Introduction
Interleukin (IL)-6 is involved in a number of
diseases, such as atherosclerosis (1,2) and
rheumatoid arthritis (3). Thus,
there are potential clinical benefits to the development of
anti-IL-6 agents as therapies for these diseases (4,5). The
first example is tocilizumab, which has been approved for the
treatment of rheumatoid arthritis (6) and others are currently in clinical
trials (7).
Endothelial lipase (EL), a member of the lipase
family, is a key enzyme with phospholipase activity that promotes
the metabolism and clearance of high-density lipoprotein (HDL)
(8,9). HDL cholesterol levels are inversely
associated with the risk of atherosclerotic cardiovascular disease
(10). Alternative functions of EL
include increasing the uptake of apolipoprotein B by endothelial
cells and the adhesion of monocytes and macrophages to endothelial
cells (11). An animal study
demonstrated that the atherosclerotic plaque area in apoE knockout
mice that overexpress EL is <70% the area of control mice
(8). Consequently, EL expression
is closely associated with the pathogenesis of atherosclerosis. EL
expression is affected by a number factors, such as inflammation,
blood pressure and an increased rate of blood flow (12–14).
However, the mechanisms that underlie the regulation of EL
expression have not been fully elucidated.
The p38 mitogen-activated protein kinase (MAPK) is a
member of the MAPK family and is involved in multiple intracellular
signal transduction (15). p65
nuclear factor (NF)-κB is a member of the NF-κB transcription
factor family and is crucial in inflammatory diseases, such as
atherosclerosis (16).
The aim of the present study was to stimulate human
umbilical vein endothelial cell (HUVEC) culture in vitro
with recombinant human interleukin-6 (rhIL-6), to examine its
effect on EL expression and its effects on the proliferation of
HUVECs. To investigate the regulatory role of p38 MAPK and p65
NF-κB in this process, HUVECs were pretreated with a p38 MAPK
inhibitor (SB203580) or an NF-κB inhibitor (pyrrolidine
dithiocarbamate, PDTC) prior to rhIL-6 treatment.
Materials and methods
Reagents
SB203580, protease inhibitor cocktail and epithelial
cell growth factor (EGF) were purchased from Sigma-Aldrich (St.
Louis, MO, USA); rhIL-6 was obtained from Peprotech (Rocky Hill,
NJ, USA). Trypsin and M199 medium were purchased from Hyclone
(Logan, UT, USA). Rabbit polyclonal anti-EL primary antibody was
purchased from Cayman Chemicals (Ann Arbor, MI, USA; cat. no.
100030). Mouse anti-Von Willebrand Factor (vWF, also known as
Factor VIII related antigen; cat. no. ab194405) was purchased from
Abcam (Cambridge, MA, USA). The mouse and rabbit
IgG-immunohistochemical SABC kit, DAB (diaminobenzidine) kit and
the MTT assay kit were purchased from Beyotime Institute of
Biotechnology (Beijing, China). Fluorescein isothiocyanate (FITC)-
and tetramethylrhodamine (TRITC)-conjugated anti-mouse IgG (cat.
nos. ZF-0312 and ZF-0313, respectively) were obtained from
Zhongshanjinqiao Biotechnology (Beijing, China); mouse
anti-proliferating cell nuclear antigen (PCNA) antibody (cat. no.
BM0104) was purchased from Boster Biological Technology, Ltd.
(Wuhan, China), and monoclonal anti-β-actin mouse primary antibody
(cat. no. AA128-1) was from Beyotime Institute of Biotechnology.
Radioimmunoprecipitation assay (RIPA) lysis buffer and the
Bicinchoninic acid (BCA) Protein Assay kit were purchased from
Beyotime Institute of Biotechnology. Fetal bovine serum (FBS) was
obtained from Yuanpeng Biotech Co. (Jinan, China). Penicillin was
from North China Pharmaceutical Co. (Shijiazhuang, China).
Streptomycin was obtained from Merro Pharmaceutical Co. (Dalian,
China). Polyvinylidene fluoride (PVDF) membranes were from
Millipore (Millipore Corporation, Bedford, MA, USA). An
electrochemiluminescence kit was purchased from Amersham (GE
Healthcare, Barrington, IL, USA).
Ethical approval
This study was approved by the Ethical Committee at
the School of Medicine, Shandong University (Jinan, China; permit
number: 200800243). Written informed consent for the donation of
the umbilical cords used in this study was obtained from the
parents of the newborn.
Cell culture
Human umbilical vein endothelial cells were obtained
from one fresh umbilical cord ~15-cm long from a newborn infant
after maternal cesarean section in the Department of Obstetrics and
Gynecology of Qilu Hospital (Jinan, China). Briefly, the umbilical
vein was washed with sterile phosphate-buffered saline (PBS) for 5
min. Trypsin-ethylenediaminetetraacetic acid (0.25%; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) was injected through the
umbilical vein and digested for 10–15 min at room temperature with
gentle shaking so that the enzyme solution was in full contact with
the vascular wall. The solution was then collected in a 50-ml
sterile tube and centrifuged at 100 x g for 10 min. The supernatant
was removed and 20% FBS cell culture media were added (Hyclone, 20%
FBS, 100 U/ml penicillin, 100 U/ml streptomycin and 3 µl/ml
EGF) to obtain the cell suspension. The cell suspension was
transferred to a 6-well culture plate and incubated in an incubator
with saturated humidity and 5% CO2 at 37°C. The media
were changed every 2–3 days. Cells were passaged after they reached
80–90% confluency. The passaged cells were continuously incubated
in 10% FBS culture media. The purity of HUVECs was determined by
immunofluorescence staining of vWF when cells had reached >90%
confluence.
Cell treatments
Cells were treated after the third passage and
divided into 4 groups treated as follows: i) Control group without
any treatment; ii) rhIL-6 (10 ng/ml); iii) SB203580 (10
µmol/l) pretreatment for 1 h+rhIL-6 (10 ng/ml); and iv) PDTC
(10 mmol/l) pretreatment for 1 h+rhIL-6 (10 ng/ml) purchased from
Beyotime Institute of Biotechnology. Cells from each group
(~2×107) were collected at 0, 4, 8, 12 and 24 h
respectively. Before being passaged and treated, coverslips were
placed in the culture plates for later use.
Immunocytochemical staining
The coverslips coated with cells were removed and
washed in sterile PBS. The cells were fixed in methanol: acetic
acid (3:1) for 10 min and washed in PBS. Then, the cells were
immunoblocked with 10% normal goat serum in 0.01 M PBS containing
0.3% Triton X-100 at room temperature for 1 h. Primary antibodies
against EL (1:100), vWF (1:100) and PCNA (1:100) were then added
and incubated in a humidified box at 4°C overnight.
Biotin-conjugated anti-rabbit IgG (for EL; cat. no. A0277; Beyotime
Institute of Biotechnology) or FITC- (for vWF cat. no. ZF-0312) and
TRITC- (for PCNA; cat. no. ZF-0313) conjugated anti-mouse IgG were
added (for PCNA and vWF) and incubated at 37°C for 1 h. Then SABC
complex was added and incubated at 37°C for 1 h according to the
manufacturer's instructions of the SABC kit and DAB color was
developed. Between each step, cells were washed twice in PBS.
Negative control staining was performed with nonspecific IgG
instead of the primary antibody. Cells were counterstained with
hematoxylin and finally sealed with Neutral balsam. For vWF and
PCNA, after IgG incubation, they were counterstained with DAPI and
sealed with anti-fade (Beyotime Institute of Biotechnology). All
coverslips were examined under an Olympus U-LH100HG microscope
(Olympus Corporation, Tokyo, Japan).
Western blotting
EL protein levels in HUVECs were detected by western
blotting. HUVECs in each group were harvested separately, washed in
cold PBS, and homogenized at 4°C in lysis buffer containing 10 mM
HEPES, pH 7.9; 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5
mM DTT, 10 mM α-glycerophosphate, 0.1 mM sodium vanadate and a
protease inhibitor cocktail. After 15 min of incubation on ice,
cell debris was removed by centrifugation at 15,000 x g for 20 min
at 4°C. Protein concentration was determined by the BCA assay with
bovine serum albumin (Boster Biological Technology, Ltd.) as a
standard. Proteins (40 µg) were separated on a 10% sodium
dodecyl sulfate-polyacrylamide gel, and then transferred to a
polyvinylidene difluoride membrane. After blocking with 5% (w/v)
fat-free milk for 1 h at room temperature, the membranes were
probed with anti-EL (1:1,500) overnight at 4°C, followed by
incubation with peroxidase-conjugated anti-rabbit IgG (1:500) for 1
h at room temperature. The interaction was monitored with an
electrochemiluminescence kit. β-Actin was used to monitor the
loading amount.
Statistical analysis
Densitometric evaluation of western blotting results
were conducted using the Quantity One software, version 4.62
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) with β-actin as an
internal control. Immunocytochemical staining results were analyzed
with Image-Pro Plus 5.0 (Media Cybernetics, Inc., Rockville, MD,
USA). Data are presented as the mean ± standard deviation of three
separate experiments. Comparisons among groups were conducted using
one-way analysis of variance (ANOVA). If the result of ANOVA was
statistically significant, then multiple comparison tests between
groups were performed using the Student-Newman-Keuls method.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Isolation and identification of
HUVECs
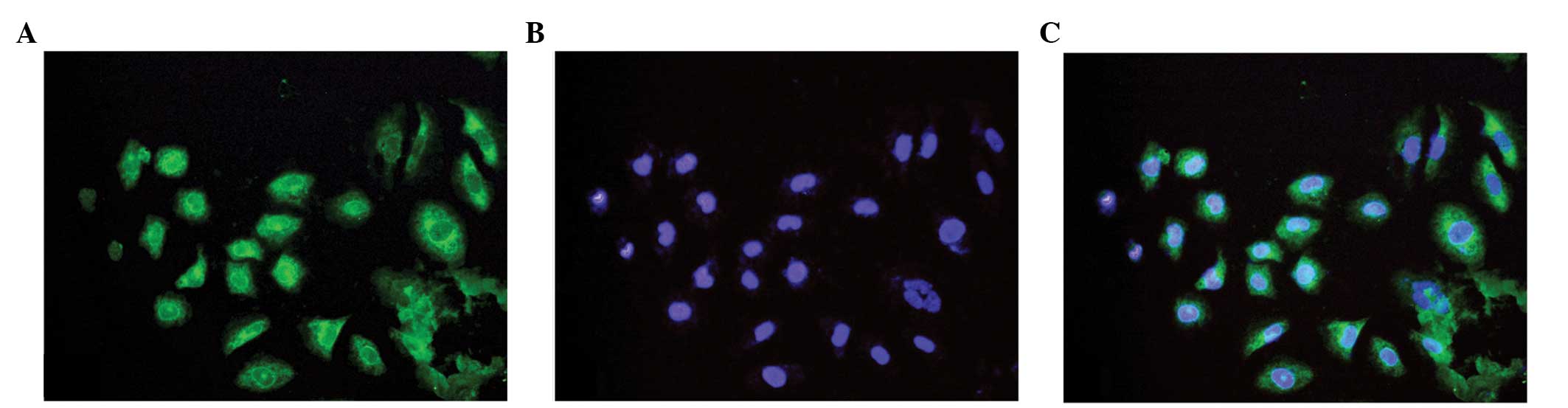
vWF is expressed in HUVECs, this the
immunofluorescence staining of vWF was performed to identify
HUVECs. The results showed that almost all cells were vWF positive
(Fig. 1).
rhIL-6 treatment increases the EL
expression in HUVECs
After HUVECs were treated with rhIL-6, EL expression
levels increased significantly from 4 h. The effect persisted at
significant levels until 12 h after treatment, and EL expression
decreased to basal levels by 24 h. The effect was most significant
at 8 h after rhIL-6 treatment. After that time, the EL expression
level began to decrease up to 24 h after treatment, when the EL
expression level returned to the basal level. The
immunocytochemical staining results are consistent with that of
western blotting (Fig. 2).
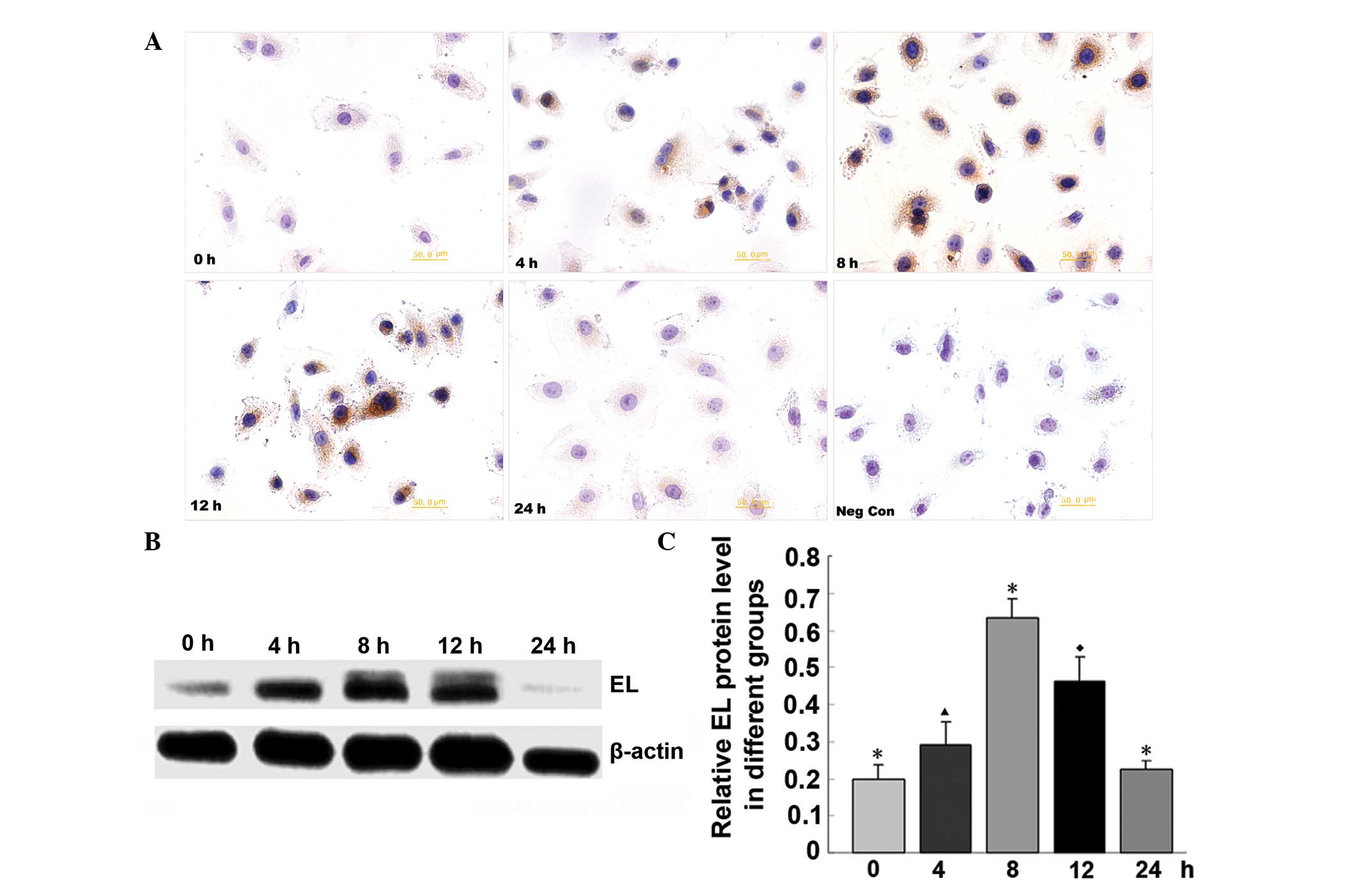 | Figure 2rhIL-6 treatment increased EL
expression in HUVECs by immunocytochemical staining and western
blotting. (A) Immunocytochemical staining of EL at 0, 4, 8, 12 and
24 h after rhIL-6 treatment and the negative control.
Magnification, ×400. (B) EL expression by western blotting at 0, 4,
8, 12 and 24 h after rhIL-6 treatment. (C) Semi-quantitative
analysis of the EL level of the western blotting results.
▲P<0.05 compared with the 0, 8, 12 and 24 h groups;
*P<0.05 compared with 0, 4, 12 and 24 h groups;
◆P<0.05 compared with 0, 4, 8 and 24 h group. rhIL-6,
recombinant human interleukin-6; EL, endothelial lipase; HUVECs,
human umbilical vein endothelial cells. |
rhIL-6 treatment promotes proliferation
of HUVECs
In order to detect the effects of increased EL on
HUVECs, immunocytostaining of PCNA and an MTT assay were performed
to detect the proliferative activity of HUVECs at 0, 4, 8, 12 and
24 h. The results showed that increased EL promoted HUVEC
proliferation (Fig. 3). Similar to
the change in the expression levels of EL, the ratio of
PCNA-positive cells was highest at 8 h after rhIL-6 treatment,
after which the ratio reduced. At 24 h after rhIL-6 treatment, the
ratio of PCNA-positive cells ratio remained higher than the
original level.
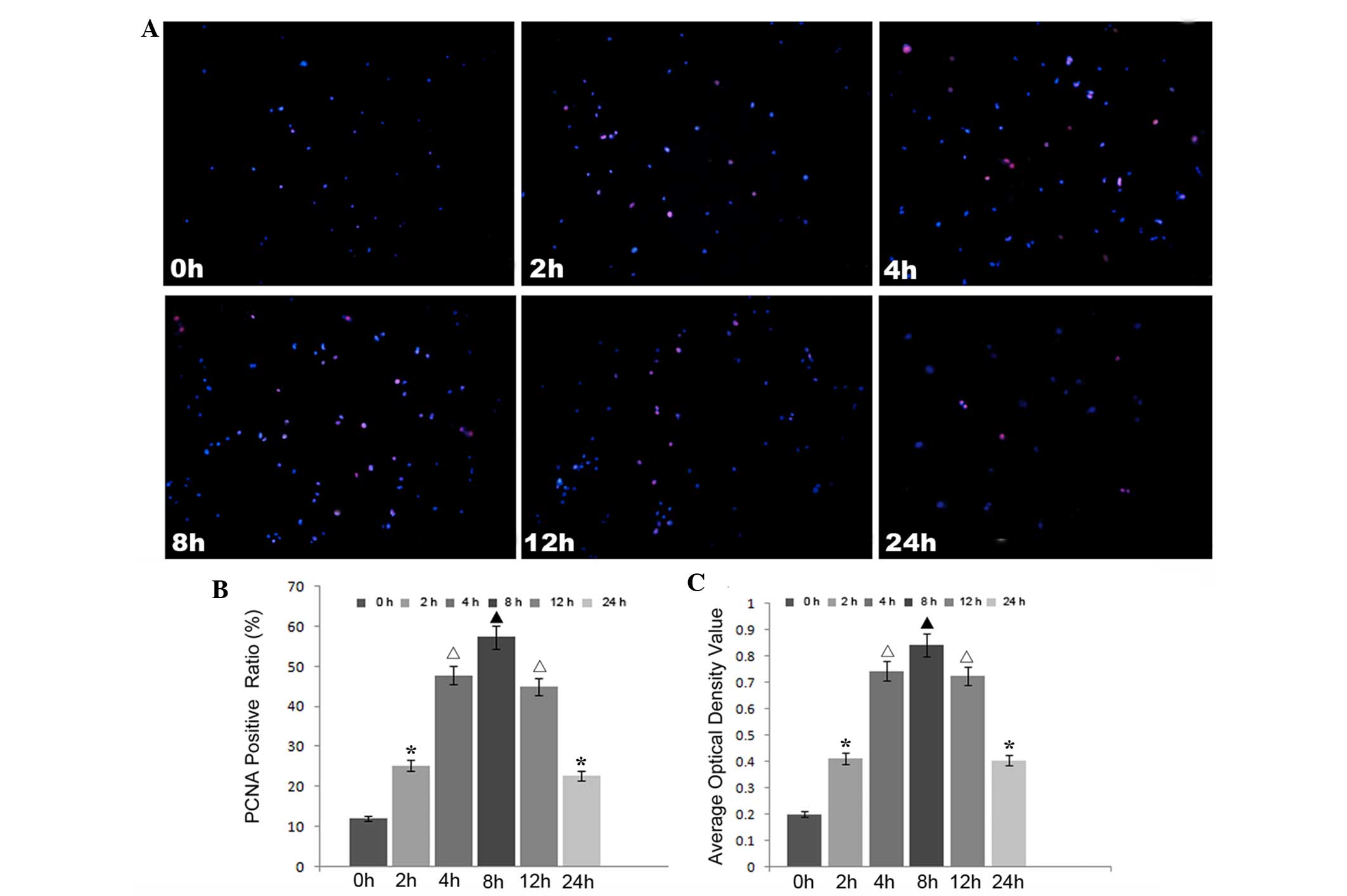 | Figure 3rhIL-6 treatment promoted the
proliferation of HUVECs. rhIL-6 treatment was shown to promote
HUVEC proliferation by immunofluorescence staining of PCNA and MTT
assays. (A) Immunofluorescence staining of PCNA in HUVECs in each
group. magnification, ×100. (B) Semi-quantitative analysis of the
immunofluorescence staining of PCNA. (C) Statistical analysis of
the MTT assays for each group. Different symbols indicate
statistical significance (P<0.05). ▲P<0.05
compared with the 0, 2, 4, 12 and 24 h groups;
*P<0.05 compared with the 0, 4, 8 and 12 h groups;
△P<0.05 compared with the 0, 2, 8 and 24 h groups.
rhIL-6, recombinant human interleukin-6; EL, endothelial lipase;
HUVECs, human umbilical vein endothelial cells; PCNA, proliferating
cell nuclear antigen. |
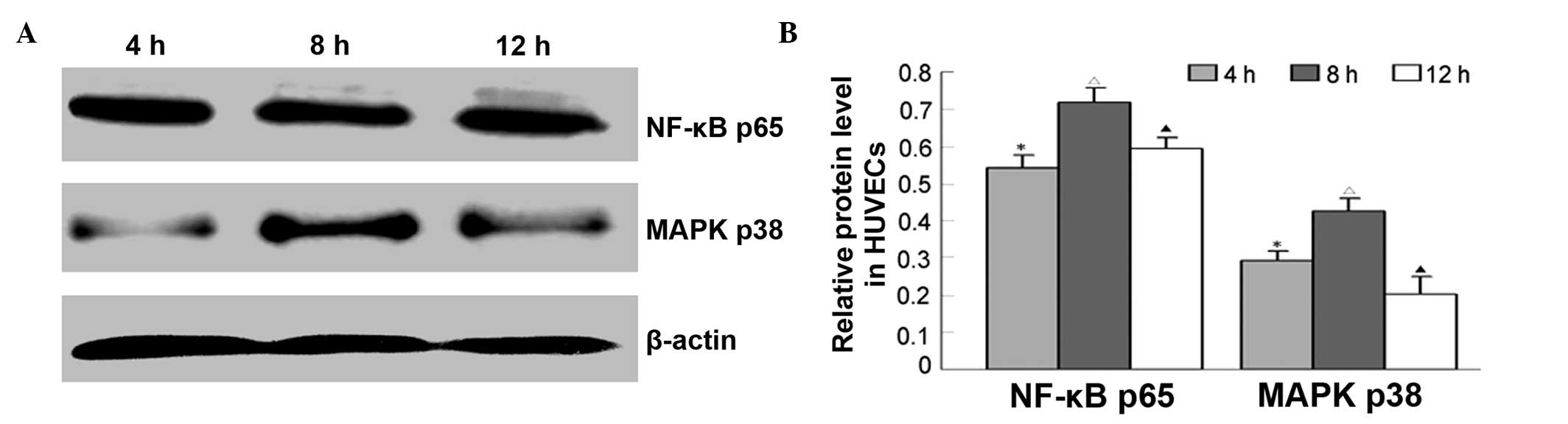
rhIL-6 treatment increases p38 MAPK and
p65 NF-κB expression in HUVECs
To determine whether IL-6 regulates EL expression
through MAPK or NF-κB signaling pathway, p38 MAPK and p65 NF-κB
protein levels at 4, 8 and 12 h were detected by western blotting.
The results showed that p38 MAPK or p65 expression changes were
consistent with that of EL. That is, after HUVECs were incubated
with rhIL-6, p38 MAPK and p65 NF-κB expression levels increased
significantly at 4, 8 and 12 h (Fig.
4).
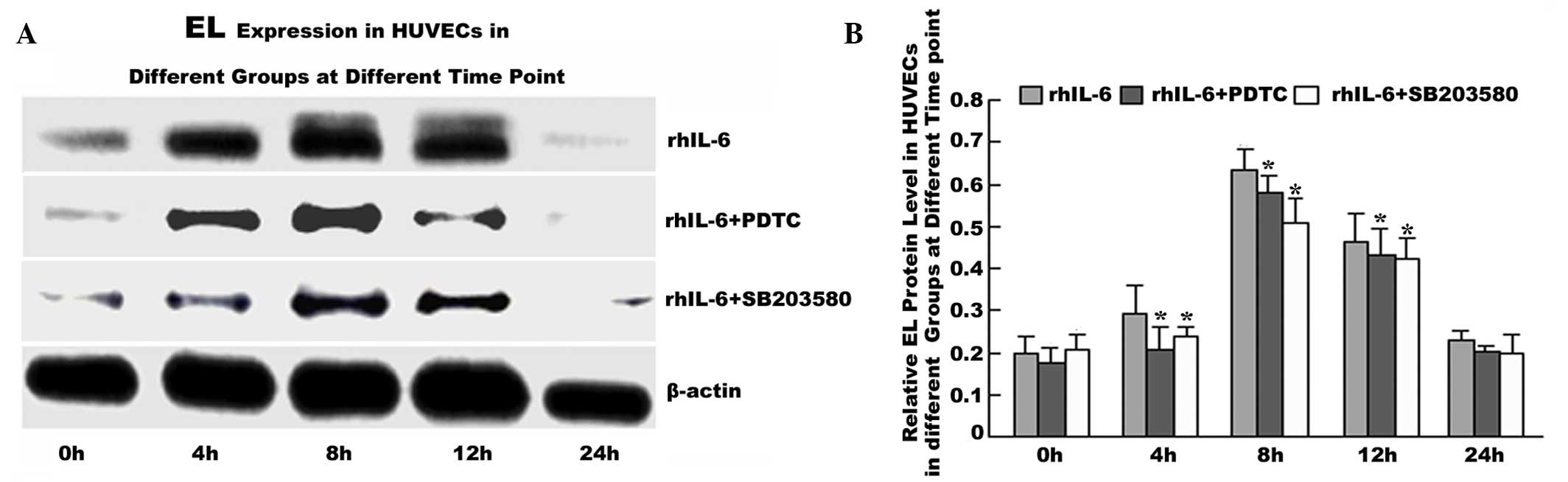
p38 MAPK inhibitor SB203580 or p65 NF-κB
inhibitor PDTC pretreatment decreases EL expression in HUVECs
To confirm that IL-6 regulates the EL level via the
MAPK p38 and P65 NF-kB signaling pathway, HUVECs were pretreated
with the p38 MAPK inhibitor SB203580 or P65 NF-kB inhibitor PDTC
for 1 h prior to rhIL-6 treatment. The results showed that EL
expression in the rhIL-6+SB203580 and rhIL-6+PDTC groups was
significantly decreased at each time point between 4 and 12 h
compared with the IL-6 group (P<0.05; Fig. 5).
Discussion
EL was identified in 1999 as part of the
triglyceride lipase family of genes (8). Structurally, EL has 45, 40 and 27%
homology with lipoprotein lipase, hepatic lipase and pancreatic
lipase, respectively. EL is a critical enzyme in high density
lipoprotein (HDL) metabolism by hydrolyzing the phospholipid
component (9). The plasma HDL
concentration rises significantly in EL gene knockout mice and the
overexpression of EL reduces the HDL level markedly while HDL is an
independent risk factor for atherosclerosis (8).
EL also has non-enzymatic activities, including
mediating the uptake of apolipoprotein B and the adhesion of
monocytes or macrophages to the endothelia, which indicates that EL
participates in the pathogenesis of atherosclerosis (11,16,17).
Recently, accumulating clinical evidence demonstrated that EL is
closely associated with coronary atherosclerosis. Badellino et
al (18) demonstrated that
serum EL concentration in patients with metabolic syndrome was
significantly higher than that in the normal population, and this
was positively correlated with the development of coronary
atherosclerosis. Furthermore, polymorphisms of the gene encoding EL
may be associated with the progression of acute coronary syndrome
(18–20). Fang et al (21) found that EL expression increases in
the endothelial cells of the vascular system in patients with
coronary heart disease and this increase is correlated with the
severity of the clinical syndrome and also increases the coronary
risk scores.
Atherosclerosis is a chronic inflammatory disease,
characterized by the interaction of inflammatory mediators and
cytokines in vascular endothelia. In 2000, it was demonstrated that
IL-1β and tumor necrosis factor-α (TNF-α) upregulates the mRNA
expression of EL in HUVECs in vitro (22). Jin et al (23) confirmed that this was partially
mediated through the NF-κB pathway. Badellino et al
(24) and Paradis et al
(25) found that IL-6, soluble
tumor necrosis factor-R, soluble intracellular adhesion molecule
and other cytokines are positively correlated with EL levels
(24). These data indicated that
inflammation has a significant effect on the expression of EL. The
present results also showed that rhIL-6 treatment increased EL
expression between 4 and 12 h and peaked 8 h after treatment. This
confirmed the time-dependent effects of IL-6 on EL expression in
endothelial cells.
MAPK is an important intracellular signal
transduction pathway. The MAPK signaling pathway can respond to
various extracellular stimuli to mediate cell growth,
differentiation and apoptosis, and is associated with endothelial
dysfunction, inflammation, hypertension and vascular remodeling
(25–28). The inhibition of p38 MAPK has been
shown to be a clinical tool against inflammatory diseases,
including chronic obstructive pulmonary disease (29). The p38 MAPK expression level
increases in atherosclerosis, which may constitute a causative
mechanism (15,29). This may be one of the mechanisms
for inflammation-induced atherosclerosis. Paravicini et al
(29) found that the development
of fibrosis can be inhibited by the inhibition of p38 MAPK by
SB203580, which suggests that the signal is transmitted via the p38
MAPK pathway.
NF-κB is an important transcriptional regulatory
factor, which responds to various cytokines. NF-κB is activated and
enters the nucleus where it combines with specific DNA motifs and
stimulates the expression of various genes. Chromatin
immunoprecipitation and electrophoretic mobility shift assays have
revealed that the EL gene has two NF-κB binding sites (30). The activation of NF-κB has also
been observed in human and experimental atherosclerosis (31,32).
Cao et al (33) found that
the progression of the instability of atherosclerotic plaques may
be mediated by the NF-κB pathway.
In the current study, HUVECs were pretreated with a
p38 MAPKs inhibitor (SB203580) or an NF-κB inhibitor (PDTC) prior
to rhIL-6 treatment and the results showed that the pretreatment of
HUVECs with either SB203580 or PDTC inhibited the IL-6-induced
upregulation of EL expression. This therefore confirmed that IL-6
can regulate the expression of EL partly via the p38 MAPK and NF-κB
signaling pathways.
In conclusion, the current study found that external
addition of rhIL-6 upregulates the expression of EL in endothelial
cells in a time-dependent manner and this upregulation can be
partially inhibited by the p38 MAPK inhibitor (SB203580) or NF-κB
inhibitor (PDTC). This in vitro study suggests that p38 MAPK
and p65 NF-κB may participate in the process of IL-6-regulated
expression of EL, and may be another mechanism by which IL-6 is
involved in the pathogenesis of atherosclerosis. Although this
study cannot be extrapolated for in vivo conditions, it
provides evidence on a possible mechanism by which IL-6 affects EL
expression in endothelial cells. Thus, this study may provide a
theoretical and experimental basis for future preventative
treatments that target these factors specifically.
Abbreviations:
|
IL-6
|
interleukin-6
|
|
rhIL-6
|
recombinant human interleukin-6
|
|
NF-κB
|
nuclear factor-κB
|
|
PDTC
|
pyrrolidine dithiocarbamate
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
EL
|
endothelial lipase
|
|
HDL
|
high density lipoprotein
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
EGF
|
epithelial growth factor
|
|
EDTA
|
ethylenediaminetetraacetic acid
|
|
FBS
|
fetal bovine serum
|
|
PBS
|
phosphate-buffered saline
|
|
DAB
|
diaminobenzidine
|
Acknowledgments
This study was supported by the Natural Science
Foundation of Shandong Province (grant nos. Y2008C47, ZR2011HM086,
ZR2014HM082 and ZR2011CM042).
References
|
1
|
Rattazzi M, Puato M, Faggin E, Bertipaglia
B, Zambon A and Pauletto P: C-reactive protein and interleukin-6 in
vascular disease: Culprits or passive bystanders? J Hypertens.
21:1787–1803. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bernberg E, Ulleryd MA, Johansson ME and
Bergström GM: Social disruption stress increases IL-6 levels and
accelerates atherosclerosis in ApoE-/- mice. Atherosclerosis.
221:359–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nishimoto N: Interleukin-6 in rheumatoid
arthritis. Curr Opin Rheumatol. 18:277–281. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barton BE: Interleukin-6 and new
strategies for the treatment of cancer, hyperproliferative diseases
and paraneoplastic syndromes. Expert Opin Ther Targets. 9:737–752.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smolen JS and Maini RN: Interleukin-6: A
new therapeutic target. Arthritis Res Ther. 8:S52006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Genovese MC, McKay JD, Nasonov EL, Mysler
EF, da Silva NA, Alecock E, Woodworth T and Gomez-Reino JJ:
Interleukin-6 receptor inhibition with tocilizumab reduces disease
activity in rheumatoid arthritis with inadequate response to
disease-modifying antirheumatic drugs: The tocilizumab in
combination with traditional disease-modifying antirheumatic drug
therapy study. Arthritis Rheum. 58:2968–2980. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Williams SC: First IL-6-blocking drug
nears approval for rare blood disorder. Nat Med. 19:11932013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaye M, Lynch KJ, Krawiec J, Marchadier D,
Maugeais C, Doan K, South V, Amin D, Perrone M and Rader DJ: A
novel endothelial-derived lipase that modulates HDL metabolism. Nat
Genet. 21:424–428. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ishida T, Choi S, Kundu RK, Hirata K,
Rubin EM, Cooper AD and Quertermous T: Endothelial lipase is a
major determinant of HDL level. J Clin Invest. 111:347–355. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gordon DJ and Rifkind BM: High-density
lipoproteins-the clinical implications of recent studies. N Engl J
Med. 321:1311–1316. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Broedl UC, Maugeais C, Millar JS, Jin W,
Moore RE, Fuki IV, Marchadier D, Glick JM and Rader DJ: Endothelial
lipase promotes the catabolism of ApoB-containing lipoproteins.
Circ Res. 94:1554–1561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ishida T, Choi SY, Kundu RK, Spin J,
Yamashita T, Hirata K, Kojima Y, Yokoyama M, Cooper AD and
Quertermous T: Endothelial lipase modulates susceptibility to
atherosclerosis in apolipoprotein-E-deficient mice. J Biol Chem.
279:45085–45092. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jin W, Sun GS, Marchadier D, Octtaviani E,
Glick JM and Rader DJ: Endothelial cells secrete triglyceride
lipase and phospholipase activities in response to cytokines as a
result of endothelial lipase. Circ Res. 92:644–650. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi SY, Hirata K, Ishida T, Quertermous T
and Cooper AD: Endothelial lipase: A new lipase on the block. J
Lipid Res. 43:1763–1769. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Monaco C, Andreakos E, Kiriakidis S, Mauri
C, Bicknell C, Foxwell B, Cheshire N, Paleolog E and Feldmann M:
Canonical pathway of nuclear factor kappa B activation selectively
regulates proinflammatory and prothrombotic responses in human
atherosclerosis. Proc Natl Acad Sci. 101:5634–5639. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Annema W and Tietge UJ: Role of hepatic
lipase and endothelial lipase in high-density lipoprotein-mediated
reverse cholesterol transport. Nat Rev Drug Discov. 4:193–205.
2005.
|
|
18
|
Badellino KO, Wolfe ML, Rielly MP and
Rader DJ: Endothelial lipase concentrations are increased in
metabolic syndrome and associated with coronary atherosclerosis.
PLoS Med. 3:e222006. View Article : Google Scholar
|
|
19
|
Cai G, He G and Qi C: The association
between endothelial lipase 2384A/C gene polymorphism and acute
coronary syndrome in a Chinese population. Mol Biol Rep.
39:9879–9884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singaraja RR, Sivapalaratnam S, Hovingh K,
Dubé MP, Castro-Perez J, Collins HL, Adelman SJ, Riwanto M, Manz J,
Hubbard B, et al: The impact of partial and complete loss of
function mutations in endothelial lipase on high-density
lipoprotein levels and functionality in humans. Circ Cardiovasc
Genet. 6:54–62. 2013. View Article : Google Scholar
|
|
21
|
Fang YQ, Huang L and Li AM: Correlation
between the expression of endothelial lipase in CECs and the
improved jeopardy score in CAD patients. Chinese Journal of
Critical Care Medicine. 28:5–8. 2008.
|
|
22
|
Hirata K, Ishida T, Matsushita H, Tsao PS
and Quertermous T: Regulated expression of endothelial cell-derived
lipase. Biochem Biophys Res Commun. 272:90–93. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin W, Sun GS, Marchadier D, Octtaviani E,
Glick JM and Rader DJ: Endothelial cells secrete triglyceride
lipase and phospholipase activities in response to cytokines as a
result of endothelial lipase. Circ Res. 92:644–650. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Badellino KO, Wolfe ML, Reilly MP and
Rader DJ: Endothelial lipase is increased in vivo by inflammation
in humans. Circulation. 117:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paradis ME, Badellino KO, Rader DJ,
Deshaies Y, Couture P, Archer WR, Bergeron N and Lamarche B:
Endothelial lipase is associated with inflammation in humans. J
Lipid Res. 47:2808–2813. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yogi A, Callera GE, Aranha AB, Antunes TT,
Graham D, McBride M, Dominiczak A and Touyz RM:
Sphingosine-1-phosphate-induced inflammation involves receptor
tyrosine kinase transactivation in vascular cells: Upregulation in
hypertension. Hypertension. 57:809–818. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gerthoffer WT: Mechanisms of vascular
smooth muscle cell migration. Circ Res. 100:607–621. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jacobsen JC, Mulvany MJ and
Holstein-Rathlou NH: A mechanism for arteriolar remodeling based on
maintenance of smooth muscle cell activation. Am J Physiol Regul
Integr Comp Physiol. 294:R1379–R1389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paravicini TM, Montezano AC, Yusuf H and
Touyz RM: Activation of vascular p38 MAPK by mechanical stretch is
independent of c-Src and NADPH oxidase: Influence of hypertension
and angiotensin II. J Am Soc Hypertension. 6:169–178. 2012.
View Article : Google Scholar
|
|
30
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|
|
31
|
Brand K, Page S, Rogler G, Bartsch A,
Brandl R, Knuechel R, Page M, Kaltschmidt C and Baeuerle PA:
Activated transcription factor nuclear factor-kappa B is present in
the atherosclerotic lesion. J Clin Invest. 97:1715–1722. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hajra L, Evans AI, Chen M, Hyduk SJ,
Collins T and Cybulsky MI: The NF-kappa B signal transduction
pathway in aortic endothelial cells is primed for activation in
regions predisposed to atherosclerotic lesion formation. Proc Natl
Acad Sci USA. 97:9052–9057. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cao Y, Zhou X, Liu H, Zhang Y, Yu X and
Liu C: The NF-κB pathway: Regulation of the instability of
atherosclerotic plaques activated by Fg, Fb and FDPs. Mol Cell
Biochem. 383:29–37. 2013. View Article : Google Scholar : PubMed/NCBI
|