Introduction
Cardiac fibrosis, as a consequence of remodeling
processes, results from cardiac adaptations to the hemodynamic
overload caused by heart failure and cardiac hypertrophy. Adaptive
remodeling processes initiate structural alterations representing
cell morphological changes and excessive deposition of
extracellular matrix (ECM) (1–3).
Substances in the ECM provide cardiomyocytes with structural,
chemical and mechanical substrates, which are essential for normal
development and responses to pathological signals. Cardiomyocytes
can sense stimuli from the ECM, which results in structural or
functional remodeling (4).
Collagen, particularly type I, is a major component of the ECM,
which forms networks in interstitial spaces among cells, and
provides structural integrity and mechanical stretching of the
heart (5). Therefore, type I
collagen may be critical in cardiac fibrosis.
Transforming growth factor (TGF)-β is a key mediator
in the pathogenesis of hypertrophic and dilative ventricular
remodeling by stimulating cardiomyocyte growth and inducing
interstitial fibrosis (6,7). In the pressure-overloaded human
heart, the upregulation of TGF-β is correlated with the degree of
fibrosis (8). The overexpression
of TGF-β in transgenic mice has been found to result in atrial
hypertrophy, which is characterized by interstitial fibrosis and
hypertrophic growth of cardiomyocytes (9). Active TGF-β binds to the
constitutively active type II receptor (TGFβRII) at the cell
surface, and subsequently transphosphorylates the cytoplasmic
domain of the type I receptor (TGFβRI) (10). As TGFβRI mediates the majority of
the cellular responses to TGF-β1 and has a predominant
role in intracellular signal transduction (11), the activation of TGF-β receptors
propagates downstream intracellular signals through Smad proteins,
which are essential components in the signaling pathway of TGF-β
(10,12–14).
Although TGF-β signaling is important in genes associated with the
ECM, whether the ECM is involved in regulating TGF-β signaling
remains to be elucidated.
In vivo, cardiomyocytes are intricately
connected to the ECM, and stretch is transduced via the
intercellular and ECM connections via proteins located on the cell
surface and subcellular adhesion complexes, and via stretch-sensing
proteins. Stretching and the expression of integrin are increased
during cardiac fibrosis (15–17).
Integrin represents a primary link between ECM ligands and directly
initiates intracellular signaling cascades (18–21).
Integrin can interact with TGF-β to enhance cardiac fibrosis
(21), suggesting that collagen
may be involved in TGF-β signaling, which regulates the homeostasis
of cardiac fibrosis. Integrin can activate focal adhesion kinase
(FAK), which results in cardiac structural and electrical changes
(22). In addition, mechanical
stretch is important in tissue morphogenesis and remodeling, and
evokes various hypertrophic responses from cell adhesion receptors
and the expression of TGF-β1, and activates
mitogen-activated protein kinases (MAPKs) in cardiomyocytes
(23–25). Accordingly, integrin and
mechanosensitive stretch channels may potentially regulate TGF-β
signaling. MAPKs have pathophysiological effects in cardiac
remodeling (26). Our previous
studies demonstrated that collagen can modulate calcium dynamics
and electrical activities of cardiomyocytes, with activation of the
renin-angiotensin and MAPK systems (27,28).
Therefore, the aims of the present study were to evaluate the
effects of collagen on TGF-β signaling and investigate the
underlying mechanisms. The results of the present study
demonstrated that collagen differentially modulated the expression
levels of TGFβRI and TGFβRII and the activity of MAPKs through
interactions of integrin with stretch signaling.
Materials and methods
Cell culture
The protocol was approved by the Department of
Medical Research of Cathay General Hospital, Taipei, Taiwan (no.
CGH-MR-10014). HL-1 cells derived from mouse atrial cardiac muscle
cells (29) were provided by Dr
Claycomb, Louisiana State University Health Sciences Center (New
Orleans, USA). The cells for various experiments were cultured in a
humidified atmosphere of 5% CO2 at 37°C in Claycomb
medium (JRH Biosciences, Lenexa, KS, USA). As described previously,
the HL-1 cells were cultured with or without (control) type I rat
tail tendon collagen (10 µg/ml; Sigma-Aldrich, St. Louis,
MO, USA) for 24 h following cell seeding (27). To investigate the roles of integrin
and stretch, the HL-1 cells were pretreated with either monoclonal
hamster anti-rat β1 integrin antibody (β1 integrin; 10
µg/ml; clone Ha2/5; #555002; BD Pharmingen, San Diego, CA,
USA), to inhibit the in vitro adhesion of CD29-expressing
cells to type I collagen, or the stretch-activated ion channel
inhibitor, gadolinium (50 µM; Sigma-Aldrich) for 30 min
prior to collagen incubation (30,31).
The cells were plated at a density of 5×105 cells/well
in 6-well culture plates.
Western blot analysis
The HL-1 cells were homogenized and lysed in RIPA
buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP40, 0.5%
sodium deoxycholate, 0.1% sodium dodecylsulfate (SDS) and protease
inhibitor cocktails (Sigma-Aldrich). The protein concentrations
were determined using Bio-Rad protein assay reagent (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Protein samples (40
µg) were separated by 10% SDS-polyacrylamide gel
electrophoresis under reducing conditions and electrophoretically
transferred onto equilibrated polyvinylidenedifluoride membranes
(Amersham Biosciences, Upsulla, Sweden). The blots were probed with
the following primary antibodies overnight at 4°C: Rabbit anti-FAK
(cat. no. 3285; 1:1,000), rabbit anti-phosphorylated FAK (cat. no.
3283; 1:1,000), rabbit anti-extracellular signal-regulated kinase
1/2 (ERK1/2; cat. no. 9102; 1:1,000), rabbit
anti-phosphorylated-ERK1/2 (cat. no. 4370; 1:1,000) rabbit
anti-c-Jun N-terminal kinase (JNK; cat. no. 9258; 1:1,000), mouse
anti-phosphorylated-JNK (cat. no. 9255; 1:1,000), rabbit anti-p38
(cat. no. 9212; 1:1,000), rabbit anti-phosphorylated-p38 (cat. no.
9211; 1:1,000), rabbit anti-Smad2/3 (cat. no. 8685; 1:1,000),
rabbit anti-phosphorylated-Smad2/3 (cat. no. 8828; 1:1,000), rabbit
anti-TGFβRII (cat. no. 11888; 1:500) (all Cell Signaling
Technology, Inc., Danvers, MA, USA); rabbit anti-TGFβRI (cat. no.
SAB1300113, Sigma-Aldrich; 1:500); and horseradish
peroxidase-conjugated goat anti-rabbit (cat. no. AP132P; 1:20,000)
or goat anti-mouse immunglobulin G (cat. no. AP124P; 1:8,000)
secondary antibodies (EMD Millipore, Billerica, MA, USA) for 1 h at
room temperature. The bound antibodies were detected with an
enhanced chemiluminescence detection system (EMD Millipore) and
analyzed with AlphaEaseFC version 4.0 software (Alpha Innotech
Corporation, San Leandro, CA, USA). The targeted bands were
normalized to mouse anti-cardiac α-sarcomeric actin (cat. no.
A2172; Sigma-Aldrich; 1:2,000) to confirm equal protein
loading.
Cell proliferation assay
Cell proliferation was analyzed using a BrdU
incorporation assay (BrdU Cell Proliferation Assay kit, Cell
Signaling Technologies, Inc.). The HL-1 cells were seeded at
1×104 cells/well in 96-well plates and left to attach
overnight. The cells were then incubated with or without type I rat
tail tendon collagen (10 µg/ml) for 24 h. The HL-1 cells
were pretreated with β1 integrin antibody (10 µg/ml),
gadolinium (50 µM), p38 MAPK inhibitor (SB203580; 3
µM; Merck Millipore, Darmstadt, Germany), ERK inhibitor
(PD98059; 50 µM; Sigma-Aldrich) or JNK inhibitor (SP600125;
50 µM; Calbiochem, San Diego, CA, USA) for 30 min prior to
collagen incubation. BrdU solution was added 4 h prior to the end
of cell treatment. Following fixing of cells and denaturing of DNA,
BrdU detection antibody and horseradish peroxidase-conjugated
secondary antibody were added. Finally, the HRP substrate,
3,3′,5,5′-tetramethylbenzidine, was added to develop color, and
BrdU incorporation was quantified by reading the absorbance at 540
nm.
Cell adhesion assay
The HL-1 cells were trypsinized and seeded at a
density of 2×104 cells/well in a 96-well plate following
treatment with type I collagen (10 µg/ml) and β1 integrin
antibody (10 µg/ml). Subsequent to attachment for 2.5 h in a
37°C incubator, the unattached cells were removed by washing with
phosphate-buffered saline. The cells were then stained with 0.1%
crystal violet in 2% ethanol in 0.1 M borate (pH 9) for 15 min.
Cell adhesion levels were evaluated by reading the absorbance of
crystal violet at 540 nm following the addition of 100 ml pure
methanol to solubilize the dye.
Statistical analysis
Continuous variables are expressed as the mean ±
standard error of the mean. The different groups of HL-1 cells were
compared using a paired t-test or repeated one-way analysis
of variance with a Tukey's post-hoc test. The effects of the β1
integrin antibody or gadolinium on collagen were evaluated using an
unpaired t-test. All statistical tests were performed using
SigmaStat 3.5 (Systat Software, Inc., San Jose, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Effects of collagen and β1 integrin in
modulating the expression of TGF-β receptors and FAK
Compared with the control, the collagen (10
µg/ml)-treated HL-1 cells exhibited significantly decreased
expression levels of TGFβRI (16±5%) and TGFβRII (21±5%) and
upregulated FAK phosphorylation, (42±9%). The β1 integrin antibody
(10 µg/ml) did not alter the expression levels of TGFβRI,
TGFβRII or the phosphorylation of FAK in the HL-1 cells. In the
presence of the β1 integrin antibody, collagen downregulated the
expression of TGFβRI, however, no effects on the expression of
TGFβRII or the phosphorylation of FAK were observed (Fig. 1A). Therefore, the β1 integrin
antibody attenuated the collagen-associated down-regulation in the
expression of TGFβRII and phosphorylation of FAK, but did not alter
the collagen-associated downregulation of TGFβRI.
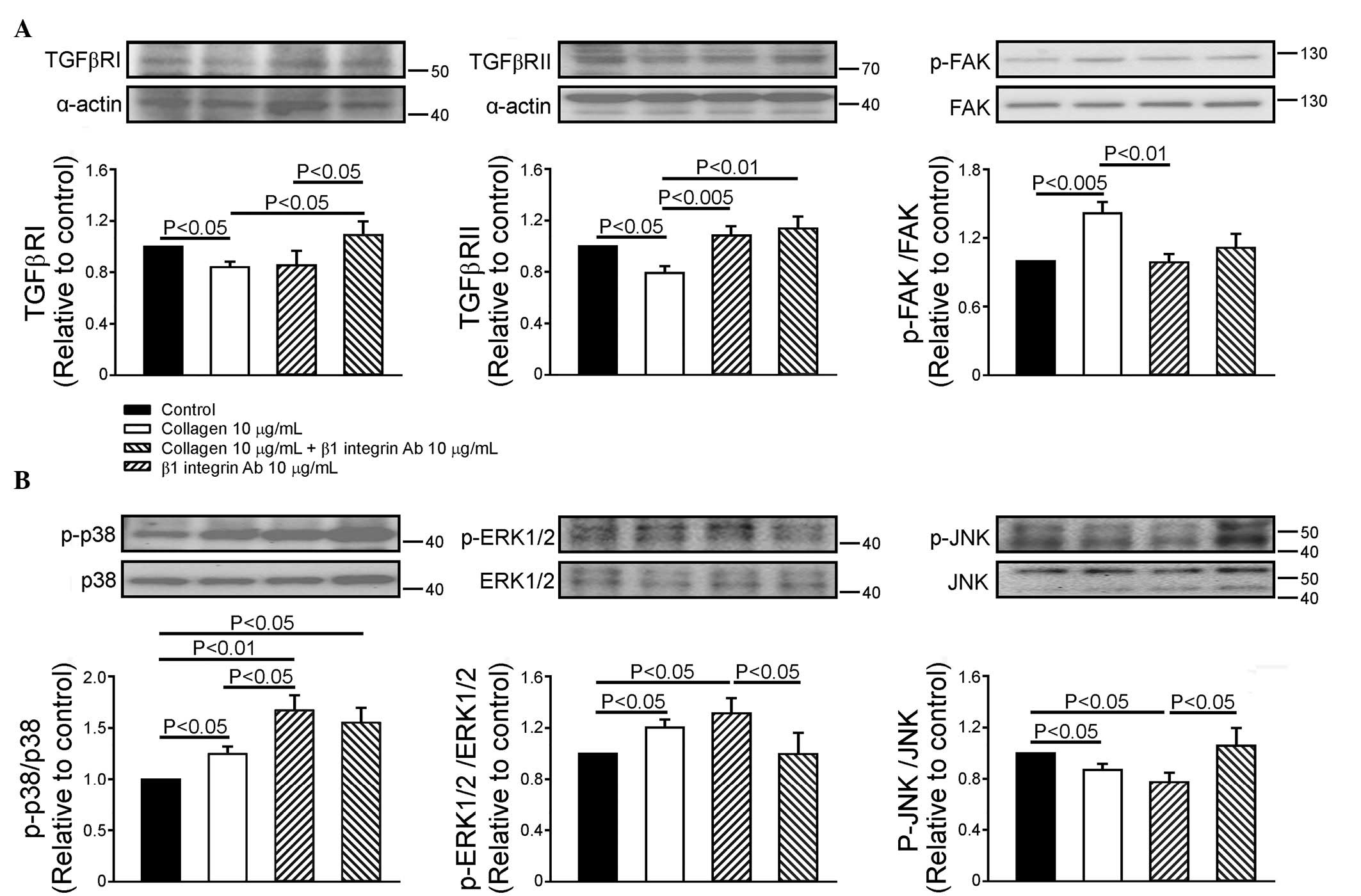 | Figure 1Effects of collagen and β1 integrin
Ab on the expression levels of TGFβRI and TGFβRII, and the
downstream signaling in HL-1 cells. (A) Representative images and
quantified data (n=7) of the expression levels of TGFβRI and
TGFβRII, and the phosphorylation ratio of FAK in the control,
collagen (10 µg/ml)-treated, collagen (10 µg/ml) with
β1 integrin Ab (10 µg/ml)-treated, and β1 integrin Ab (10
µg/ml)-treated groups. (B) Representative image and
quantified data (n=6) of the phosphorylation ratio of p38, ERK1/2
and JNK in the control, collagen (10 µg/ml)-treated,
collagen (10 µg/ml) with β1 integrin Ab (10
µg/ml)-treated and β1 integrin Ab (10 µg/ml)-treated
groups. Data are presented as the mean ± standard error of the
mean. TGFβR, transforming growth factor β receptor; FAK, focal
adhesion kinase; ERK1/2, extracellular-regulated kinase 1/2; JNK,
Jun N-terminal kinase; Ab, antibody; p-, phosphorylated. |
Effects of collagen and β1 integrin in
the modulation of MAPK phosphorylation
As shown in Fig.
1B, the collagen (10 µg/ml)-treated HL-1 cells exhibited
increased phosphorylation of p38 (25±7%) and ERK1/2 (20±6%), but
decreased the phosphorylation of JNK (13±5%). The β1 integrin
antibody increased the phosphorylation of p38 by 55±15% in the HL-1
cells, but did not affect the phosphorylation of ERK1/2 or JNK.
Therefore, β1 integrin was expected to increase the level of
phosphorylated p38. In the presence of the β1 integrin antibody,
collagen did not alter the phosphorylation of p38, however, the
phosphorylation of ERK1/2 was increased and that of JNK was
decreased. Accordingly, the collagen-induced changes in the levels
of phosphorylated ERK1/2 and JNK were not altered by the presence
of the β1 integrin antibody, which suggested that integrin may not
completely modulate the collagen effects of collagen on MAPKs.
Effect of mechanical stretch in
collagen-mediated expression of the TGF-β receptor and Smad2/3
Compared with the control, gadolinium (50 µM)
decreased the expression of TGFβRI by 38±12% and TGFβRII by 26±6%.
In the presence of gadolinium, collagen did not alter the
expression levels of TGFβRI or TGFβRII. However, gadolinium
decreased the collagen-induced downregulation of TGFβRI by 25±7%.
Collagen decreased the phosphorylation of Smad2/3 by 17±5%,
compared with the control. Gadolinium (50 µM) decreased the
phosphorylation of Smad2/3 in the HL-1 cells by 24±6%, and
decreased the phosphorylation of Smad2/3 by 23±6% in the presence
of collagen (Fig. 2A).
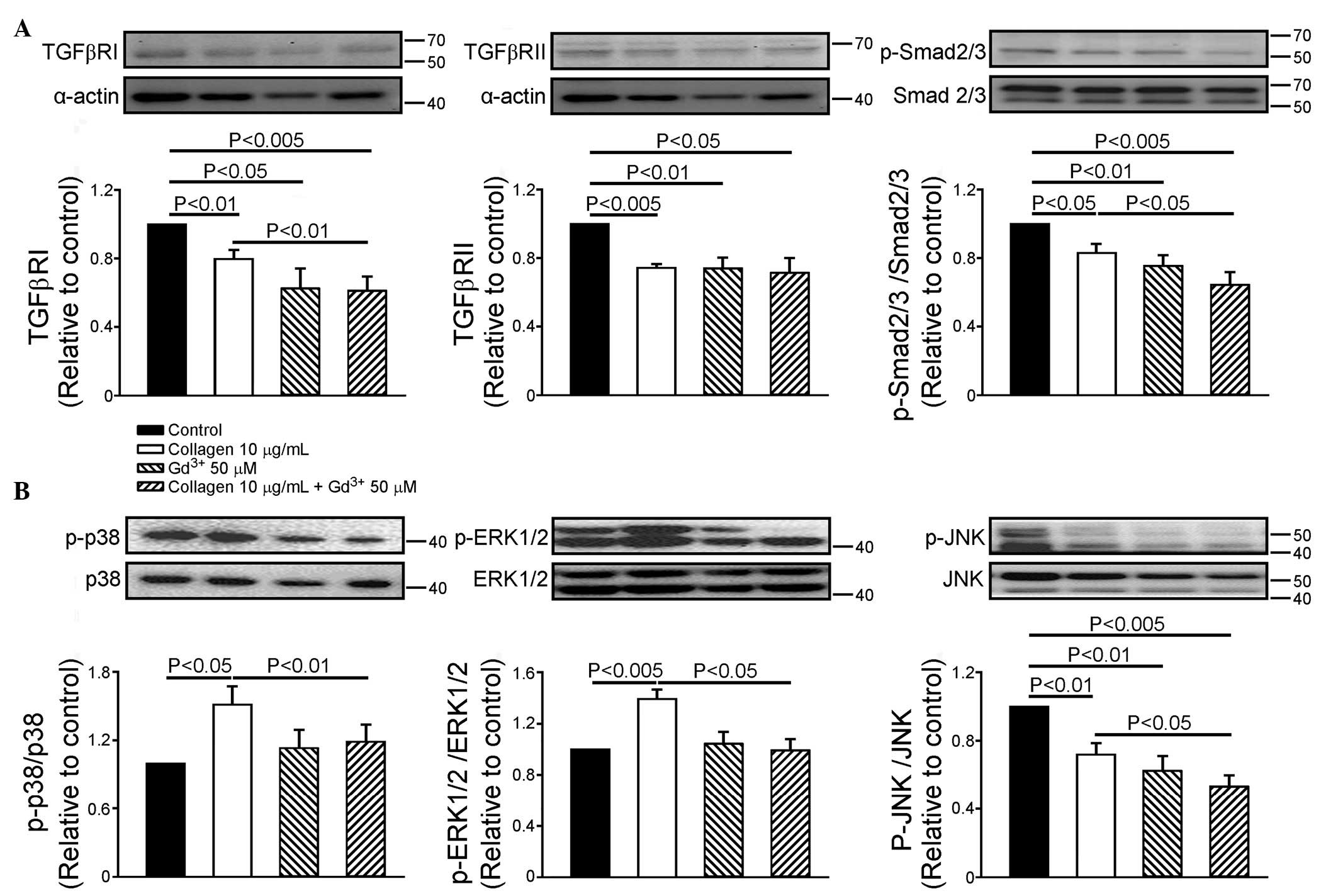 | Figure 2Effects of Gd3+ on TGFβRI,
TGFβRII and Smad2/3, and the phosphorylation of mitogen-activated
protein kinases. (A) Representative images and quantified data of
TGFβRI, TGFβRII and Smad2/3 (n=7) and (B) phosphorylation ratios of
p38, ERK1/2 and JNK (n=6) in the control, collagen (10
µg/ml)-treated, Gd3+ (50 µM)-treated and
collagen (10 µg/ml) with Gd3+ (50
µM)-treated groups. Data are presented as the mean ±
standard error of the mean. Gd3+, gadolinium; TGFβR,
transforming growth factor β receptor; ERK1/2,
extracellular-regulated kinase 1/2; JNK, Jun N-terminal kinase;
Smad, small mothers against decapentaplegic; Ab, antibody; p-,
phosphorylated. |
Effect of mechanical stretch on the
collagen-mediated phosphorylation of MAPKs
Compared with the control cells, the gadolinium (50
µM)-treated HL-1 cells exhibited decreased JNK
phosphorylation by 37±6%, however, the phosphorylation of p38 or
ERK1/2 were not affected. In the presence of gadolinium, collagen
did not alter the phosphorylation of p38, ERK or JNK. However,
gadolinium further decreased the collagen-induced downregulated
phosphorylation of JNK by 25±8%, and reversed the collagen-induced
upregulated phosphorylation of p38 and ERK1/2, compared with the
collagen-treated HL-1 cells (Fig.
2B).
Effects of β1 integrin and mechanical
stretch on the collagen-mediated expression of TGF-β receptor and
Smad2/3
As shown in Fig.
3A, the β1 integrin antibody (10 µg/ml) and gadolinium
(50 µM) decreased the expression of TGFβRII by 32±6%
(P<0.05, vs. control), however, no change in the expression of
TGFβRI was observed in the HL-1 cells. In the presence of the β1
integrin antibody and gadolinium, collagen downregulated TGFβRI by
66±23% and upregulated TGFβRII by 26±8%. Compared with the
gadolinium-treated cells (Fig.
2A), the HL-1 cells treated with the β1 integrin antibody and
gadolinium (Fig. 3A) increased the
expression of TGFβRI by 149±77% (P<0.05), however, the
expression of TGFβRII was not affected (data not shown). The
phosphorylation of Smad2/3 in the cells treated with the β1
integrin antibody and gadolinium did not differ from that of the
control. The effects of the β1 integrin antibody and gadolinium on
the phosphorylation of Smad2/3 were similar to those on the
expression of TGFβRI.
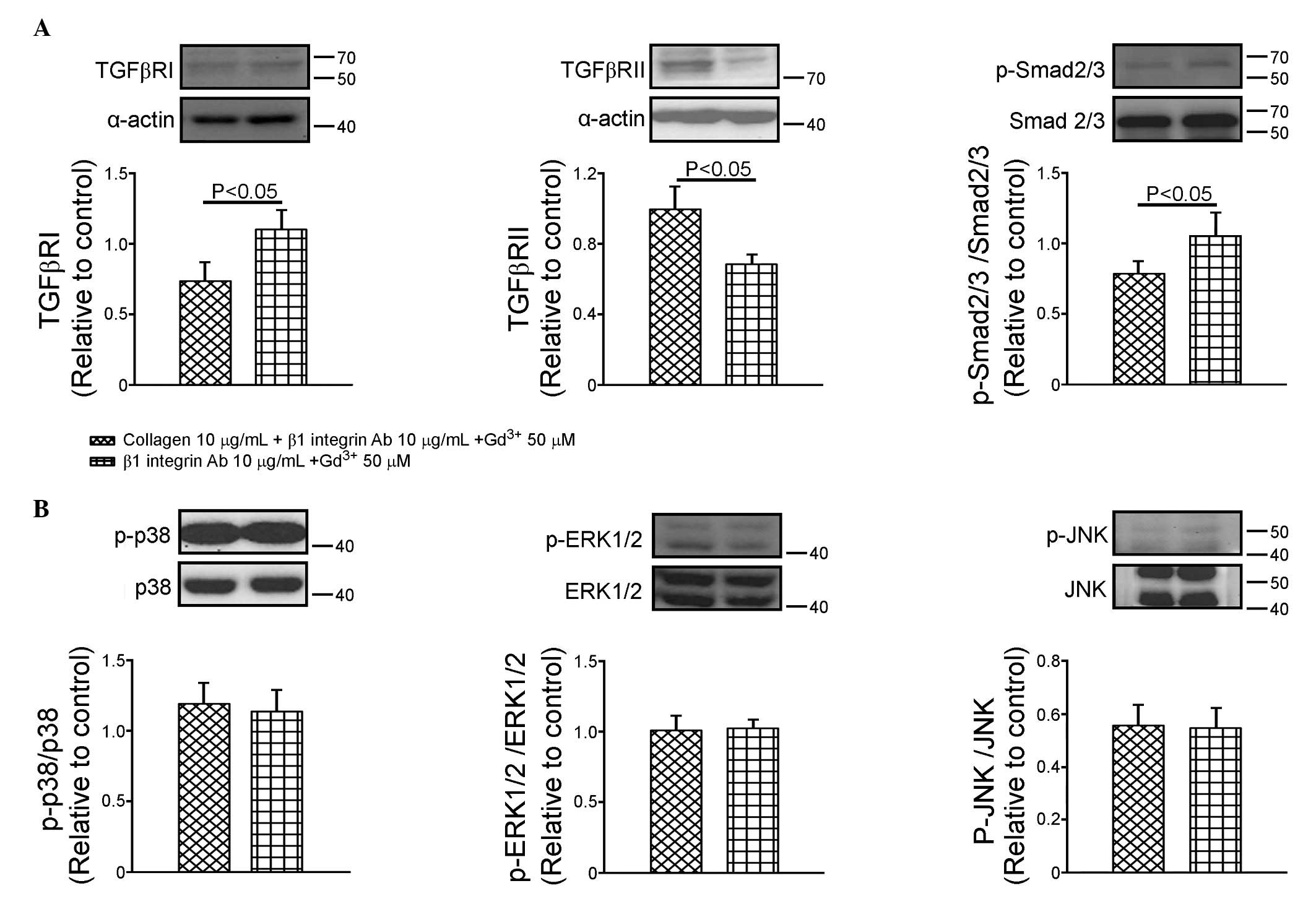 | Figure 3Effects of β1 integrin Ab and
Gd3+ on the expression of TGFβRI, TGFβRII and Smad2/3
and the phosphorylation of mitogen-activated protein kinases. (A)
Representative images and quantified data (n=7) of TGFβRI, TGFβRII
and Smad2/3 in the groups treated with collagen, β1 integrin Ab (10
µg/ml) and Gd3+ (50 µM), and with β1
integrin Ab (10 µg/ml) and Gd3+ (50 µM).
(B) Representative images and quantified data (n=6) of the
phosphorylation ratios of p38, ERK1/2 and JNK in the groups treated
with the collagen, β1 integrin Ab (10 µg/ml) and
Gd3+ (50 µM), and with β1 integrin Ab (10
µg/ml) and Gd3+ (50 µM). Data are
presented as the mean ± standard error of the mean.
Gd3+, gadolinium; TGFβR, transforming growth factor β
receptor; FAK, focal adhesion kinase; ERK1/2,
extracellular-regulated kinase 1/2; JNK, Jun N-terminal kinase; Ab,
antibody; p-, phosphorylated. |
Effects of β1 integrin and mechanical
stretch in the collagen-mediated expression of MAPKs
Compared with the control HL-1 cells, β1 integrin
antibody (10 µg/ml) and gadolinium (50 µM) decreased
the phosphorylation of JNK by 45±8% (P<0.005), but did not alter
the phosphorylation of p38 or ERK1/2 (Fig. 3B). In the presence of β1 integrin
antibody and gadolinium, no changes in the phosphorylation of p38,
ERK1/2 or JNK were observed, compared with the gadolinium-treated
cells and the cells concomitantly treated with collagen.
Effects of β1 integrin and mechanical
stretch on the proliferation and adhesion of collagen-treated HL-1
cells
To investigate the role of integrin and mechanical
stretch on cell proliferation, the collagen (10
µg/ml)-treated HL-1 cells were incubated with or without β1
integrin antibody (10 µg/ml) or gadolinium (50 µM).
As shown in Fig. 4A, collagen
increased cell proliferation, and this collagen-induced cell
proliferation was inhibited by β1 integrin antibody, but not by
gadolinium. In addition, collagen increased cell adhesion by 16±7%,
which was also inhibited by β1 integrin antibody (Fig. 4B). These results suggested that
collagen promoted cardiomyocyte proliferation and adhesion through
β1 integrin signaling.
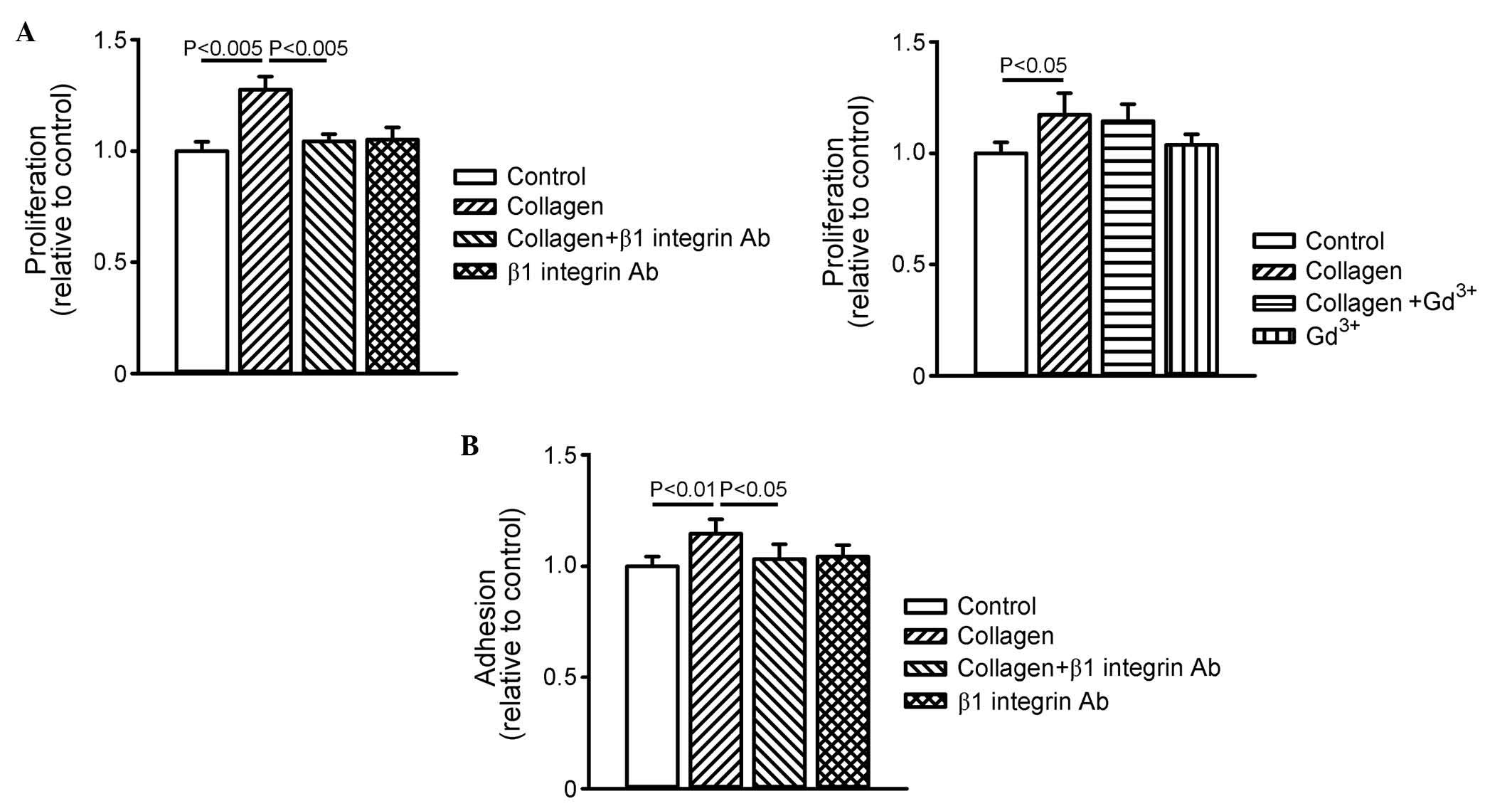 | Figure 4Effects of collagen, β1 integrin Ab
and Gd3+ on cell proliferation and adhesion. (A) Cell
proliferation was assessed by BrdU absorbance at 450 nm. The graph
on the left shows data (n=9) of cell proliferation in the control,
collagen (10 µg/ml)-treated, collagen (10 µg/ml) with
β1 integrin Ab (10 µg/ml)-treated, and β1 integrin Ab (10
µg/ml)-treated groups. The graph on the right shows data
(n=9) of cell proliferation in the control, collagen (10
µg/ml)-treated, collagen with Gd3+ (50
µM)-treated, and Gd3+ (50 µM)-treated
groups. (B) Cell adhesion was assessed by crystal violet absorbance
at 540 nm in the control, collagen (10 µg/ml)-treated,
collagen (10 µg/ml) with β1 integrin Ab (10
µg/ml)-treated, and β1 integrin Ab (10 µg/ml)-treated
groups. Data are presented as the mean ± standard error of the mean
(n=9). Gd3+, gadolinium; TGFβR, transforming growth
factor β receptor; Ab, antibody. |
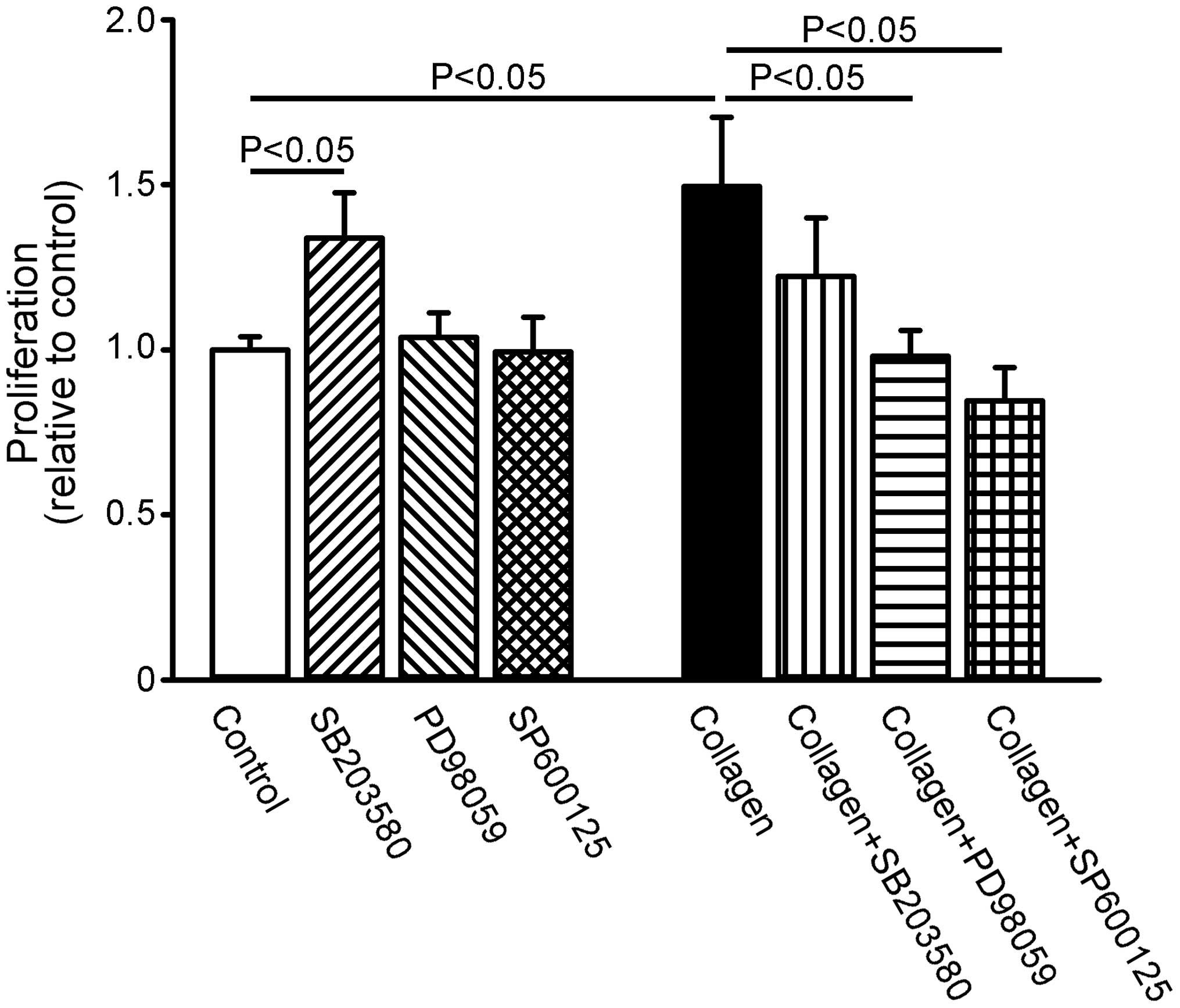
Effect of MAPK inhibition on the
proliferation of collagen-treated HL-1 cells
Compared with the control HL-1 cells, SB203580 (3
µM) increased cell proliferation by 33±11%, whereas PD98059
(50 µM) and SP600125 (50 µM) did not alter cell
proliferation (Fig. 5). However,
in the presence of collagen, PD98059 and SP600125, but not
SB203580, inhibited the collagen-induced increase in cell
proliferation.
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate that collagen directly modulated the
expression of TGF-β receptors and their downstream signaling
molecules. TGF-β is an important cytokine in the fibrotic responses
of several organs, including the heart (6,7). The
present study demonstrated that collagen accumulation during
fibrosis can downregulate the expression of TGF-β receptors. TGF-β
enhances the production of the ECM from cardiac fibroblasts, which
can induce fibrosis (6,32). Although the underlying mechanisms
remain to be fully elucidated, sustained TGF-β exposure has been
shown to suppress the expression of TGF-β receptors (33). Therefore, an increase or decrease
of collagen leads to the downregulation or upregulation of TGF-β
receptor signaling, which may, at least in part, result in the
known feedback loop between TGF-β and TGF-β receptors. In addition,
the ECM has been reported to modulate MAPKs in cell function and
differentiation (28,34,35).
Collagen was found to increase the phosphorylation of p38 and
ERK1/2 in the present study, which is compatible with previous
findings in cardiac remodeling (36,37).
The present study found that the collagen-induced
down-regulation of TGFβRII was inhibited by the integrin antibody.
A previous study demonstrated that the interaction of type I
collagen with α2β1 integrin causes downregulation of the TGF-β
receptor via the activation of FAK and its diverse downstream
signals, and this was found to be abrogated by treatment that
inactivated FAK (35). The
integrin antibody decreased the collagen-activated phosphorylation
of FAK, suggesting that FAK uniquely mediates matrix-integrin
interactions in cellular processes. However, integrin did not
regulate the effects of collagen on TGFβRI, suggesting that
collagen differentially modulates the expression of TGFβRI and
TGFβRII.
The ECM has a regulatory role in several cellular
processes, including cell growth, adhesion, division and
differentiation. However, whether collagen regulates cardiomyocyte
proliferation and adhesion remains to be elucidated. The ECM
secreted from cardiac fibroblasts has been reported to promote
cardiomyocyte proliferation through β1 integrin signaling (38). Integrins are a major class of ECM
receptors for cell adhesion (20).
The present study showed that collagen-binding β1 integrin is
involved in mediating cell proliferation and adhesion, which
suggested that β1 integrin may be responsible for cardiomyocyte
proliferation during cardiac fibrosis. Although the inhibition of
p38 enables the proliferation of cardiomyocytes (39), however, collagen-induced cell
proliferation was inhibited by the inhibition of ERK in the present
study. These results suggested that integrin and MAPK signaling in
the cardiomyocytes may have been responsible for the effects of
collagen on cardiomyocyte proliferation.
Mechanical stretch induces the release of TGF-β and
is coupled with intracellular signals, which are responsible for
cardiac hypertrophy (40,41). In the present study, it was found
that gadolinium enhanced the downregulation of the TGFβRI, but not
TGFβRII. The activation of TGFβRI leads to the phosphorylation of
Smad2 and Smad3, and regulates transcription (13,14).
In the present study, collagen down-regulated the phosphorylation
of Smad2/3 in parallel with the expression of TGFβRI, which
suggested that collagen was involved in the Smad-dependent pathway
through TGFβRI. The MAPK pathway may be involved in the mechanical
stretching-induced hypertrophic response (42,43).
In the present study, it was found that gadolinium decreased the
collagen-induced phosphorylation of p38 and ERK1/2. As gadolinium
only reversed the effects of collagen on the activity of MAPKs in
cells without the presence of the β1-integrin antibody, it is
possible that collagen activated the stretch-induced MAPK signaling
pathway independently of integrin-FAK signaling (44). However, collagen and gadolinium
showed synergistic effects in inhibiting the expression of TGFβRI
and phosphorylation of Smad2/3. These results suggested the
presence of cross-talk between collagen and mechanical stretch in
the TGFβRI/Smad signaling pathways. The present study demonstrated
that collagen regulated the expression of TGF-β receptors and
phosphorylation of MAPKs through at least two distinct mechanisms.
Collagen regulated the expression of TGFβRII through cross-talk
between integrin-FAK signaling and mechanical stretch, whereas the
collagen-induced expression of TGFβRI and activation of MAPKs were
predominantly mediated by mechanical stretch. In addition, collagen
may modulate the phosphorylation of JNK beyond the mechanisms
identified in the present study. In the present in vitro
study, an atrial cell line from mice was used, and these highly
purified cells were placed on an artificial collagen matrix, which
may or may not be reflective of human atrial cells. The findings
suggested that coordinated actions occurred among the ECM, integrin
and mechanical stretch in response to cardiac fibrosis.
In conclusion, the present study demonstrated that
collagen was involved in altering intracellular signaling in a
positive and negative manner. The observations indicated that
collagen modulated the expression of TGF-β receptors and the
phosphorylation of MAPKs through interactions with integrin and
mechanical stretch.
Acknowledgments
This study was supported by grants (grant nos.
NSC101-2314-B-040-017-MY2, NSC102-2314-B-016-029-MY2,
NSC102-2325-B-010-005, NSC102-2628-B-038-002-MY3 and
103-2314-B-281-005-MY2) from the National Science Council of Taiwan
(grant nos. 102CGH-TMU-04, CGH-MR-A10219, CGH-MR-A10221,
CGH-MR-A10222, CGH-MR-A10305 and 105CGH-TMU-08) from Cathay General
Hospital (Taipei, Taiwan) and (grant nos. 103swf05, 103-wf-eva-02,
104-wf-eva-01, 104swf02, and 105-wf-eva-14) from Wan Fang Hospital,
Taipei Medical University (Taipei, Taiwan).
References
|
1
|
Sugden PH and Clerk A: Cellular mechanisms
of cardiac hypertrophy. J Mol Med (Berl). 76:725–746. 1998.
View Article : Google Scholar
|
|
2
|
Manabe I, Shindo T and Nagai R: Gene
expression in fibroblasts and fibrosis: Involvement in cardiac
hypertrophy. Circ Res. 91:1103–1113. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swynghedauw B: Molecular mechanisms of
myocardial remodeling. Physiol Rev. 79:215–262. 1999.PubMed/NCBI
|
|
4
|
Bissell MJ, Hall HG and Parry G: How does
the extracellular matrix direct gene expression? J Theor Biol.
99:31–68. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pelouch V, Dixon IM, Golfman L, Beamish RE
and Dhalla NS: Role of extracellular matrix proteins in heart
function. Mol Cell Biochem. 129:101–120. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Border WA and Noble NA: Transforming
growth factor beta in tissue fibrosis. N Engl J Med. 331:1286–1292.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lijnen PJ, Petrov VV and Fagard RH:
Induction of cardiac fibrosis by transforming growth factor-beta
(1). Mol Genet Metab. 71:418–435. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hein S, Arnon E, Kostin S, Schönburg M,
Elsässer A, Polyakova V, Bauer EP, Klövekorn WP and Schaper J:
Progression from compensated hypertrophy to failure in the
pressure-overloaded human heart: Structural deterioration and
compensatory mechanisms. Circulation. 107:984–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakajima H, Nakajima HO, Salcher O, Dittiè
AS, Dembowsky K, Jing S and Field LJ: Atrial but not ventricular
fibrosis in mice expressing a mutant transforming growth
factor-beta (1) transgene in the heart. Circ Res. 86:571–579. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wrana JL, Attisano L, Cárcamo J, Zentella
A, Doody J, Laiho M, Wang XF and Massague J: TGF beta signals
through a heteromeric protein kinase receptor complex. Cell.
71:1003–1014. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen R, Halder G, Zhang Z and Mardon G:
Signaling by the TGF-beta homolog decapentaplegic functions
reiteratively within the network of genes controlling retinal cell
fate determination in Drosophila. Development. 126:935–943.
1999.PubMed/NCBI
|
|
13
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dennler S, Itoh S, Vivien D, ten Dijke P,
Huet S and Gauthier JM: Direct binding of Smad3 and Smad4 to
critical TGF beta-inducible elements in the promoter of human
plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar
|
|
16
|
Ross RS and Borg TK: Integrins and the
myocardium. Circ Res. 88:1112–1119. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McNicholas-Bevensee CM, DeAndrade KB,
Bradley WE, Dell'Italia LJ, Lucchesi PA and Bevensee MO: Activation
of gadolinium-sensitive ion channels in cardiomyocytes in early
adaptive stages of volume overload-induced heart failure.
Cardiovasc Res. 72:262–270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clark EA and Brugge JS: Integrins and
signal transduction pathways: The road taken. Science. 268:233–239.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schlaepfer DD, Hanks SK, Hunter T and van
der Geer P: Integrin-mediated signal transduction linked to Ras
pathway by GRB2 binding to focal adhesion kinase. Nature.
372:786–791. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Margadant C and Sonnenberg A:
Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing.
EMBO Rep. 11:97–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parsons JT, Schaller MD, Hildebrand J, Leu
TH, Richardson A and Otey C: Focal adhesion kinase: Structure and
signalling. J Cell Sci Suppl. 18:109–113. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aikawa R, Nagai T, Kudoh S, Zou Y, Tanaka
M, Tamura M, Akazawa H, Takano H, Nagai R and Komuro I: Integrins
play a critical role in mechanical stress-induced p38 MAPK
activation. Hypertension. 39:233–238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Komuro I, Kudo S, Yamazaki T, Zou Y,
Shiojima I and Yazaki Y: Mechanical stretch activates the
stress-activated protein kinases in cardiac myocytes. FASEB J.
10:631–636. 1996.PubMed/NCBI
|
|
25
|
Blaauw E, van Nieuwenhoven FA, Willemsen
P, Delhaas T, Prinzen FW, Snoeckx LH, van Bilsen M and van der
Vusse GJ: Stretch-induced hypertrophy of isolated adult rabbit
cardiomyocytes. Am J Physiol Heart Circ Physiol. 299:H780–H787.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Asrih M, Mach F, Nencioni A, Dallegri F,
Quercioli A and Montecucco F: Role of mitogen-activated protein
kinase pathways in multifactorial adverse cardiac remodeling
associated with metabolic syndrome. Mediators Inflamm.
2013:3672452013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu YY, Chen YC, Kao YH, Wu TJ, Chen SA and
Chen YJ: Extracellular matrix of collagen modulates intracellular
calcium handling and electrophysiological characteristics of HL-1
cardiomyocytes with activation of angiotensin II type 1 receptor. J
Card Fail. 17:82–90. 2011. View Article : Google Scholar
|
|
28
|
Lu YY, Chen YC, Kao YH, Chen SA and Chen
YJ: Extracellular matrix of collagen modulates arrhythmogenic
activity of pulmonary veins through p38 MAPK activation. J Mol Cell
Cardiol. 59:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Claycomb WC, Lanson NA Jr, Stallworth BS,
Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ Jr: HL-1 cells: A
cardiac muscle cell line that contracts and retains phenotypic
characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA.
95:2979–2984. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hou G, Mulholland D, Gronska MA and
Bendeck MP: Type VIII collagen stimulates smooth muscle cell
migration and matrix metalloproteinase synthesis after arterial
injury. Am J Pathol. 156:467–476. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pascarel C, Hongo K, Cazorla O, White E
and Le Guennec JY: Different effects of gadolinium on I (KR), I
(KS) and I (K1) in guinea-pig isolated ventricular myocytes. Br J
Pharmacol. 124:356–360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eghbali M, Tomek R, Sukhatme VP, Woods C
and Bhambi B: Differential effects of transforming growth
factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts.
Regulation of fibrillar collagen mRNAs and expression of early
transcription factors. Circ Res. 69:483–490. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim KK, Ji C, Chang W, Wells RG, Gundberg
CM, McCarthy TL and Centrella M: Repetitive exposure to TGF-beta
suppresses TGF-beta type I receptor expression by differentiated
osteoblasts. Gene. 379:175–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morino N, Mimura T, Hamasaki K, Tobe K,
Ueki K, Kikuchi K, Takehara K, Kadowaki T, Yazaki Y and Nojima Y:
Matrix/integrin interaction activates the mitogen-activated protein
kinase, p44erk-1 and p42erk-2. J Biol Chem. 270:269–273. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takeuchi Y, Suzawa M, Kikuchi T, Nishida
E, Fujita T and Matsumoto T: Differentiation and transforming
growth factor-beta receptor down-regulation by collagen-alpha2beta1
integrin interaction is mediated by focal adhesion kinase and its
downstream signals in murine osteoblastic cells. J Biol Chem.
272:29309–29316. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sadoshima J and Izumo S: Mechanical
stretch rapidly activates multiple signal transduction pathways in
cardiac myocytes: Potential involvement of an autocrine/paracrine
mechanism. EMBO J. 12:1681–1692. 1993.PubMed/NCBI
|
|
37
|
Kudoh S, Komuro I, Hiroi Y, Zou Y, Harada
K, Sugaya T, Takekoshi N, Murakami K, Kadowaki T and Yazaki Y:
Mechanical stretch induces hypertrophic responses in cardiac
myocytes of angiotensin II type 1a receptor knockout mice. J Biol
Chem. 273:24037–24043. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ieda M, Tsuchihashi T, Ivey KN, Ross RS,
Hong TT, Shaw RM and Srivastava D: Cardiac fibroblasts regulate
myocardial proliferation through beta1 integrin signaling. Dev
Cell. 16:233–244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Engel FB, Schebesta M, Duong MT, Lu G, Ren
S, Madwed JB, Jiang H, Wang Y and Keating MT: p38 MAP kinase
inhibition enables proliferation of adult mammalian cardiomyocytes.
Genes Dev. 19:1175–1187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sadoshima J, Jahn L, Takahashi T, Kulik TJ
and Izumo S: Molecular characterization of the stretch-induced
adaptation of cultured cardiac cells. An in vitro model of
load-induced cardiac hypertrophy. J Biol Chem. 267:10551–11560.
1992.PubMed/NCBI
|
|
41
|
Takahashi N, Calderone A, Izzo NJ Jr, Mäki
TM, Marsh JD and Colucci WS: Hypertrophic stimuli induce
transforming growth factor-beta 1 expression in rat ventricular
myocytes. J Clin Invest. 94:1470–1476. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Huang S, Sah VP, Ross J Jr, Brown
JH, Han J and Chien KR: Cardiac muscle cell hypertrophy and
apoptosis induced by distinct members of the p38 mitogen-activated
protein kinase family. J Biol Chem. 273:2161–2168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seko Y, Takahashi N, Tobe K, Kadowaki T
and Yazaki Y: Pulsatile stretch activates mitogen-activated protein
kinase (MAPK) family members and focal adhesion kinase (p125 (FAK))
in cultured rat cardiac myocytes. Biochem Biophys Res Commun.
259:8–14. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hsu HJ, Lee CF, Locke A, Vanderzyl SQ and
Kaunas R: Stretch-induced stress fiber remodeling and the
activations of JNK and ERK depend on mechanical strain rate, but
not FAK. PLoS One. 5:e124702010. View Article : Google Scholar : PubMed/NCBI
|