Introduction
Phosphatidylinositol-4,5-bisphosphate 3-kinase
(PI3K) and protein kinase B (AKT) are involved in major
pro-survival signaling pathways in cancer cells. While excessive
proliferation and survival of aberrant cells is suppressed in
normal homeostasis, in tumor cells, this is overridden by the
upregulation of antiapoptotic proteins and other cell
death-associated factors. In particular, AKT regulates cell
survival, metabolism, invasion and metastasis, and is also pivotal
role in antiapoptotic and antiproliferative signaling in response
to essential cytokines. AKT-dependent cell survival is mediated
through the induction of antiapoptotic gene expression (1), including members of the forkhead and
B-cell lymphoma (BCL)-2 family, as well as the nuclear factor
(NF)-κB. It is known that the BCL-2 family of proteins regulates
apoptosis through modulating mitochondrial permeability (2). In addition, AKT-dependent degradation
of the inhibitor of NF-κB leads to NF-κB translocation from the
cytoplasm to the nucleus where it promotes cell survival by
regulating the expression levels of target genes. AKT can also
inhibit p53, which is a tumor suppressor protein that induces
apoptosis, senescence and cell cycle arrest (3).
Consequently, drugs designed to target AKT have
become an important and viable approach in the development of
cancer therapeutics with a number of AKT inhibitors already being
used in clinical trials. Additionally, it has become increasingly
apparent that inhibiting AKT via the inhibition of upstream signals
can be equally as significant as direct targeting of this kinase.
More specifically, tumor necrosis factor receptor-associated factor
(TRAF)6, which is an E3 ubiquitin ligase, has been shown to
ubiquitylate and activate AKT (4),
and it also serves an essential role in regulating the NF-κB and
mitogen-activated protein kinase signaling cascades. TRAF6 consists
of a conserved C-terminal domain that regulates interactions with
upstream signaling proteins, and an N-terminal domain that
comprises the core of the ubiquitin ligase catalytic domain
(5). In the presence of E2
ubiquitin-conjugating enzymes, TRAF6 catalyzes the transfer of
polyubiquitin chains to the substrates (6). Notably, TRAF6 mediates K63-directed
ubiquitylation of AKT, and this type of modification is associated
with functional changes in the target substrate in contrast to
classical K48-directed ubiquitylation, which leads to substrate
degradation.
Additionally, in the context of insulin-like growth
factor-1 stimulation, overexpression of TRAF6 enhances AKT
phosphorylation and activation. By contrast, AKT phosphorylation at
Thr-308 and Ser-473 was inhibited in TRAF6-deficient mouse
embryonic fibroblasts (4). TRAF6
is recruited by the phosphorylated discoidina neuropilin-like
membrane protein discoidin, CUB and LCCL domain-containing protein
2, leading to increased E3 ubiquitin ligase activity and subsequent
activation of AKT, and accelerating epidermal growth factor
receptor-driven tumorigenesis (7).
As a result of the close association between TRAF6 and AKT, the
present study speculated that AKT can be indirectly targeted by
developing small molecules that can bind to TRAF6 and disrupt its
interaction with AKT, and that this may be a promising approach in
cancer therapeutics. To determine this, the present study performed
a computational docking study to screen small molecules, which can
bind to TRAF6 in regions critical for its interaction with AKT
(residues 50–159, RING and zinc finger domains). The present study
found that quinine, a member of well-known Cinchona
alkaloids, has a high affinity for TRAF6 and may indeed induce
apoptosis, as well as reduce cell viability. The underlying
mechanism by which quinine inhibited AKT activation was also
examined using a range of ex vivo cellular assays.
Materials and methods
Materials
A549 human lung adenocarcinoma and HeLa cervical
cancer cell lines were kindly provided by Dr. Yaozhou Zhang
(Tianjin International Joint Academy of Biomedicine, Tianjin,
China). RPMI-1640 medium and fetal bovine serum (FBS) were obtained
from Tianjin HOPE Biotechnology Co., Ltd. (Tianjin, China). Rabbit
polyclonal antibodies against BCL-2 (cat. no. 2876),
BCL-2-associated X protein (BAX; cat. no. 2772), total AKT (cat.
no. 4691), phosphorylated (p-)AKT (Thr308; cat. no. 4056s) and
p-AKT (Ser473; cat. no. 4051s) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Rabbit polyclonal antibody
against β-actin (cat. no. PR0255) was purchased from Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing, China).
Horseradish peroxidase (HRP)-conjugated goat poly-clonal antibody
against rabbit and mouse (cat. no. M21003) was obtained from
Shanghai Abmart Biotechnology Co., Ltd. (Shanghai, China).
Radioimmunoprecipitation assay (RIPA) buffer and protease inhibitor
cocktail were purchased from Beijing Kangwei Biotechnology Co.,
Ltd. (Beijing, China). Phosphatase inhibitor cocktail and
lipopolysaccharide (LPS) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Quinine was purchased from Tianjin Xiensi
Biochemical Technology Co., Ltd. (Tianjin, China). Cis-platinum was
purchased from Jiangsu Hansoh Pharmaceutical Co., Ltd. (Tianjin,
China). Protein A sepharose beads were purchased from Beijing
Kangwei Biotechnology Co., Ltd. (Pierce, Rockford, IL, USA). All
chemicals and solvents were of analytical grade.
Virtual screening
A three-dimensional (3D) structure library
containing 1,792 compounds was established for the virtual screen.
Structures of these compounds were generated using a
two-dimensional (2D)/3D editor-sketcher and were minimized to a
local energy minimum using the CHARMm-like force field implemented
within with Catalyst 4.11 software (8). The 3D structure of TRAF6 (residues
50–159, RING and zinc finger domains) was retrieved from the
Protein Data Bank as the docking receptor (http://www.rcsb.org/pdb; accession no. 3HCT). Both the
TRAF6 crystal structure and candidate ligands were prepared using
AutoDock Tools v.1.5.2 software (http://autodock.scripps.edu).
For virtual screening, flexible ligands and rigid
receptor docking calculations were performed with the AutoDock4.10
software package using the following parameters: The Lamarckian
genetic algorithm; a population size of 300; energy evaluations of
25,000,000; search runs of 100. The docking area was defined as a
dimension of 60×60×60 points with grid spacing of 0.375 Å. The grid
box was centered on the binding site of the ubiquitin-conjugating
E2 enzyme, Ubc13. The results were ranked, according to the binding
free energy. The conformations from the docking experiments were
analyzed using Chimera software (9), which also identified the hydrophobic
interactions between the receptor and the ligands.
Cell culture
A549 and HeLa cells were cultured in RPMI-1640
medium, supplemented with 10% FBS at 37°C in a humidified
atmosphere of 5% CO2.
Cell viability assay
The effect of quinine on HeLa and A549 cell
viability and proliferation was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, 200 µl HeLa or A549 cell suspensions were
seeded into 96-well plates at densities of 4 and 3×103
cells/well, respectively. The cells were allowed to adhere
overnight and were subsequently treated with LPS (20 µg/ml)
in the absence or presence of quinine at various concentrations (1,
5, 10, 25, 50, 100, 250 and 500 µM). Cis-platinum (16.7
µM) was used as a positive control. Cell viability was
assessed by adding 20 µl of 5 mg/ml MTT to each well and
incubating for 4–6 h. The medium was replaced with 150 µl
dimethyl sulfoxide, which was stirred to dissolve the formazan
crystals. The absorbance (A) was measured at 490 nm using an ST-360
microplate reader (Shanghai Kehua Bio-engineering Co., Ltd.,
Shanghai, China). The survival rate of each group of cells was
calculated relative to the vehicle-treated control cells
(designated as 100%). Cell inhibition ratio was calculated using
the following formula: Cell inhibition ration =
(Acontrol − Atreated / Acontrol) ×
100.
Detection of apoptosis by flow
cytometry
Apoptosis was assessed by flow cytometry using an
Apoptosis Detection kit (Becton-Dickinson, Franklin Lakes, NJ,
USA), according to the manufacturer's protocol. Briefly, HeLa cells
were seeded into a 6 cm plate at a density of 6×105
cells/well and allowed to attach overnight. The medium was replaced
with fresh medium containing various concentrations of quinine
(100, 150 or 200 µM) for 48 h at 37°C. The cells were
collected and resuspended in 1X binding buffer at a concentration
of 1×106 cells/ml, and 100 µl of the cell
suspension was transferred to a 5 ml culture tube, to which 5
µl annexin V-fluorescein isothiocyanate (FITC) and propidium
iodide (PI) were added (from the Apoptosis Detection kit). The
mixture was incubated in the dark for 15 min at room temperature
(25°C). The apoptotic index was immediately determined with a
FACSCalibur instrument (BD Biosciences, Franklin Lakes, NJ, USA).
Cell Quest Pro software v5.1 (BD Biosciences) was used to analyze
flow cytometry data.
Western blot analysis
Western blotting was performed according to a
standard protocol. Briefly, HeLa and A549 cells at 60–70%
confluence were incubated with 150 µM quinine. The cells
were rinsed with ice-cold phosphate-buffered saline and resuspended
in RIPA buffer, supplemented with phenylmethylsulfonyl fluoride and
phosphatase inhibitors. Following an incubation on ice for 30 min,
the lysates were centrifuged at 12,000 × g for 10 min and the
supernatant was collected. Protein concentrations were determined
using a bicinchoninic acid protein assay kit (CWBIO, Beijing,
China) and 40 µg protein was resolved by 3% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis. The proteins were
transferred onto a polyvinylidene difluoride membrane (EMD
Millipore, Bedford, MA, USA), which was blocked with a solution of
5% non-fat dry milk or 5% bovine serum albumin in Tris-buffered
saline (10 mM Tris-HCl and 150 mM NaCl) with 0.05% Tween-20. After
2 h, the membranes were washed and incubated with primary
antibodies against β-actin (1:1,500), AKT (1:1,000), p-AKT
(1:1,000), BCL-2 (1:1,000), BAX (1:1,000), and TRAF6 (1:1,000)
overnight at 4°C, followed by washing and incubation with
HRP-conjugated goat anti-rabbit or anti-mouse immunoglobulin G
(1:10,000) for 1 h at 25°C. Antibody binding was detected using an
enhanced chemiluminescence kit (CWBIO), according to the
manufacturer's protocol. Image J software v.1.48u (National
Institutes of Health, Bethesda, MD, USA) was used to quantify the
intensity of protein bands.
Statistical analysis
The data are presented as the mean ± standard
deviation of multiple independent experiments. One-way analysis of
variance was used to compare group means, followed by Dunnett's
t-test, using StataSE12 software (Stata Corp., College Station, TX,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Selection of quinine using computational
virtual screening
The 1,792 compounds were docked into the binding
pocket of TRAF6 using AutoDock 4.10 software, and the top 50
compounds were selected with the lowest estimated inhibition
constant. Subsequently, binding conformations for each of these
selected compounds were exported and evaluated manually, according
to the estimated binding free energy and hydrophobic interactions.
Quinine (ΔG = −6.8 kcal/mol) was selected based on these
calculations and purchased for further experimental testing.
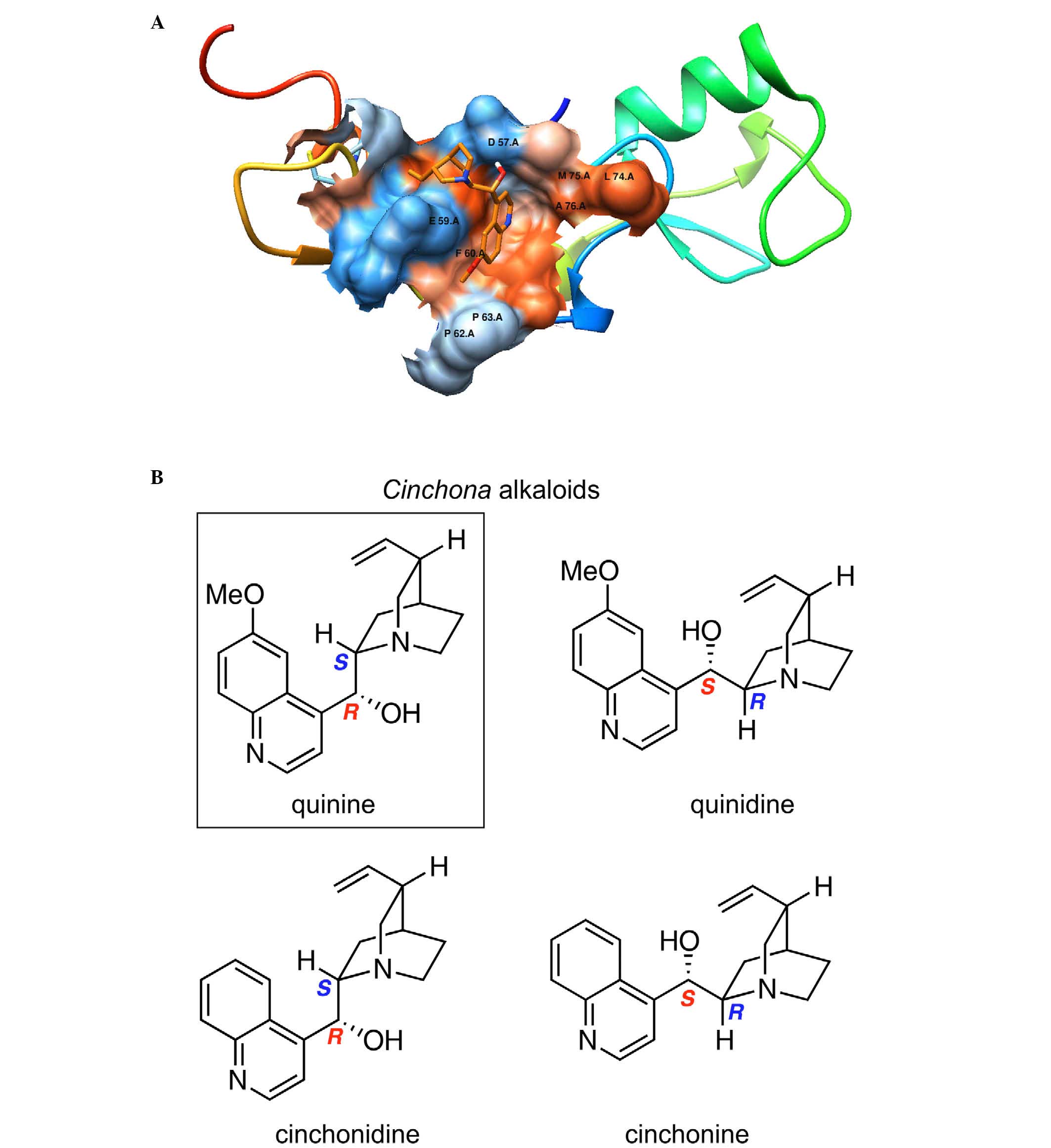
Fig. 1 demonstrated
the binding mode of quinine: The oxygen atom of the hydroxyl group
of quinine forms H-bonds to the OD1 atom of residue Asp-57; the
quinoline ring is in π–π stacking interaction with Phe-60 and also
in good hydrophobic interactions with the surrounding residues,
Phe-60, Pro-63 and Leu-64.
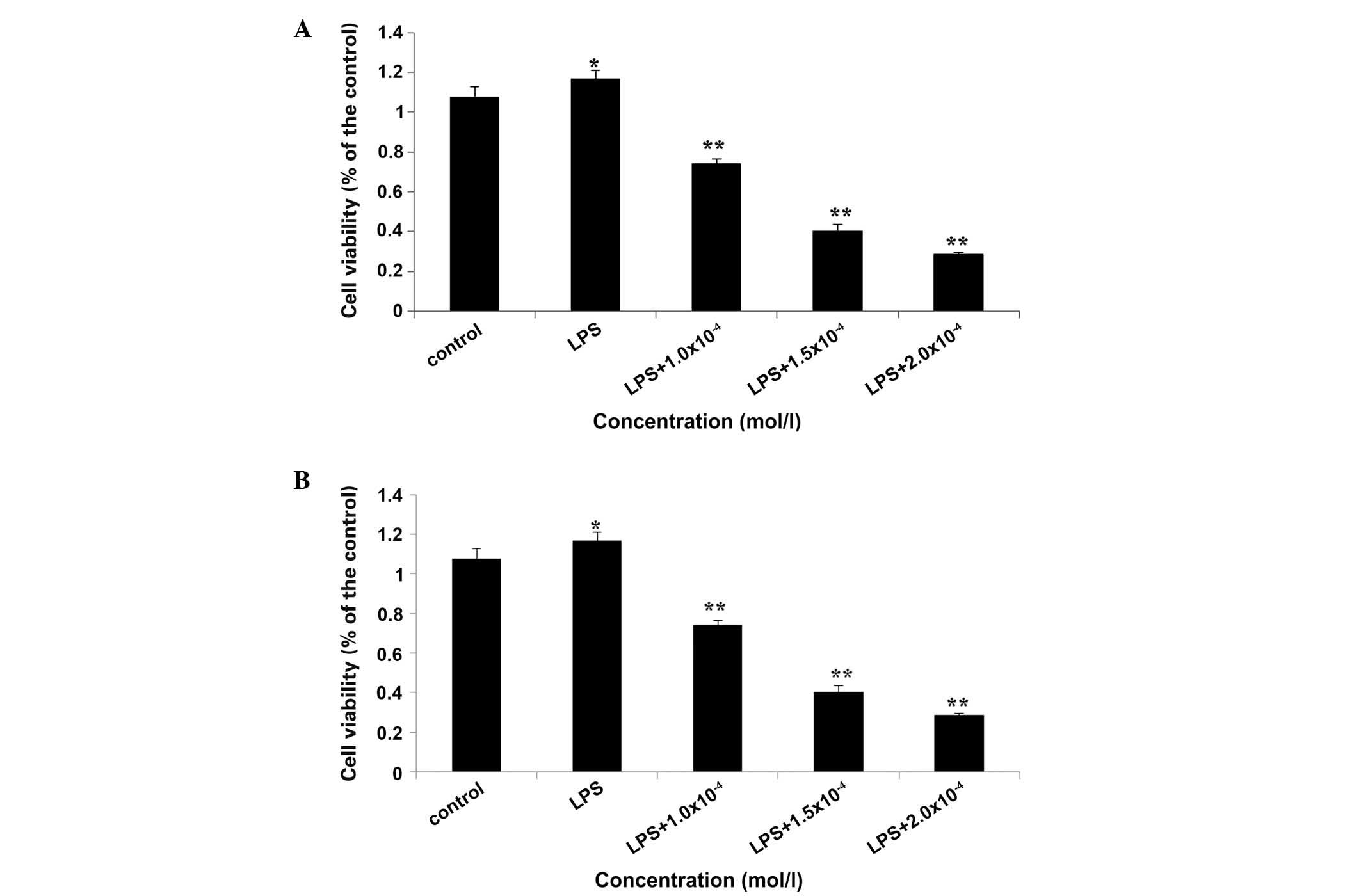
Quinine suppresses the growth of A549 and
HeLa cells
As a result of the molecular docking results, which
suggested quinine binds to TRAF6 and affects its function, the
present study therefore used HeLa and A549 cell lines that express
high levels of endogenous TRAF6 (5,10) in
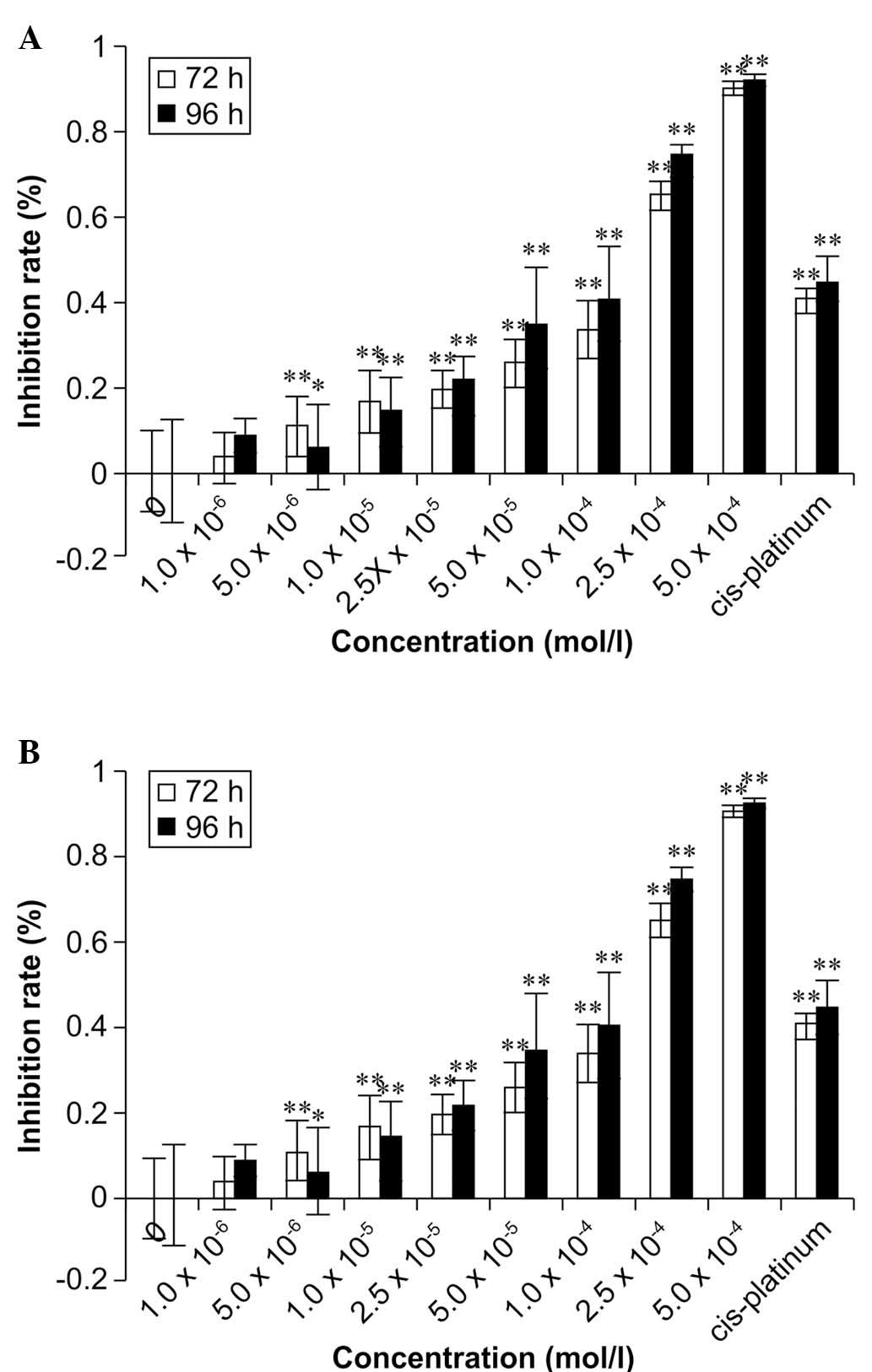
the following experiments. As shown in Fig. 2A, quinine inhibited HeLa cell
proliferation in a concentration- and time-dependent manner, with
concentrations >5 µM markedly reducing cell viability. An
increase in the concentration between 10 and 500 µM
increased the inhibition rate from 12.80 to 82.53%, when treated
for 72 h. Similarly, prolonged incubation with quinine for 96 h
increased the rate of inhibition from 17.67 to 89.73% when the dose
was increased between 10 and 500 µM. These effects were also
reproduced in the A549 cells (Fig.
2B).
Quinine induces apoptosis in HeLa
cells
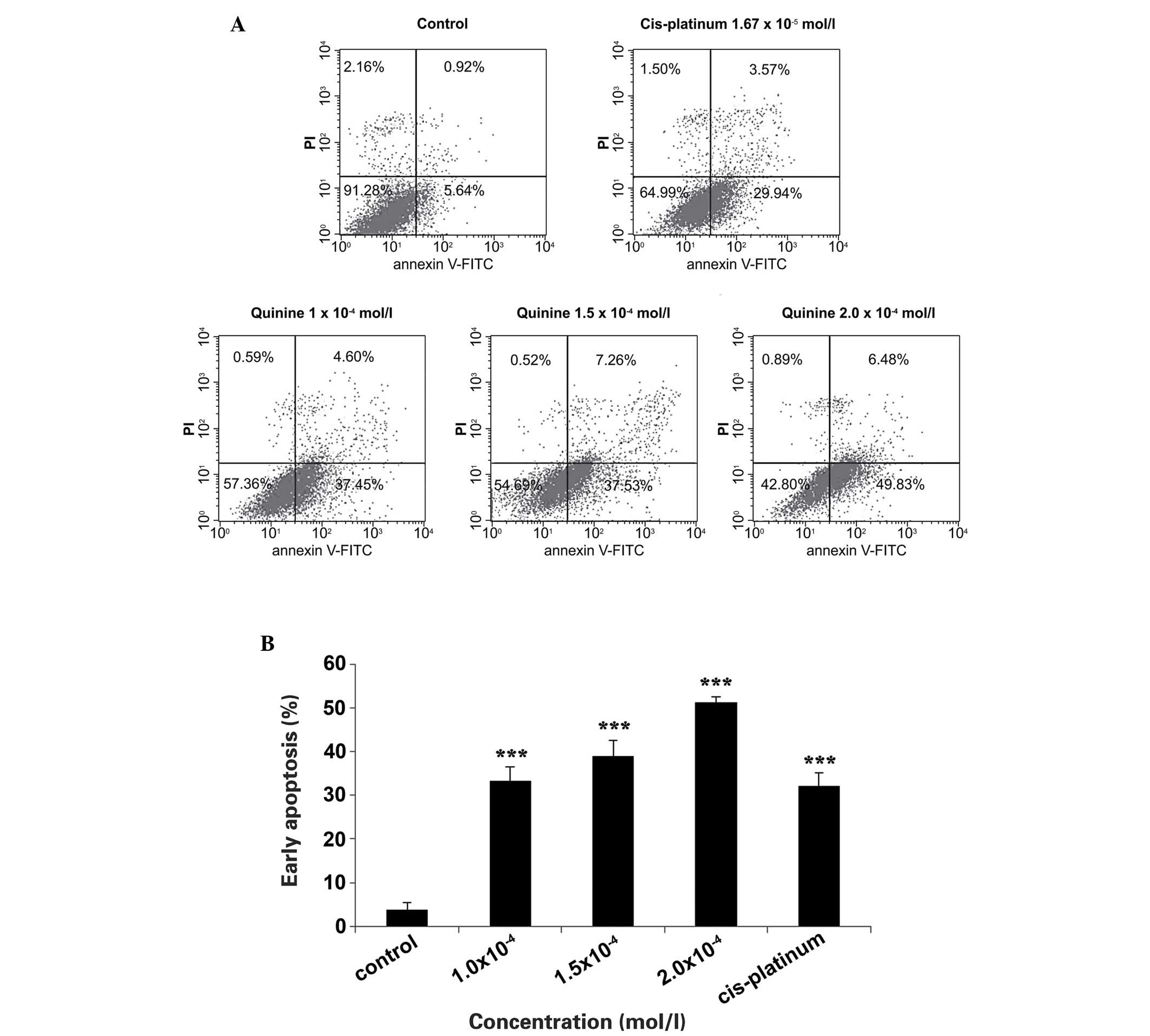
The present study next investigated whether the
decreased cell viability following quinine treatment was due to the
occurrence of apoptosis. To confirm this hypothesis, annexin V-FITC
and PI staining were used to discriminate between cells undergoing
early apoptosis (annexin V+/PI−) and necrosis (PI+). The percentage
of apoptotic cells increased in a dose-dependent manner upon
quinine treatment (Fig. 3). The
fraction of viable cells decreased from 91.28 down to 42.80%
following treatment with 200 µM quinine. This was
simultaneously accompanied with an increase (5.64 to 49.83%) in the
fraction of early apoptotic cells in the population. Treatment with
the positive control, Cis-platinum, reduced the number of viable
cells to 64.99% and was accompanied by a concomitant increase in
the fraction of early apoptotic cells to 29.94%. By contrast, the
percentage of PI+ cells increased from 0.92 to 6.48% upon treatment
with 200 µM quinine. These results indicated that quinine
induces cell apoptosis, rather than necrosis.
Quinine-induced apoptosis activates BAX
in HeLa and A549 cells
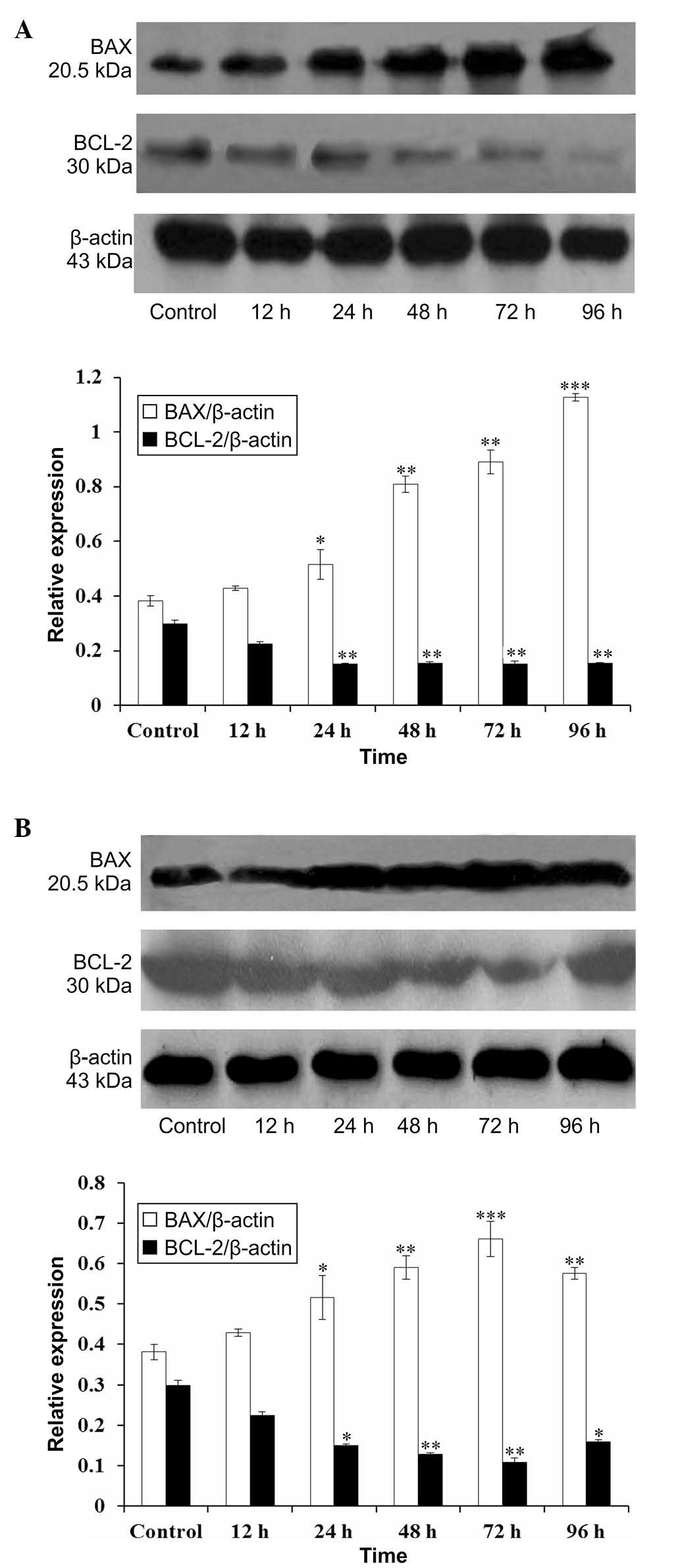
The above results showed that quinine application
resulted in the inhibition of HeLa and A549 cell growth and
proliferation, and induced early apoptosis. Since the BCL-2 family
serves a critical role in the execution of programmed cell death,
the present study assessed whether the apoptosis induced by quinine
was associated with changes in the levels of either BCL-2 or BAX
(Fig. 4). Indeed, the quinine
treatment led to an increase in the levels of the pro-apoptotic
factor, BAX, in a time-dependent manner, as well as a decrease in
the expression of BCL-2. These results suggested that quinine
exposure is responsible for inducing apoptosis, which underlies the
reduced viability of HeLa and A549 cells.
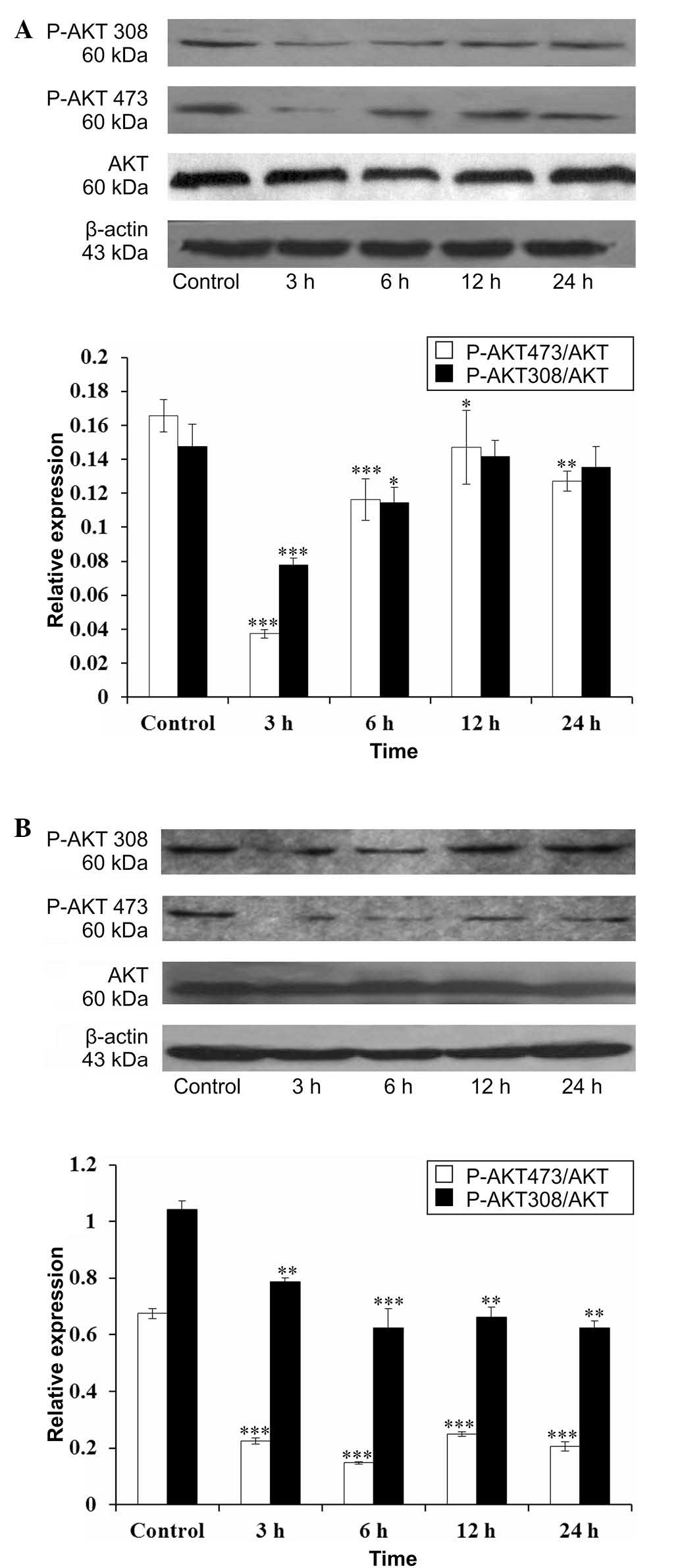
Quinine treatment regulates the
phosphorylation of AKT
To elucidate the underlying molecular mechanism in
which quinine induces apoptosis, the phosphorylation status of AKT
was analyzed in these two cell lines following LPS treatment. The
present study decided to incorporate LPS here, since it is a robust
inducer of AKT activity in HeLa and A549 cells (11,12).
Stimulation of HeLa and A549 cells with LPS for 3 h resulted in an
increase in p-AKT levels, and this effect was inhibited by quinine
treatment (Fig. 5). In addition,
HeLa and A549 cell proliferation was enhanced by treatment with
LPS, which was abrogated in the presence of quinine (Fig. 6). These results indicated that
quinine induced apoptosis via the activation of AKT.
Discussion
Cancer has been the top health threat for over a
century and is second only to cardiovascular diseases in mortality.
AKT is activated in >50% of human cancer cell lines. The AKT
signaling pathway has been linked to a variety of cellular
processes, including proliferation and migration, and it also
serves a role in response to anticancer therapeutics (13,14).
An important step in AKT activation is its phosphorylation, and
this activation is subsequently enhanced by TRAF6-dependent
ubiquitination. Yang et al (4) demonstrated that the formation of a
stable complex between AKT and TRAF6 is responsible for the
enhanced activation. Previous evidence suggested that TRAF6 is
associated with tumors, since TRAF6 depletion in osteosarcoma cells
was found to reduce NF-κB expression and the survival rate for
cancer patients overexpressing TRAF6 is relatively low (5,10–17).
Therefore, targeting TRAF6-mediated AKT activation may constitute
an excellent therapeutic strategy for the treatment of cancer.
The present study used computational methods to
select small molecules, which can bind with TRAF6, and pursued
ex vivo assays to evaluate their antitumor activities and to
investigate possible mechanisms of action. Firstly, the
computational docking data revealed that quinine may be an
excellent ligand for TRAF6, thereby representing a potential
candidate for modulating TRAF6-mediated AKT activation.
Subsequently, the antitumor activity of quinine was assessed and it
was revealed that this compound inhibited the growth of HeLa and
A549 cells in a dose-dependent manner. Secondly, it is well-known
that apoptosis is closely associated with the initiation,
progression and metastasis of cancer, as well as the elimination of
malignant or infected cells (18–21).
Apoptosis provides a vital homeostatic mechanism for maintaining an
appropriate number of cells in the body through a balance of cell
division and cell death (22).
Consequently, the present study evaluated the cytotoxic and
apoptotic effects of quinine through appropriately selected cell
lines and found that quinine caused a significant loss of cell
viability in HeLa and A549 cells in a concentration- and
time-dependent manner. Under these experimental conditions, the
loss of cell viability following quinine treatment was
predominantly due to the induction of apoptosis.
Next, to further investigate the underlying
mechanism of quinine-induced apoptosis, western blotting was used
to analyze the expression levels of BCL-2 and BAX in HeLa and A549
cells. Quinine was demonstrated to suppress the expression of
BCL-2, whilst stimulating that of BAX in a time-dependent manner.
These results supported that quinine induces apoptosis in HeLa and
A549 cells via modulation of these key apoptosis-regulating
proteins. It has been previously suggested that mitochondria serve
a key role in cell apoptosis, since numerous BCL-2 family proteins
interact with the mitochondrial outer membrane (23,24).
The mutual antagonism that exists between subsets of BCL-2 family
members, including BCL-2 and BAX, directly impacts the
mitochondrial membrane permeability, which ultimately determines
whether cytochrome c is released in order to activate
caspases and induce apoptosis (25).
AKT can also directly or indirectly affect
mitochondria-dependent apoptosis via the modulation of BCL-2 family
proteins. Previous evidence demonstrates that phosphorylation of
AKT can inactivate BAX (26,27).
Others have found that activated AKT can phosphorylate
BCL-2-associated death domain (BAD) at Ser136, and the association
of p-BAD with 14-3-3 proteins can inhibit apoptosis by preventing
p-BAD from interacting with the antiapoptotic BCL-2 family members,
including BCL-2 or BCL-extra large (28). These collective observations along
with the present results indicated that activated AKT can attenuate
mitochondria-dependent apoptosis by inactivating BAD, BAX and other
forkhead transcription factors, and that quinine can counter this
process by inhibiting the activation of AKT.
Additionally, while serving a critical role in the
regulation of cell survival and apoptosis in a variety of cell
types (29–31), AKT activation is accomplished via
the phosphorylation at Ser473 in the C-terminal hydrophobic region
by phosphoinositide-dependent kinase (PDK)-2 and at Thr308 in the
catalytic domain by PDK-1 (32).
Through computational virtual docking, the present study quickly
identified appropriate ligands, including quinine, which can bind
effectively with TRAF6, and subsequently, detected the effect of
quinine on the activity of AKT. These experimental results
demonstrated that quinine can inhibit the phosphorylation of AKT at
both Thr-308 and Ser-473, thereby suggesting that quinine-induced
apoptosis is likely a result of inhibition of AKT
phosphorylation.
In conclusion, the present study demonstrated that
quinine can reduce cell viability and induce apoptosis in HeLa and
A549 cells. These novel activities were associated with the
inhibition of AKT activation by inhibiting its phosphorylation at
Thr-308 and Ser-473, as well as AKT-induced suppression of BCL-2
and upregulation of BAX (Fig. 7).
Further investigations are currently underway to determine whether
quinine is a viable anticancer agent and whether TRAF6 can serve as
a therapeutic target for developing novel anticancer drugs.
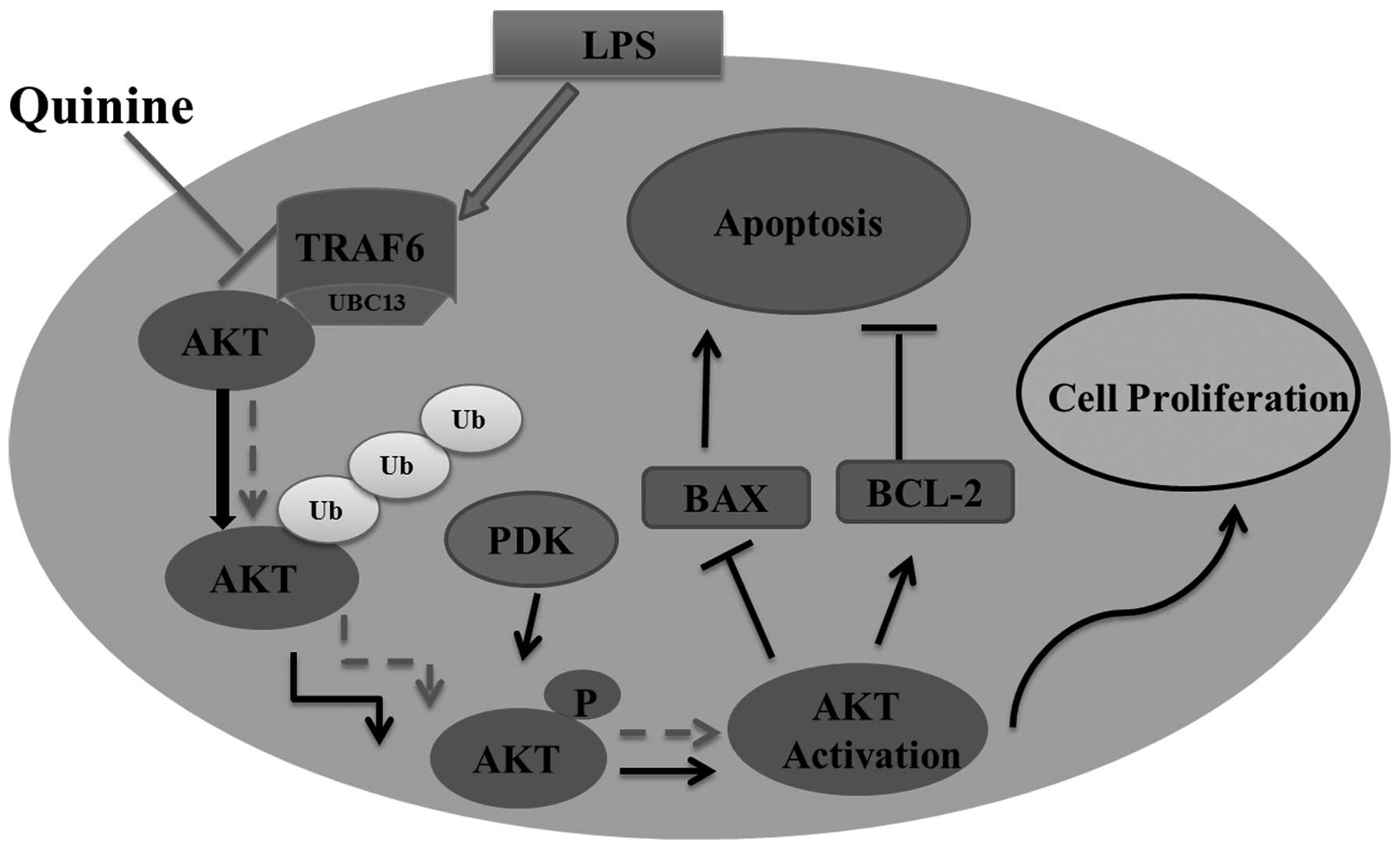 | Figure 7Model for the inhibition of AKT
signaling by quinine. Stimuli, including the bacterial endotoxin
LPS leads to the activation of the E3 ubiquitin ligase, TRAF6, via
a series of cytokines. Through conjugation of K63-linked ubiquitin
chains, TRAF6 enhances AKT activity. Two major consequences of AKT
signaling are the suppression of apoptosis, via the modulation of
BCL-2 family members, and enhanced proliferation. Positive
regulation of AKT by TRAF6 was inhibited by the antimalarial
compound, quinine. The consequent suppression of BCL-2 and
upregulation of BAX sensitizes cells to apoptosis. LPS,
lipopoly-saccharide; TRAF6, tumor necrosis factor
receptor-associated factor 6; AKT, protein kinase B; BCL, B-cell
lymphoma; BAX, Bcl-2-associated X protein; p, phosphate; Ub,
ubiquitin; UBC, Ub C. |
Acknowledgments
The preset study was supported by the Tianjin
Research Program of Applied Basic Science and Advanced Technology
(no. 14JCZDJC33600). The authors would like to thank The Scripps
Research Institute for making available the AutoDock docking
program and supporting literature, and the PDB database for free
access to the X-ray crystal structure of TRAF6.
Abbreviations:
|
BCL
|
B-cell lymphoma
|
|
BAD
|
BCL-2-associated death domain
|
|
BAX
|
BCL-2-associated X protein
|
|
Co-IP
|
co-immunoprecipitation
|
|
FBS
|
fetal bovine serum
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
HRP
|
horseradish peroxidase
|
|
LPS
|
lipopolysaccharide
|
|
NF-κB
|
nuclear factor-κB
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PDK
|
phosphoinositide-dependent kinase
|
|
RIPA
|
radioimmunoprecipitation assay
|
|
TRAF6
|
tumor necrosis factor
receptor-associated factor 6
|
References
|
1
|
Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ,
Lee H, Kim K, Kim J, An IS, Kim J, et al: Akt is negatively
regulated by the MULAN E3 ligase. Cell Res. 22:873–885. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai J, Yang J and Jones DP: Mitochondrial
control of apoptosis: The role of cytochrome c. Biochim Biophys
Acta. 1366:139–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeong SJ, Pise-Masison CA, Radonovich MF,
Park HU and Brady JN: Activated AKT regulates NF-kappaB activation,
p53 inhibition and cell survival in HTLV-1-transformed cells.
Oncogene. 24:6719–6728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang WL, Wang J, Chan CH, Lee SW, Campos
AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG and Lin HK: The
E3 ligase TRAF6 regulates Akt ubiquitination and activation.
Science. 325:1134–1138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhong L, Cao F and You Q: Effect of TRAF6
on the biological behavior of human lung adenocarcinoma cell. Tumor
Biol. 34:231–239. 2013. View Article : Google Scholar
|
|
6
|
Deng L, Wang C, Spencer E, Yang L, Braun
A, You J, Slaughter C, Pickart C and Chen ZJ: Activation of the
IkappaB kinase complex by TRAF6 requires a dimeric
ubiquitin-conjugating enzyme complex and a unique polyubiquitin
chain. Cell. 103:351–361. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng H, Lopez GY, Kim CK, Alvarez A,
Duncan CG, Nishikawa R, Nagane M, Su AJ, Auron PE, Hedberg ML, et
al: EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates
AKT-promoted tumorigenesis. J Clin Invest. 124:3741–3756. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang DH, Qu WL, Shi LQ and Wei J:
Molecular docking and pharmacophore model studies of Rho kinase
inhibitors. Mol Simulat. 37:488–494. 2011. View Article : Google Scholar
|
|
9
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF chimera-A
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hartupee J, Liu C, Novotny M, Sun D, Li X
and Hamilton TA: IL-17 signaling for mRNA stabilization does not
require TNF receptor-associated factor 6. J Immunol. 182:1660–1666.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elumalai P, Arunkumar R, Benson CS,
Sharmila G and Arunakaran J: Nimbolide inhibits IGF-I-mediated
PI3K/Akt and MAPK signalling in human breast cancer cell lines
(MCF-7 and MDA-MB-231). Cell Biochem Funct. 32:476–484.
2014.PubMed/NCBI
|
|
12
|
Schnetzke U, Fischer M, Kuhn AK,
Spies-Weisshart B, Zirm E, Hochhaus A, Müller JP and Scholl S: The
E3 ubiquitin ligase TRAF6 inhibits LPS-induced AKT activation in
FLT3-ITD-positive MV4-11 AML cells. J Cancer Res Clin Oncol.
139:605–615. 2013. View Article : Google Scholar
|
|
13
|
Gills JJ and Dennis PA: The development of
phosphatidylinositol ether lipid analogues as inhibitors of the
serine/threonine kinase, Akt. Expert Opin Investig Drugs.
13:787–797. 2004. View Article : Google Scholar
|
|
14
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishimura M and Naito S: Tissue-specific
mRNA expression profiles of human toll-like receptors and related
genes. Biol Pharm Bull. 28:886–892. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mao R, Fan Y, Mou Y, Zhang H, Fu S and
Yang J: TAK1 lysine 158 is required for TGF-β-induced
TRAF6-mediated Smad-independent IKK/NF-κB and JNK/AP-1 activation.
Cell Signal. 23:222–227. 2011. View Article : Google Scholar
|
|
17
|
Chantzoura E, Prinarakis E, Panagopoulos
D, Mosialos G and Spyrou G: Glutaredoxin-1 regulates TRAF6
activation and the IL-1 receptor/TLR4 signalling. Biochem Biophys
Res Commun. 403:335–339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Golbano JM, Lóppez-Aparicio P, Recio MN
and Pérez-Albarsanz MA: Finasteride induces apoptosis via Bcl-2,
Bcl-xL, Bax and caspase-3 proteins in LNCaP human prostate cancer
cell line. Int J Oncol. 32:919–924. 2008.PubMed/NCBI
|
|
19
|
Alonso-Castro AJ, Ortiz-Sánchez E,
García-Regalado A, Ruiz G, Núñez-Martínez JM, González-Sánchez I,
Quintanar-Jurado V, Morales-Sánchez E, Dominguez F, López-Toledo G,
et al: Kaempferitrin induces apoptosis via intrinsic pathway in
HeLa cells and exerts antitumor effects. J Ethnopharmacol.
145:476–489. 2013. View Article : Google Scholar
|
|
20
|
Xu C, Wu A, Zhu H, Fang H, Xu L, Ye J and
Shen J: Melatonin is involved in the apoptosis and necrosis of
pancreatic cancer cell line SW-1990 via modulating of Bcl-2/Bax
balance. Biomed Pharmacother. 67:133–139. 2013. View Article : Google Scholar
|
|
21
|
Costa LS, Telles CB, Oliveira RM, Nobre
LT, Dantas-Santos N, Camara RB, Costa MS, Almeida-Lima J,
Melo-Silveira RF, Albuquerque IR, et al: Heterofucan from Sargassum
filipendula induces apoptosis in HeLa Cells. Mar Drugs. 9:603–614.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu W and Kavanagh JJ: Anticancer therapy
targeting the apoptotic pathway. Lancet Oncol. 4:721–729. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: A
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang L, Xing D and Chen MJ: Bim(L)
displacing Bcl-x(L) promotes bax translocation during TNF
alpha-induced apoptosis. Apoptosis. 13:950–958. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gardai SJ, Hildeman DA, Frankel SK,
Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL and
Henson PM: Phosphorylation of Bax Ser184 by Akt regulates its
activity and apoptosis in neutrophils. J Biol Chem.
279:21085–21095. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Linseman DA, Butts BD, Precht TA, Phelps
RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML and
Heidenreich KA: Glycogen synthase kinase-3 beta phosphorylates bax
and promotes its mitochondrial localization during neuronal
apoptosis. J Neurosci. 24:9993–10002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zha J, Harada H, Yang E, Jockel J and
Korsmeyer SJ: Serine phosphorylation of death agonist BAD in
response to survival factor results in binding to 14-3-3 not
BCL-XL. Cell. 87:619–628. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marte BM and Downward J: PKB/Akt:
Connecting phosphoinositide 3-kinase to cell survival and beyond.
Trends Biochem Sci. 22:355–358. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scheid MP and Woodgett JR: Unravelling the
activation mechanisms of protein kinase B/Akt. FEBS Lett.
546:108–112. 2003. View Article : Google Scholar : PubMed/NCBI
|