, the upper
side of the sample wells was removed with a razor blade. The bottom
right-hand corner of the gel was notched for orientation purposes,
and the gel was placed in 1X transfer buffer (Sigma-Aldrich). PVDF
membranes (EMD Millipore, Billerica, MA, USA) were sliced,
according to the size of the gel, and incubated in 95% methanol for
~1 min on a rocker at room temperature. The methanol was removed
and the membrane was equilibrated in 1X transfer buffer
(Sigma-Aldrich; 400 ml methanol, 200 ml 10X transfer buffer and
1,400 ml water). The membrane was subjected to 100 V (constant
voltage) for 1 h at 4°C. The membrane was washed with 10 ml
Tris-buffered saline (TBS) buffer [Sigma-Aldrich; 1.22 g Tris (10
mM) and 8.78 g NaCl (150 mM) to 1 liter distilled water and pH was
adjusted to 7.5 with HCl] and 5% blocking buffer (Sigma-Aldrich;
0.5 g bovine serum albumin in TBS and Tween 20 buffer to a final
volume of 10 ml); the membrane was gently agitated for ≥1 h. The 5%
blocking buffer was removed and the membrane was rinsed three
times, with TBST (5 min per wash).
The primary antibodies used were as follows: Rabbit
monoclonal anti-human Bcl-2 (cat. no. 2780), rabbit monoclonal
anti-human HSP90 (cat. no. 4874), rabbit anti-human
phosphorylated-p38 (cat. no. 9211), and rabbit anti-human total p38
(cat. no. 9212; all 1:1,000 dilution), and all were obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Mouse
monoclonal anti-human HSP27 (cat. no. 12215; 1:1,000 dilution) was
obtained from Cayman Chemistry Company (Ann Arbor, MI, USA) and
rabbit anti-GAPDH (cat. no. sc-25778; 1:1,000 dilution) served as
an internal control, and was obtained from Santa Cruz
Biotechnology, Inc. (Dallas, Tx, USA). These primary antibodies
were added at the appropriate dilution to 10 ml 5% blocking buffer
and agitated gently for ≥1 h. The first antibody solution was
discarded and the membrane was washed twice for 10 min with TBST
buffer. The horseradish peroxidase (HRP)-conjugated secondary
antibodies [goat anti-rabbit (cat. no. 172-1019) or goat anti-mouse
(cat. no. 170-6515) IgG-HRP (all 1:1,000 dilution; Bio-Rad
Laboratories, Inc.)] were added at the appropriate dilution to 5 ml
5% blocking buffer and agitated gently for ≥1 h. The secondary
antibody solution was discarded and the membrane was washed twice
for 10 min with TBST buffer. The PVDF membranes were subsequently
developed using a chemiluminescence kit [Westzol®
(plus); Intron Biotechnology, Inc., Seoul, South Korea]. The bands
were quantified using densitometry and GelQuant Pro software
version 1.0 (MicroChemi; FroggaBio Inc., Toronto, ON, Canada).
Statistical analysis
All data are presented as the mean ± standard
deviation and were analyzed using SPSS 15.0 (SPSS Inc., Chicago,
IL, USA). For multi-group comparisons, analysis of variance
followed by Newman-Keuls tests were performed. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of heat stress and LPS on
caspase-3 activity, DNA fragmentation and the MTT assay
The cells were treated with heat stress, LPS or the
combination of heat stress pretreatment followed by LPS. Caspase-3
activity, DNA fragmentation and cell viability were detected as
indicators of cellular apoptosis. The results revealed that LPS
increased caspase-3 activity, DNA fragmentation and cell viability;
and these effects were rescued by heat stress pretreatment
(Fig. 1). These findings indicated
that heat stress pretreatment inhibited LPS-induced apoptosis in
the HUVECs.
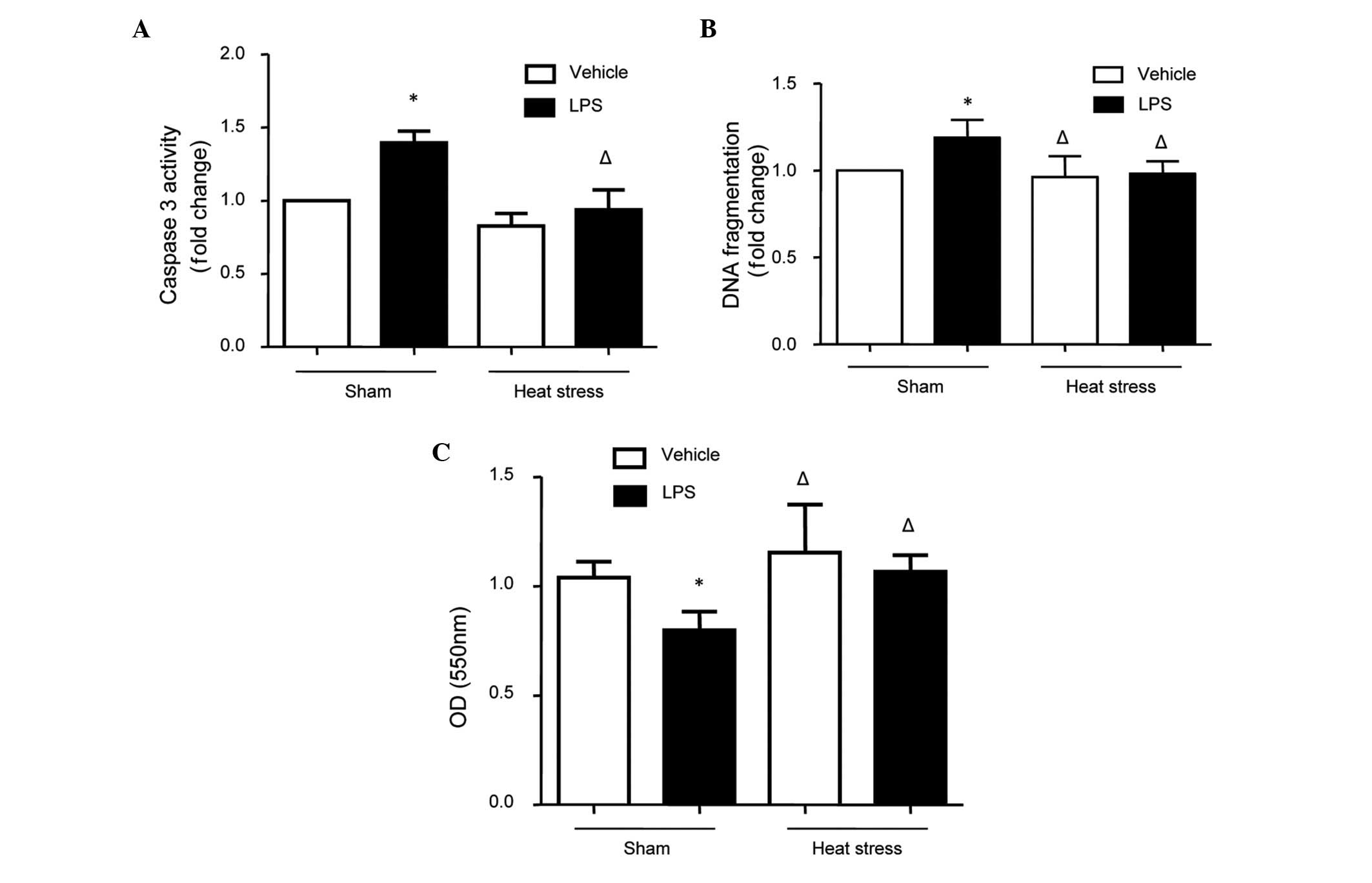 | Figure 1Changes in caspase-3 activity, DNA
fragmentation and the cell viability in human umbilical vein
endothelial cells stimulated by heat stress followed by LPS. The
cells were treated with either heat stress (43°C for 1 h, followed
by 37°C for 23 h), LPS (1 µg/ml for 24 h) or the combination
of heat stress (43°C for 1 h), followed by LPS (1 µg/ml for
23 h). (A) Caspase-3 activities, (B) DNA fragmentation and (C) cell
viability, determined using an MTT assay, were measured. The
results indicated that cellular apoptosis was increased, as
indicated by high levels of caspase-3 activity and DNA
fragmentation, and low OD values in the MTT assay. The data are
presented as the mean ± standard deviation from five independent
experiments. *P<0.05, vs. sham vehicle group;
ΔP<0.05, vs. sham LPS group. LPS, lipopolysaccharide; MTT, 3-(4,
5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide; OD,
optical density. |
Effects of heat stress and LPS on the
expression levels of HSP27, HSP90 and Bcl-2
The treatments described above were performed, and
the expression levels of HSP27, HSP90 and Bcl-2 were determined
using Western blot analysis. As shown in Fig. 2, LPS decreased the expression
levels of HSP27, HSP90 and Bcl-2, and heat stress significantly
increased the expression levels of these proteins under normal and
LPS-stimulated conditions (Fig. 2A and
B). As protective proteins, the changes in the expression
levels of these three proteins suggested that they may be involved
in the protective role of heat stress against LPS-induced HUVEC
apoptosis.
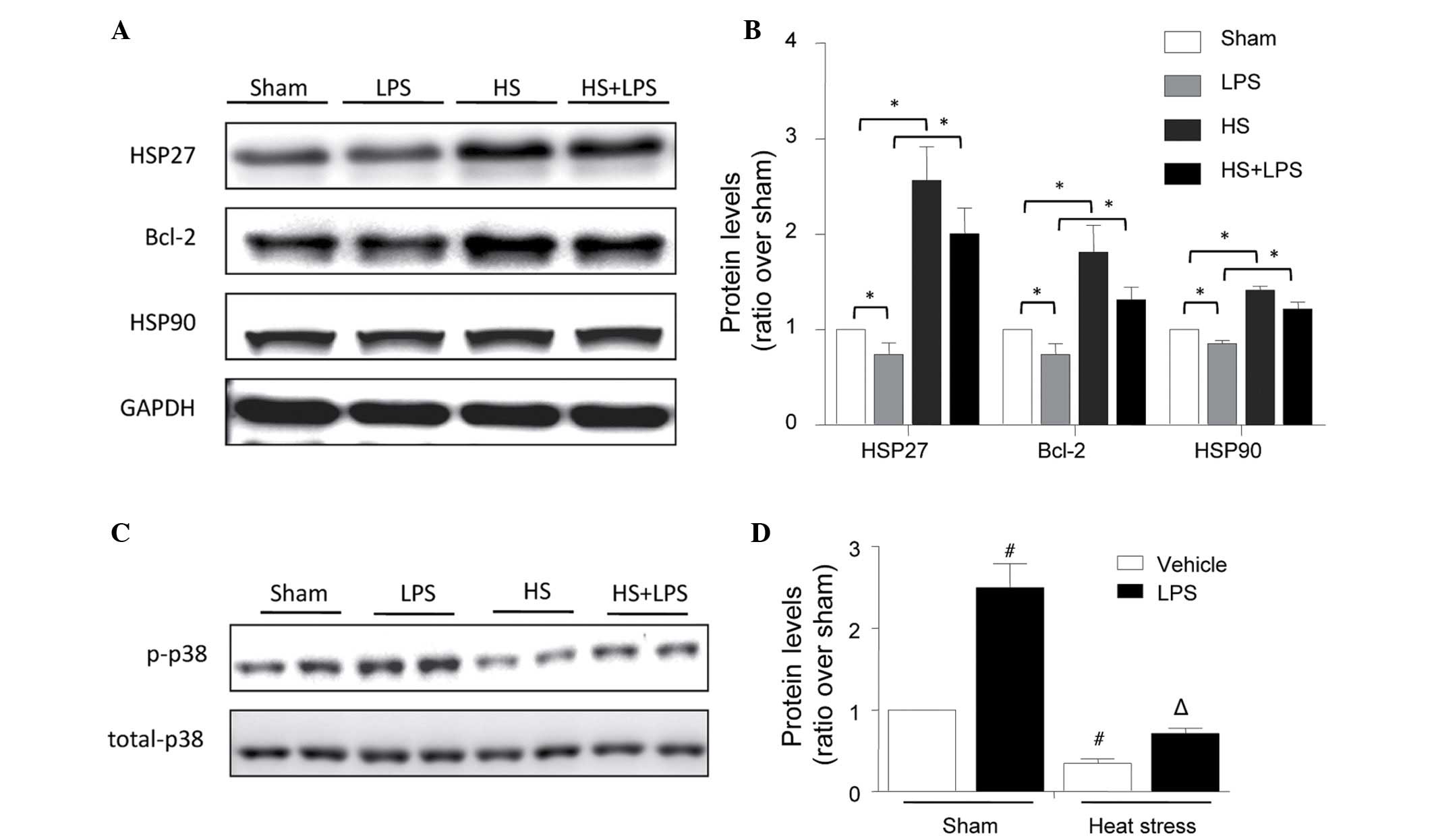 | Figure 2Changes in the expression levels of
HSP27, Bcl-2 and HSP90, and p-p38 phosphorylation in human
umbilical vein endothelial cells stimulated with heat stress and
LPS. The cells were treated with heat stress (43°C for 1 h,
followed by 37°C for 23 h), LPS (1 µg/ml for 24 h) or the
combination of heat stress (43°C for 1 h), followed by LPS (1
µg/ml for 23 h). (A) Expression levels of HSP27, HSP90 and
Bcl-2 were determined using Western blot analysis. (B)
Corresponding bands were quantified using densitometry
(protein/GAPDH) and are presented as fold changes relative to the
sham group. (C) Expression levels of total and phosphorylated p38
were determined using Western blot analysis. (D) Corresponding
bands were quantified using densitometry (phosphorylated/total
protein) and are presented as fold changes relative to the sham
group. The data are presented as the mean ± standard deviation from
three independent experiments. *P<0.05, between the
indicated groups; #P<0.05, vs. sham vehicle group;
ΔP<0.05, vs. sham LPS group. HSP, heat shock protein;
Bcl-2, B-cell lymphoma 2; p-p38, phosphorylated p38; LPS,
lipopolysaccharide; HS, heat stress. |
Effects of heat stress and LPS on the
phosphorylation of p38
To investigate the protective mechanisms underlying
the effect of heat stress pretreatment against LPS-induced
endothelial apoptosis in more detail, the expression levels of
total and phosphorylated p38 were determined using Western blot
analysis. Similar to the observed changes in cellular apoptosis,
LPS increased the phosphorylation of p38, and heat stress decreased
the baseline level and LPS-induced high phosphorylation level of
p38 (Fig. 2C and D).
Roles of p38 in heat stress and
LPS-stimulation of apoptosis in HUVECs
The specific inhibitor of p38, SB203580, was used to
further determine the role of p38 in heat stress and the subsequent
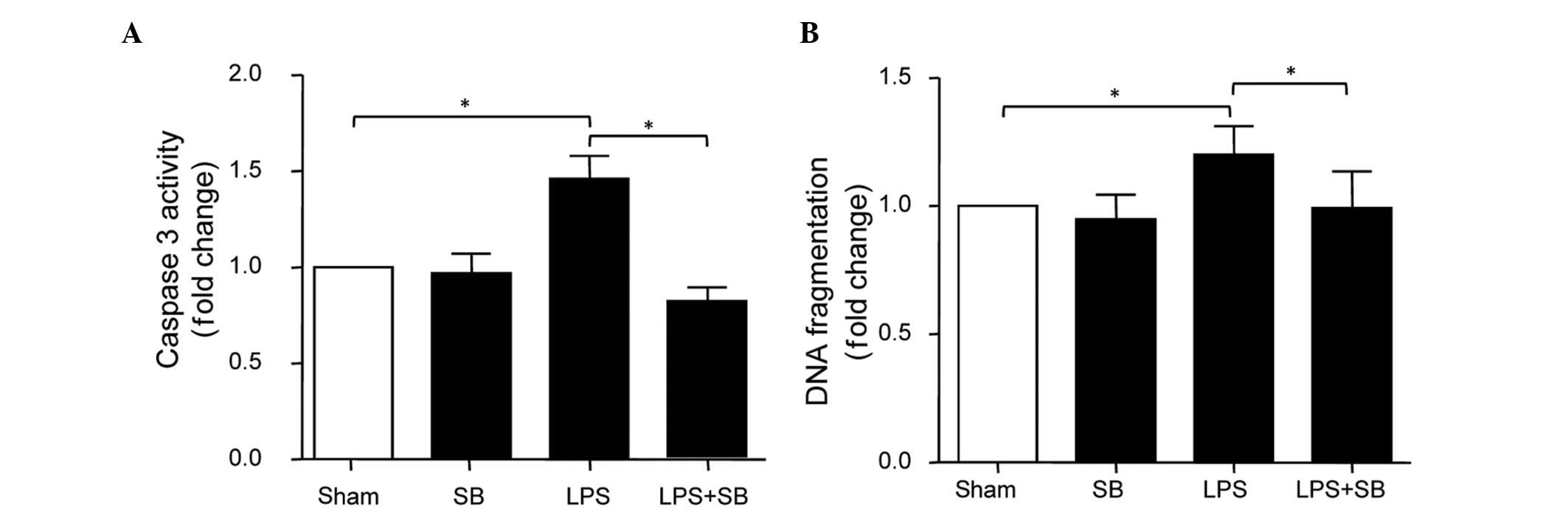
LPS-induced apoptosis of HUVECs. The results revealed that SB203580
decreased LPS-induced caspase-3 activation. In addition, SB203580
reduced the LPS-induced elevation in DNA fragmentation (Fig. 3). No changes in DNA fragmentation
were found following the SB203580 and heat stress treatment (data
not shown). Based on these data, it was concluded that heat stress
pretreatment inhibited LPS-induced apoptosis by attenuating the
activation of p38 MAPK.
Role of p38 MAPK in the expression levels
of HSP27, HSP90 and Bcl-2
The results described above demonstrated that heat
stress pretreatment inhibited LPS-induced apoptosis by attenuating
the activation of p38 MAPK, and that heat stress also increased the
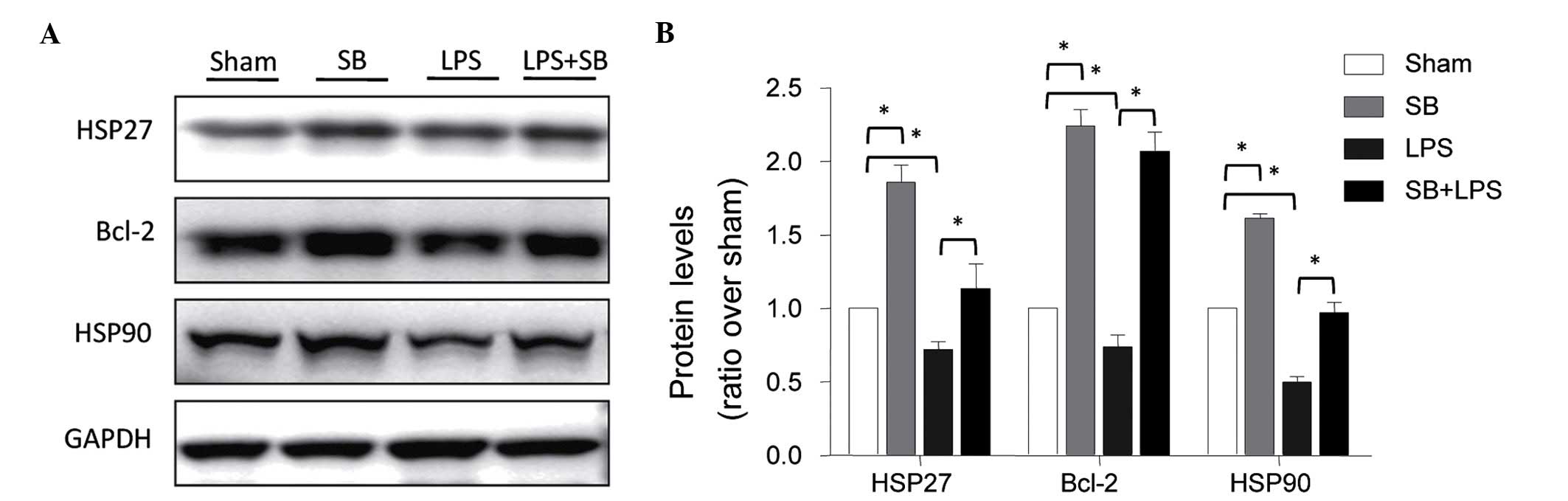
expression levels of HSP27, HSP90 and Bcl-2. To further determine
the role of p38 activation on the expression levels of HSP27, HSP90
and Bcl-2 in the HUVECs, the cells were pretreated with SB203580
for 1 h, and then stimulated with LPS. The expression levels of
HSP27, HSP90 and Bcl-2 were determined using Western blot analysis.
The results revealed that SB203580 had a similar effect as heat
stress by directly increasing the expression levels of these
proteins and inhibiting the LPS-induced downregulation of these
proteins (Fig. 4).
Discussion
In the present study, the in vivo
micro-environment of direct heat attack followed by stimulation
with gut-derived endotoxemia during heatstroke was investigated
using an in vitro model of HUVECs, which were stimulated
with heat stress and LPS. This was performed to investigate the
possible changes in the vascular endothelium and the associated
signaling pathways. The present study demonstrated that LPS
activated p38 MAPK, which then increased endothelial apoptosis, as
indicated by the observed high level of caspase-3 activity,
increased levels of DNA fragmentation and decreased cellular
viabilities. Heat stress pretreatment inhibited LPS-induced
apoptosis by attenuating p38 MAPK, and further increasing the
expression levels of Bcl-2, HSP27 and HSP90.
Roles of heat stress and LPS in
apoptosis
The apoptosis of vascular endothelial cells has been
associated with impairments of endothelial function and organ
injury during sepsis, and LPS-induced caspase-3 activation and
apoptosis in the endothelium have previously been reported,
including in our previous investigations (24,25).
In the present study, similar results were found, and revealed that
LPS increased apoptosis, as indicated by the elevated caspase-3
activity, DNA fragmentation and decreased cell viability in the
HUVECs. However, heat stress pretreatment exerted effects, which
were opposite to those of LPS. The heat, which induces heatstroke
is known to be directly cytotoxic, and studies in cell lines and
animal models have suggested that heat can directly induce tissue
injury (2,26). However, as a physical stress, the
protective role of heat stress has also been reported, which is
similar to the protective effect of hypoxia in
ischemia/reperfusion-induced apoptosis. For example, the protective
role of heat shock on cardiomyocytes, following injury of the cells
by ischemia/reperfusion has been well reviewed (27). In addition, it has been observed
that heat stress preconditioning can prevent the endothelial
coronary dysfunction that is induced by ischemia and reperfusion
(28,29). The varied nature of the findings
described above may be due to the severity and timing of heat
stress, and the heat tolerances of different cell lines. (30–32).
In conclusion, the evidence from the present study regarding
caspase-3 activation and high levels of DNA fragmentation
demonstrated that heat stress pretreatment decreased LPS-induced
apoptosis in the HUVECs via the caspase-3 pathway.
Role of the p38-HSP/Bcl-2 pathway in heat
stress and LPS-induced apoptosis
Heat stress alone had a protective effect against
LPS-induced endothelial apoptosis; thus the present study aimed to
determine how this effect was mediated. In the present study, three
protective proteins, HSP27, HSP90 and Bcl-2, were selected as
candidates for roles in the protective effects of heat stress
against apoptosis.
The results of the present study revealed that LPS
decreased the expression levels of HSP27, HSP90 and Bcl-2, and that
heat stress pretreatment significantly increased the expression
levels of HSP27, HSP90 and Bcl-2. The possible protective roles of
HSP27 and HSP90 have been observed in several studies, including
those of heat stress-induced intestinal epithelial apoptosis
(33) and LPS-induced endothelial
barrier dysfunction (34). In
addition, HSP90 inhibits cell apoptosis by inhibiting the activity
of proapoptotic kinase apoptosis signal-regulating kinase 1
(17). The results of the present
study reinforce the cytoprotective mechanisms of the chaperoning
HSPs. The founding member of the Bcl-2 family of regulator proteins
is Bcl-2, and cell death is regulated by either the induction
(anti-apoptotic) or inhibition (pro-apoptotic) of Bcl-2. The Bcl-2
protein is specifically considered to be an important
anti-apoptotic protein, and represents an important apoptotic
pathway (19). In the present
study, the expression levels of Bcl-2 were decreased by LPS
stimulation and increased by heat stress, which indicated that the
heat stress pretreatment-induced decrease in LPS-induced HUVEC
apoptosis was mediated through the anti-apoptotic Bcl-2
pathway.
MAPKs include three well-characterized subfamilies
of protein kinases: Extracellular signal-regulated kinases
(ERK1/2), p38 kinases and c-Jun NH2-terminal kinases (JNK1/2/3).
The activations of each of the three subfamilies have been
implicated in gene expression during pathological and physiological
conditions, and p38 MAPK has been found to affect a multitude of
cellular events, including cell growth and death, differentiation
and inflammation, in response to oxidative stress and LPS (20,21).
The present study also demonstrated that LPS increased p38
activation, and that heat stress decreased baseline and LPS-induced
high phosphorylation levels of p38. In addition, the specific
inhibitor of p38, SB203580, attenuated LPS-induced apoptosis in the
HUVECs. Similarly, it has been reported that LPS stimulation
induces p38 MAPK phosphorylation in HMVEC-Ls (35).
Following the observation in the present study that
HSP27 and HSP90 may have functions in heat stress, the subsequent
step in further investigating the signaling pathway involved in the
protective effects of heat stress against apoptosis was to
determine the association between these proteins and the activation
of p38. To investigate this, SB203580 was used, and it was found
that pretreatment with SB203580 had the same effects as heat stress
on the LPS-induced downregulation of HSP27 and HSP90. In addition,
SB203580 was found to rescue the LPS-induced downregulation of
Bcl-2; this finding suggested that LPS-induced HUVEC apoptosis and
the protective role of heat stress were mediated through the
p38-HSP/Bcl-2 pathway.
In conclusion, the present study demonstrated that
heat stress pretreatment decreased LPS-induced Bcl-2-associated
apoptosis by attenuating the activation of p38, which increased the
expression levels of HSP27 and HSP90 in the HUVECs. These findings
provide novel evidence that, in conditions of sepsis, heat stress
pre-treatment may be useful as a therapeutic strategy for the
prevention of endothelial injury. During this process, p38 may be a
potential therapeutic target for the treatment of endothelial
dysfunction and organ injury. However, further in vivo
investigations, particularly those involving gene-knockout animal
models, are warranted to clarify these roles.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81101467), the
Project Team of Natural Science Foundation of Guangdong Province
(grant no. s2013030013217), the Project of Medical Research of PLA
(grant no. BWS12J108), the Guangdong Province Science and
Technology Planning Project of China (grant no. 2102B031800416) and
the Heart & Stroke Foundation of Canada (grant no. T6717;
awarded to Professor Tianqing Peng of the University of Western
Ontario, London, Canada). Professor Tianqing Peng is a recipient of
the New Investigator Award from the Canadian Institutes of Health
Research.
Abbreviations:
|
LPS
|
lipopolysaccharides
|
|
HS
|
heat stress
|
|
HUVECs
|
human umbilical vascular endothelial
cells
|
|
SIRS
|
systemic inflammatory response
syndrome
|
|
MAPK
|
mitogen-activated protein kinase
|
|
HSP
|
heat shock protein
|
|
Bcl-2
|
B-cell lymphoma 2
|
References
|
1
|
Varghese GM, John G, Thomas K, Abraham OC
and Mathai D: Predictors of multi-organ dysfunction in heatstroke.
Emerg Med J. 22:185–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leon LR and Helwig BG: Heat stroke: Role
of the systemic inflammatory response. J Appl Physiol (1985).
109:1980–1988. 2010. View Article : Google Scholar
|
|
4
|
Leon LR, Blaha MD and DuBose DA: Time
course of cytokine, corticosterone and tissue injury responses in
mice during heat strain recovery. J Appl Physiol (1985).
100:1400–1409. 2006. View Article : Google Scholar
|
|
5
|
Lim CL and Mackinnon LT: The roles of
exercise-induced immune system disturbances in the pathology of
heat stroke: The dual pathway model of heat stroke. Sports Med.
36:39–64. 2006. View Article : Google Scholar
|
|
6
|
Bouchama A, Roberts G, Al Mohanna F,
El-Sayed R, Lach B, Chollet-Martin S, Ollivier V, Al Baradei R,
Loualich A, Nakeeb S, et al: Inflammatory, hemostatic and clinical
changes in a baboon experimental model for heat stroke. J Appl
Physiol (1985). 98:697–705. 2005. View Article : Google Scholar
|
|
7
|
Lambert GP: Intestinal barrier
dysfunction, endotoxemia and gastrointestinal symptoms: The 'canary
in the coal mine' during exercise-heat stress? Med Sport Sci.
53:61–73. 2008. View Article : Google Scholar
|
|
8
|
de Pablo R, Monserrat J, Reyes E, Díaz D,
Rodríguez-Zapata M, de la Hera A, Prieto A and Álvarez-Mon M:
Circulating sICAM-1 and sE-Selectin as biomarker of infection and
prognosis in patients with systemic inflammatory response syndrome.
Eur J Intern Med. 24:132–138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
TIba T, Gando S, Murata A, Kushimoto S,
Saitoh D, Eguchi Y, Ohtomo Y, Okamoto K, Koseki K, Mayumi T, et al:
Predicting the severity of systemic inflammatory response syndrome
(SIRS)-associated coagulopathy with hemostatic molecular markers
and vascular endothelial injury markers. J Trauma. 63:1093–1098.
2007. View Article : Google Scholar
|
|
10
|
Roberts GT, Ghebeh H, Chishti MA,
Al-Mohanna F, El-Sayed R, Al-Mohanna F and Bouchama A:
Microvascular injury, thrombosis, inflammation and apoptosis in the
pathogenesis of heatstroke a study in baboon model. Arterioscl
Throm Vasc Biol. 28:1130–1136. 2008. View Article : Google Scholar
|
|
11
|
Bouchama A, Hammami MM, Haq A, Jackson J
and al-Sedairy S: Evidence for endothelial cell activation/injury
in heatstroke. Crit Care Med. 24:1173–1178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gray CC, Amrani M and Yacoub MH: Heat
stress proteins and myocardial protection: Experimental model or
potential clinical tool? Int J Biochem Cell Biol. 31:559–573. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugimoto N, Shido O, Matsuzaki K, Katakura
M, Hitomi Y, Tanaka M, Sawaki T, Fujita Y, Kawanami T, Masaki Y, et
al: Long-term heat exposure prevents hypoxia-induced apoptosis in
mouse fibroblast cells. Cell Biochem Biophys. 70:301–307. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gill RR, Gbur CJ Jr, Fisher BJ, Hess ML,
Fowler AA III, Kukreja RC and Sholley MM: Heat shock provides
delayed protection against oxidative injury in cultured human
umbilical vein endothelial cells. J Mol Cell Cardiol. 30:2739–2749.
1998. View Article : Google Scholar
|
|
15
|
Brinton MR, Tagge CA, Stewart RJ, Cheung
AK, Shiu YT and Christensen DA: Thermal sensitivity of endothelial
cells on synthetic vascular graft material. Int J Hyperthermia.
28:163–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mestril R and Dillmann WH: Heat shock
proteins and protection against myocardial ischemia. J Mol Cell
Cardiol. 27:45–52. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa
WC and Min W: Hsp90-Akt phosphorylates ASK1 and inhibits
ASK1-mediated apoptosis. Oncogene. 24:3954–3963. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kabakov AE, Budagova KR, Bryantsev AL and
Latchman DS: Heat shock protein 70 or heat shock protein 27
overexpressed in human endothelial cells during posthypoxic
reoxygenation can protect from delayed apoptosis. Cell Stress
Chaperones. 8:335–347. 2003. View Article : Google Scholar
|
|
19
|
Zakeri Z and Lockshin RA: Cell death:
History and future. Adv Exp Med Biol. 615:1–11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ono K and Han J: The p38 signal
transduction pathway: Activation and function. Cell Signal.
12:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng T, Lu X and Feng Q: NADH oxidase
signaling induces cyclooxygenase-2 expression during
lipopolysaccharide stimulation in cardiomyocytes. FASEB J.
19:293–295. 2005.
|
|
22
|
Mizuguchi S, Stephen J, Bihari R, Markovic
N, Suehiro S, Capretta A, Potter RF and Cepinskas G: CORM-3-derived
CO modulates polymorphonuclear leukocyte migration across the
vascular endothelium by reducing levels of cell surface-bound
elastase. Am J Physiol Heart Circ Physiol. 297:H920–H929. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yoshida N, Granger DN, Anderson DC,
Rothlein R, Lane C and Kvietys PR: Anoxia/reoxygenation-induced
neutrophil adherence to cultured endothelial cells. Am J Physiol.
262:H1891–H1898. 1992.PubMed/NCBI
|
|
24
|
Suzuki K, Murakami T, Kuwahara-Arai K,
Tamura H, Hiramatsu K and Nagaoka I: Human anti-microbial
cathelicidin peptide LL-37 suppresses the LPS-induced apoptosis of
endothelial cells. Int Immunol. 23:185–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu H, Li X, Li Y, Wang L, Mehta S, Feng Q,
Chen R and Peng T: Calpain-1 induces apoptosis in pulmonary
microvascular endothelial cells under septic conditions. Microvasc
Res. 78:33–39. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moulin M and Arrigo AP: Long lasting heat
shock stimulation of TRAIL-induced apoptosis in transformed T
lymphocytes. Exp Cell Res. 312:1765–1784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Joyeux-Faure M, Arnaud C, Godin-Ribuot D
and Ribuot C: Heat stress preconditioning and delayed myocardial
protection: What is new? Cardiovasc Res. 60:469–477. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Joyeux M, Bouchard JF, Lamontagne D,
Godin-Ribuot D and Ribuot C: Heat stress-induced protection of
endothelial function against ischaemic injury is abolished by
ATP-sensitive potassium channel blockade in the isolated rat heart.
Br J Pharmacol. 130:345–350. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amrani M, Corbett J, Allen NJ, O'Shea J,
Boateng SY, May AJ, Dunn MJ and Yacoub MH: Induction of heat-shock
proteins enhances myocardial and endothelial functional recovery
after prolonged cardioplegic arrest. Ann Thorac Surg. 57:157–160.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu DZ, Lu Q, Swank GM and Deitch EA:
Effect of heat shock and endotoxin stress on enterocyte viability
apoptosis and function varies based on whether the cells are
exposed to heat shock or endotoxin first. Arch Surg. 131:1222–1228.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Furusawa Y, Tabuchi Y, Wada S, Takasaki I,
Ohtsuka K and Kondo T: Identification of biological functions and
gene networks regulated by heat stress in U937 human lymphoma
cells. Int J Mol Med. 28:143–151. 2011.PubMed/NCBI
|
|
32
|
Heidemann SM and Glibetic M: Heat stress
protects against lung injury in the neutropenic, endotoxemic rat.
Inflammation. 29:47–53. 2005. View Article : Google Scholar
|
|
33
|
Baird CH, Niederlechner S, Beck R,
Kallweit AR and Wischmeyer PE: L-Threonine induces heat shock
protein expression and decreases apoptosis in heat-stressed
intestinal epithelial cells. Nutrition. 29:1404–1411. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hirano S, Rees RS, Yancy SL, Welsh MJ,
Remick DG, Yamada T, Hata J and Gilmont RR: Endothelial barrier
dysfunction caused by LPS correlates with phosphorylation of HSP27
in vivo. Cell Biol Toxicol. 20:1–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mizumura K, Gon Y, Kumasawa F, Onose A,
Maruoka S, Matsumoto K, Hayashi S, Kobayashi T and Hashimoto S:
Apoptosis signal-regulating kinase 1-mediated signaling pathway
regulates lipopolysaccharide-induced tissue factor expression in
pulmonary microvasculature. Int Immunopharmacol. 10:1062–1067.
2010. View Article : Google Scholar : PubMed/NCBI
|