Introduction
Diabetes mellitus is one of the most prevalent
diseases worldwide, and is associated with a number of
microvascular complications, including retinopathy, neuropathy and
nephropathy, and macrovascular complications, including ischemic
heart disease, cerebrovascular disease and peripheral vascular
diseases (1,2). Advanced glycation end products (AGEs)
have been recognized as an important inducer in diabetes and a
number of age-related vasculopathies (3). AGEs are formed by the non-enzymatic
glycation reaction of reducing sugars and proteins, nucleic acids
and lipids (4). There is
increasing evidence demonstrating that AGEs are intimately involved
in the pathophysiology of diabetic cardiovascular disease by
stimulating oxidative stress, pro-inflammatory effects and
apoptotic responses, and modulating vascular stiffness (5–7).
There is a significant difference in atherosclerotic
vascular disease between women and men, possibly due to men lacking
the protection afforded to women by estrogen (8). For decades, high levels of
testosterone have been considered to be detrimental to the
cardiovascular system following reports of sudden high-dose
anabolic steroid abuse-associated mortality and the higher male
incidence of coronary artery diseases (9,10).
However, several studies have presented alternative results. In
men, as testosterone levels fall with increasing age, the incidence
of coronary heart disease increases (11,12).
Furthermore, low plasma testosterone levels in men are associated
with other known coronary heart disease risk factors, including
hypertension, obesity, increased levels of fibrinogen,
hyperinsulinemia, diabetes mellitus and adverse lipid profiles
(13). However, the effects of the
administration of testosterone on atherogenesis are controversial.
Testosterone has been reported to increase the extent of
atherosclerosis (14), however,
its administration has also been reported to cause a decrease in
atherosclerosis in low density lipoprotein-receptor knockout mice
(15). Similarly, androgens appear
to have an anti-atherogenic effect in men (16). Testosterone is the most abundant
androgen in males, with a physiological plasma level of 22.7±4.3
nM, and it declines progressively with increasing age (17). Previous studies have shown that
testosterone at physiological concentrations may have a beneficial
effect on the prevention of thrombosis development (18), stimulation of endothelial cell
proliferation (19) and activation
of endothelial nitric oxide synthase (NOS) (20).
However, whether and how physiological testosterone
inhibits the deleterious effects of AGEs on human umbilical vein
endothelial cells (HUVECs) remains to be elucidated. Therefore, the
present study aimed to investigate the effects of physiological
testosterone on AGE-induced injury in HUVECs.
Materials and methods
Materials
Testosterone, bovine serum albumin (BSA), D-glucose,
trypsin/EDTA solution and DMSO were purchased from Sigma-Aldrich;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The MTS
CellTiter 96 Aqueous kit was purchased from Promega (Madison, WI,
USA). The Annexin V-fluorescein isothiocyanate (FITC)/propidium
iodide (PI) apoptosis detection kit was obtained from BioVision,
Inc. (Milpitas, CA, USA). Assay kits for the measurement of NO
production, NOS activity, and superoxide dismutase (SOD),
glutathione peroxidase (GSH-Px) and malondialdehyde (MDA)
concentrations were purchased from Jiancheng Bioengineering
Research Institute (Nanjing, China). The tumor necrosis factor
(TNF)-α concentration assay kit was obtained from R&D Systems
(Minneapolis, MN, USA).
Preparation of AGE-BSA in vitro
The AGEs were prepared as previously described by Xu
et al (21). Briefly, BSA
was incubated under sterile conditions with 50 mM D-glucose in 5%
CO2/95% air at 37°C in the dark for 12 weeks. The
unincorporated glucose molecules were removed by dialysis overnight
against 0.01 M phosphate-buffered saline (PBS). The success of the
AGE preparation was determined using a Spectra Max Gemini EM
fluorescence reader (Molecular Devices LLC, Sunnyvale, CA, USA).
The measurements were performed in triplicate at the excitation
wavelength of 370 nm, and the emission peak was observed at 460 nm.
The AGEs were stored at −20°C until use. BSA incubated without
D-glucose under the same conditions was used as the negative
control.
Cell culture
HUVECs were obtained from ScienCell Research
Laboratories (Carlsbad, CA, USA). The cells were cultured in
endothelial cell growth media with 5% supplemental fetal bovine
serum, 1X endothelial cell growth supplement, 10 U/ml penicillin
and 10 µg/ml streptomycin medium and all components were obtained
from ScienCell Research Laboratories) in humidified air, 5% CO2 at
37°C. The cells of the third to fifth passages were used, when
specific characteristics of endothelial cells were identified by
morphological observation. The HUVECs were separately cultivated at
a density of 105/ml in culture medium containing
testosterone at concentrations of 3 nM, 30 nM, 3 µM and 30 µM.
Following 1 h of incubation with testosterone, a sample of each
cell culture (density 105/ml) was treated with 200 mg/ml
AGEs or unmodified BSA. The cell culture was maintained in
humidified 5% CO2 atmosphere at 37°C. At 48 h post-AGE
induction, samples from these two culture groups were collected for
measurements.
Cell viability analysis
The CellTiter 96 Aqueous kit was used to assess cell
viability, according to the manufacturer's protocol, using MTS
reagent. Briefly, the cells were plated at a density of
1.5×104 cells/well in a 96-well plate and incubated in
growth media for 18 h. The cells were further incubated with or
without testosterone for 1 h, followed by stimulation with 200
µg/ml of AGEs or unmodified BSA. The cell culture was maintained in
humidified 5% CO2 atmosphere at 37°C. After 48 h of
culture, MTS solution was added to each well for 3 h, and light
absorbance was detected at 490 nm.
Apoptosis assay
To quantify apoptosis, the cells were evaluated by
double staining with FITC-conjugated Annexin V and PI, according to
the manufacturer's protocol. The cells were washed twice with PBS
and stained with Annexin V and PI for 20 min at room temperature in
the dark. The level of apoptosis was determined by measuring the
fluorescence of the cells with a FacsCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA). The viable cells (annexin-V
negative, PI negative), cells in early apoptosis (annexin-V
positive, PI negative) and cells in late apoptosis or necrosis
(annexin-V positive, PI positive) were identified and counted. Data
analysis was performed with CellQuest version 3.3
(Becton-Dickinson).
Evaluation of oxidative stress and
pro-inflammatory parameters
For the measurement of oxidative stress and
pro-inflammatory parameters in the medium, the HUVECs were seeded
at density of 105/well stimulated with BSA (200 µg/ml),
AGEs (200 µg/ml), testosterone (30 nM) or AGEs+testosterone (30
nM). Following incubation for 48 h with humidified 5%
CO2 atmosphere at 37°C, the culture supernatant was
collected. The production of NO, activity of NOS, and
concentrations of SOD, GSH-Px and MDA were determined using
commercially available assay kits. An enzyme-linked immunosorbent
assay (ELISA) for TNF-α was performed on cell culture supernatants
using a commercial assay. All procedures were performed according
to the manufacturer's protocols.
Western blot analysis
The activation of JAK2 and STAT3 in the HUVECs were
assayed using western blot analysis. Following treatment, the cells
were extracted using lysis buffer containing 50 mM Tris-Cl, 2.5 mM
EGTA, 1 mM EDTA, 10 mM NaF, 1% deoxycorticosterone, 1% Triton
X-100, 1 mM phenyl methylsulfonyl fluoride and 2 mM Na3VO4. Protein
samples from the HUVEC cells were quantified by the bovine serum
albumin protein assay kit (Thermo Fisher Scientific, Inc.). The
proteins (30 µg) were separated using 4–12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis, and were subsequently
transferred onto an Immun-Blot PVDF membrane. The membrane was then
incubated with primary antibodies overnight at 4°C. The blots were
blocked with 4% BSA for 1 h at room temperature and then probed
with the primary antibodies against rabbit primary antibodies
[Anti-JAK2 (cat. no. 3230), anti-phosphorylated-JAK2 (cat. no.
3771), anti-STAT3 (cat. no. 8768) and anti-phosphorylated-STAT3
(cat. no. 9145) and anti-β-actin (cat. no. 4967); 1:1,000 dilution;
Cell Signaling Technology, Inc.] overnight at 4°C. Following three
washes, the blots were subsequently incubated with horseradish
peroxidase-labeled anti-rabbit antibody (cat. no. 7074; 1:1,000;
Cell Signaling Technology, Inc.) for 1 h at room temperature. The
specific proteins were detected using Super Signal West Pico
Chemiluminescent Substrate kit (Thermo Fisher Scientific, Inc.).
The band intensities were measured using Quantity One software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and normalized to
total protein.
Statistical analysis
Data are presented as the mean ± standard error of
the mean obtained from three independent experiments. One-way
analysis of variance followed by Tukey's HSD post-hoc comparisons
were used to determine the differences among multiple groups. SPSS
14.0 software (SPSS, Inc., Chicago, IL, USA) was used. P<0.05
was considered to indicate a statistically significant
difference.
Results
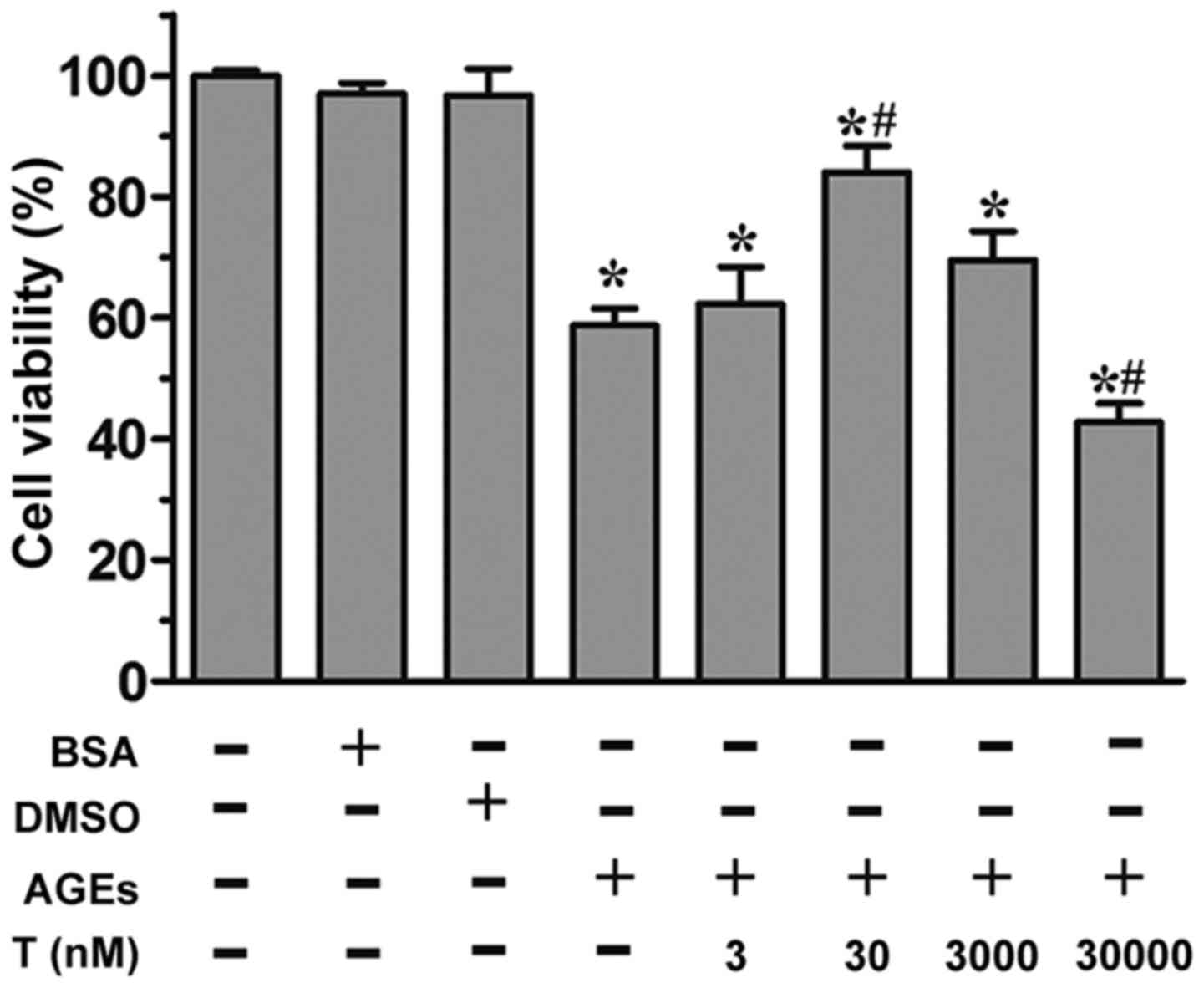
Effect of testosterone on the
viability of HUVECs exposed to AGEs
As revealed by the MTS assay, the incubation of
HUVECs with AGEs (200 µg/ml) led to a significant reduction in cell
proliferation (P<0.05, vs. control group; Fig. 1). By contrast, in the presence of
30 nM testosterone, a significant increase (P<0.05) in cell
viability was observed, compared with the AGE group, although this
effect was not observed at the 3 nM or 3 µM concentrations of
testosterone. At a supraphysiological concentration (30 µM) of
testosterone, cell viability was markedly reduced (P<0.05),
compared with the control group and AGE group. Neither the
unmodified BSA or DMSO affected cell viability (P>0.05),
compared with the control group. Collectively, these data
demonstrated that the physiological concentration of 30 nM
testosterone alleviated AGE-induced cell death.
Effect of testosterone on the
apoptosis of HUVECs exposed to AGEs
To evaluate whether the proliferative effect induced
by the physiological concentration (30 nM) of testosterone was due
to its anti-apoptotic activity, the early-stage apoptotic cells
were analyzed using Annexin V/PI double staining. Flow cytometric
analysis showed that the apoptotic rate was significantly elevated
by AGE treatment, compared with BSA treatment (P<0.05; Fig. 2A-C). The effects of testosterone on
AGE-induced apoptosis was then examined. Pretreatment of the HUVECs
with 30 nM testosterone prevented AGE-induced cytotoxicity,
compared with the AGE group (P<0.05), however, no significant
differences were observed at lower or higher testosterone
concentrations (P>0.05), compared with the AGE group (Fig. 2A and D-G). At the
supraphysiological concentration of testosterone (Fig. 2F and G), the percentage of
apoptotic cells was significantly increased (P<0.05, vs. AGE
group). These results suggested that the physiological
concentration of testosterone protected the HUVECs from AGE-induced
apoptosis.
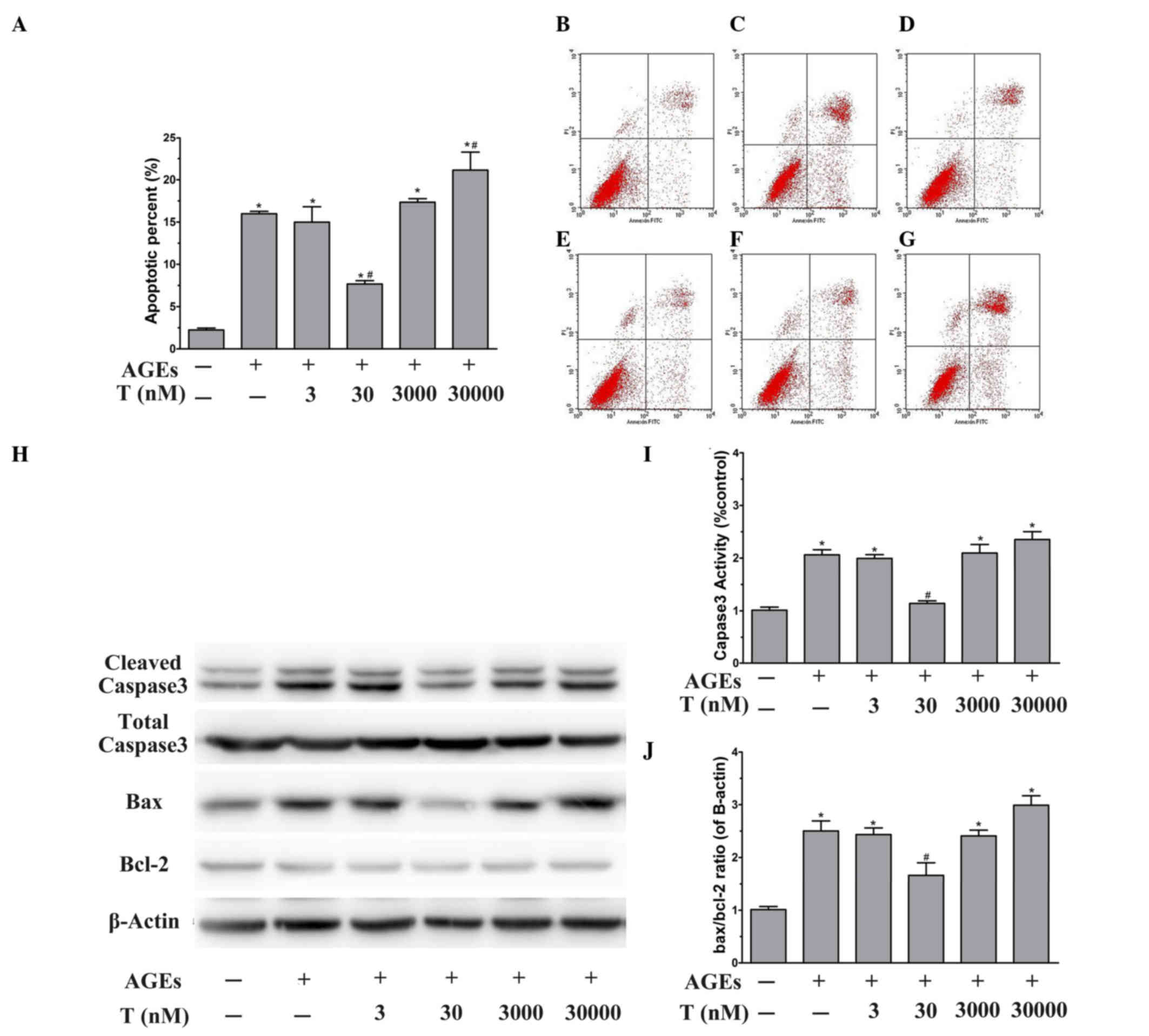 | Figure 2.Effect of testosterone on the
apoptosis of HUVECs exposed to AGEs. Following stimulation and
harvesting of the cells, the levels of apoptosis, vs. necrosis were
quantified using AnnexinV-FITC and PI staining with flow cytometric
analysis. The representative dot plots are shown. Flow cytometry
profiles show Annexin-V-FITC staining on the x-axis and PI on the
y-axis. The lower right quadrant represents the percentage of early
apoptotic cells in each condition. (A) Apoptosis was evaluated
following treatment of the HUVECs with (B) unmodified BSA, (C) AGEs
or (D) 3 nM, (E) 30 nM, (F) 3 µM or (G) 30 µM testosterone. (H)
Representative western blot images and quantitative immunoblot
analysis of (I) cleaved caspase3 normalized to total caspase3 and
the (J) Bax/Bcl-2 ratio, normalized to β-actin. Data are shown as
the mean ± standard error of the mean of triplicate experiments.
*P<0.05, vs. BSA; #P<0.05, vs. AGEs. HUVECs, human
umbilical vein endothelial cells; AGEs, advanced glycation end
products; FITC, fluorescein isothiocyanate; PI, propidium iodide;
BSA, bovine serum albumin; T, testosterone; Bcl-2, B cell
lymphoma-2; Bax, Bcl-2-associated X protein. |
To further examine the molecular mechanisms
underlying the anti-apoptotic effect of physiological testosterone,
the present study investigated the protein expression levels of
caspase-3, Bax and Bcl-2 using western blot analysis. As shown in
Fig. 2H and I, treatment of the
HUVECs with AGEs for 48 h led to a significant increase in the
activation of caspase-3 (P<0.05, vs. BSA group). In addition,
the presence of AGEs significantly increased the Bax/Bcl-2 ratio,
compared with that in the BSA group (P<0.05; Fig. 2H and J). Of note, pretreatment of
the HUVECs with 30 nM testosterone decreased the activity of
caspase-3 and the Bax/Bcl-2 ratio stimulated by AGEs (P<0.05,
vs. AGE group). However, 3 nM and 3 µM testosterone had no effect
on the AGE-induced activity of caspase-3 or upregulation of the
Bax/Bcl-2 ratio (P>0.05, vs. AGE group). Pretreatment with 30 µM
testosterone led to significant increases in the activation of
caspase-3 and the Bax/Bcl-2 ratio (P<0.05, vs. AGE group). These
data suggested that the protective effect of the physiological
concentration (30 nM) of testosterone against AGE-induced apoptosis
may be mediated by caspase-3 and Bax/Bcl-2.
Effect of physiological testosterone
on AGE-induced oxidative stress
A common indication of endothelial cell dysfunction
is enhanced oxidation, and AGEs have been associated with this
(22). Therefore, the present
study investigated the concentrations of MDA and NO, and the
activities of SOD, GSH-Px and eNOS to further confirm the
protective effects of physiological testosterone on AGE-induced
injury in HUVECs. The level of NO (Fig. 3A) and activity of NOS (Fig. 3B) in the HUVECs exposed to AGEs
were found to be significantly decreased, compared with those in
the BSA group. However, pre-treatment with 30 nM testosterone
increased the levels of these indicators (P<0.05, vs. AGE
group). Treatment of the HUVECs with AGEs caused a significant
decrease in the activities of SOD (Fig. 3C) and GSH-Px (Fig. 3D), compared with the BSA group
(P<0.05). Additionally, pre-incubation with 30 nM testosterone
attenuated the changes in the activities of SOD and GSH-Px
(P<0.05, vs. AGE group). As shown in Fig. 3E, the concentration of MDA in the
cells treated with AGEs was significantly higher, compared with
that in the BSA group (P<0.05). By contrast, there was a
significant decrease in the concentration of MDA when the HUVECs
were pre-treated with 30 nM testosterone (P<0.05, vs. AGE
group). In the presence of 30 nM testosterone without AGEs, none of
the parameters were significantly different, compared with those of
the BSA group. Collectively, these results suggested that a
physiological concentration (30 nM) of testosterone may act as an
anti-oxidative agent by protecting HUVECs against AGE-induced
oxidative stress.
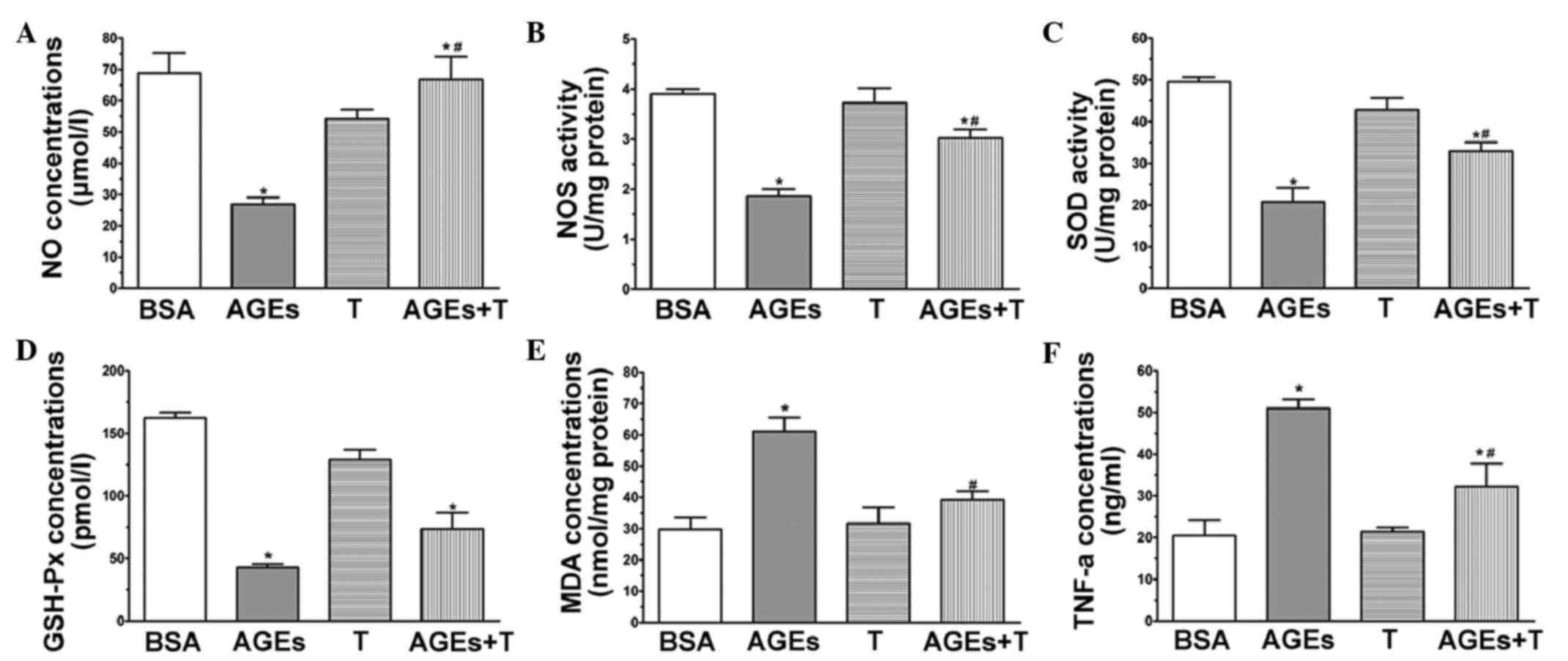 | Figure 3.Physiological testosterone suppresses
AGE-induced oxidative stress and secretion of TNF-α in human
umbilical vein endothelial cells. The images show quantitative
assessments of (A) NO concentration, (B) NOS activity, (C) SOD
activity, (D) GSH-Px concentration, (E) MDA concentration and (F)
TNF-α concentration were performed using specific ELISA following
48 h exposure to advanced glycation end products (AGEs) with or
without 30 nM testosterone. Data are shown as the mean ± standard
error of the mean of triplicate experiments. *P<0.05, vs. BSA;
#P<0.05, vs. AGEs. NO, nitric oxide; NOS, nitric
oxide synthase; SOD, superoxide dismutase; MDA, malondialdehyde;
GSH-Px, glutathione peroxidase; TNF-α, tumor necrosis factors-α;
AGEs, advanced glycation end product; BSA, bovine serum albumin; T,
testosterone. |
Effect of physiological testosterone
on the AGE-induced secretion of TNF-α
Endothelial cells are producers of and targets for
cytokines. It has been established that AGEs can stimulate the
production of TNF-α in HUVECs (23). In order to investigate whether a
physiological concentration (30 nM) of testosterone can suppress
the AGE-induced secretion of TNF-α, the levels of TNF-α were
measured in the media of cultured HUVECs using an ELISA kit.
Following the application of AGEs for 48 h, the level of TNF-α
increased significantly, compared with that of the BSA group, as
shown in Fig. 3F (P<0.05),
whereas the level of TNF-α was markedly suppressed by treatment
with 30 nM testosterone (P<0.05, vs. AGE group).
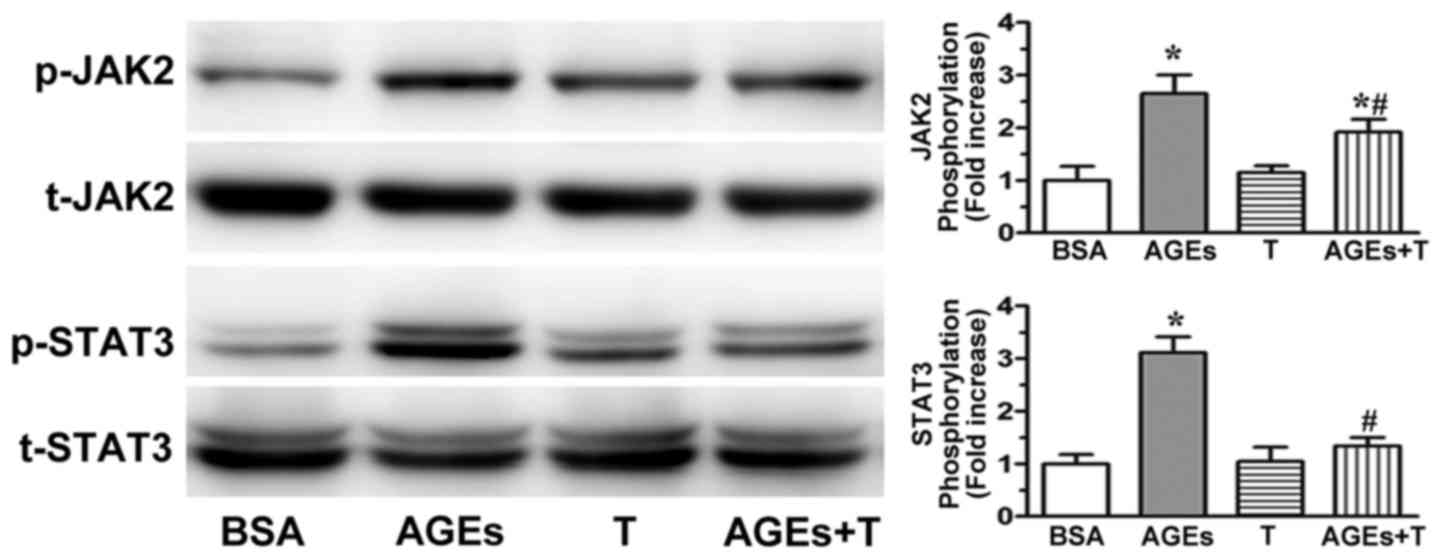
Effect of physiological testosterone
on AGE-induced JAK2/STAT3 pathway activation
AGE-induced endothelial dysfunction has been
significantly correlated with JAK2/STAT3 activation (24,25).
In the present study, western blot analysis was used to examine
whether physiological testosterone ameliorated the AGE-induced
JAK2/STAT3 pathway activation. As shown in Fig. 4, the activation of JAK2 and STAT3
were significantly increased following incubation with AGEs.
However, this process was reduced by pre-incubating the cells with
30 nM testosterone. These results indicated that the physiological
concentration of 30 nM testosterone may attenuate AGE-induced
endothelial dysfunction via the JAK2/STAT3 pathway.
Discussion
The most important characteristics linked to
diabetic vascular complications include high levels of circulating
glucose, increased oxidative stress and the accumulation of AGEs
(26). Various studies have
demonstrated that AGEs are pivotal in several diabetes-associated
vasculopathies (27,28). In the present study, it was
established that AGEs caused apoptosis in HUVECs, induced the
expression of oxidative stress and pro-inflammatory markers and
activated JAK2/STAT3 signaling. The present study revealed that a
physiological concentration of testosterone alleviated AGE-induced
apoptosis, oxidative stress and inflammation in the HUVECs. In
addition, the results showed that testosterone at a
supraphysiological concentration increased the injury induced by
AGEs.
It is widely reported that AGEs induce apoptosis
significantly in HUVECs (7,29).
However, whether testosterone can inhibit this AGE-induced
apoptosis remains to be elucidated. In the present in vitro
study, the results of the MTS assay showed that 30 nM testosterone
effectively attenuated the reduction in cell viability induced by
AGEs. By contrast, at higher concentrations, testosterone had a
dose-dependent cytotoxic effect on the endothelial cells. As the
MTS assay does not discriminate between necrosis and apoptosis,
Annexin V/PI analysis was performed using flow cytometry. The
pretreatment of HUVECs with 30 nM testosterone led to a marked
reduction in the number of apoptotic cells. However, 30 µM
testosterone promoted the apoptosis induced by AGEs. Testosterone
at physiological plasma levels of 22.7±4.3 nM exhibit progressive
age-related decline, whereas the incidence of coronary vascular
diseases has been observed to increase with age (18). Previous studies have supported the
beneficial effects of physiological testosterone on endothelial
cell function in vivo and in vitro (17,18).
Animal studies have shown that the replacement of testosterone
prevents aortic cholesterol accumulation in cholesterol-fed,
orchidectomized male rabbits (30)
and in low-density lipoprotein receptor knockout mice (15). Furthermore, the replacement of
physiological testosterone inhibits fatty streak formation in the
testicular feminized mouse (31).
In a previous in vitro study by Jin et al (18), physiological concentrations of
testosterone were suggested to have a beneficial effect on the
hemostatic system through enhancement of anticoagulant activity
resulting from stimulating the expression of tissue factor pathway
inhibitor and tissue plasminogen activator, and inhibiting the
secretion of plasminogen activator inhibitor-1 by the endothelium.
The present study also aimed to elucidate the mechanisms underlying
the inhibition of apoptosis by testosterone. The AGE-induced
apoptosis involved the activation of caspase-3, which appeared to
be important for the progression of apoptotic cell death.
Additionally, the present study revealed that the ratio of Bax, an
apoptosis promoter, to Bcl-2, an apoptosis inhibitor, was markedly
upregulated in endothelial cells stimulated by AGEs. The present
study also demonstrated that 30 nM testosterone significantly
decreased the Bax/Bcl-2 ratio and attenuated the AGE-induced
activation of caspase-3.
A variety of factors, including reactive oxygen
species (ROS), lipid oxidation enzymes and inflammatory cytokines,
can result in vascular endothelial cell damage (32). ROS affects lipids and leads to
lipid peroxidation, which consequently produces MDA. MDA may
combine with proteins, amino acids and other cellular components,
changing the structure of phospholipids. However, the antioxidants,
SOD and GSH-Px, may also be involved, in which SOD may cooperate
with GSH-Px to remove free radicals (33). In the present study, AGEs
significantly decreased the activities of SOD and GSH-Px, but
increased the contents of MDA. Following pre-treatment with 3 nM
testosterone, the HUVECs were observed to resist damage from AGEs,
reduce the content of MDA, and increase the activities of SOD and
GSH-Px. NOS, one of the key active enzymes in maintaining the
physiological function of endothelial cells, showed the capacity to
remove free radicals in vivo. In addition, NOS is an enzyme
involved in the formation of NO, which is in turn responsible for
vasodilatation, blood pressure regulation, cardiac contractility,
and the mediation of immunity during bacterial infections and
inflammation (34). Compared with
the control group, the expression levels of NO and NOS in the AGE
group were found to be lower. However, their expression levels
increased following pre-treatment with 30 nM testosterone. These
results suggested that the ameliorating effect of physiological
testosterone on AGE-induced apoptosis occurs partly through the
regulation of the anti-oxidation and NO pathway. AGEs can also
alter vessel wall homeostasis in a pro-atherogenic manner through
the release of inflammatory cytokines (5). As reported in a previous study
(23), the secretion of TNF-α was
significantly enhanced following incubation with AGEs in the
HUVECs. However, 30 nM of testosterone was more effective in
protecting the cells against the increase in TNF-α induced by
AGEs.
JAK2 and STAT3 are factors known to transduce
signals initiated by several growth factors and cytokines. The
activated JAK2 tyrosine-phosphorylate and the latent cytoplasmic
STAT3 are involved in AGE-induced cell damage (35). In the present study, it was found
that the activation of JAK2 and STAT3 were associated with the
AGEs. In addition, physiological testosterone significantly
inhibited the activation of JAK2 and STAT3. These results suggested
that inhibition of JAK2/STAT3 signaling activity may be useful for
alleviating AGE-induced cell damage.
In conclusion, the results of the present study
showed that AGEs induced endothelial cell apoptosis in vitro
and that this change was prevented by pretreatment with
physiological testosterone. It was suggested that downregulating
the Bax/Bcl-2 ratio and inhibiting the activation of caspase-3 by
physiological testosterone may protect the HUVECs from AGE-induced
apoptosis. Physiological concentrations of testosterone were also
found to suppress oxidative stress, the expression of inflammatory
cytokines and the activation of JAK2/STAT3 induced by AGEs. These
findings demonstrated the beneficial effect of physiological
testosterone on AGE-induced damage in HUVECs.
Acknowledgements
This study was supported by the Natural Science
Foundation of Zhejiang Province of China (grant no. LQ14H070001)
and the Administration of Traditional Chinese Medicine of Zhejiang
Province (grant no. 2015ZA058).
References
|
1
|
Rahman S, Rahman T, Ismail AA and Rashid
AR: Diabetes-associated macrovasculopathy: Pathophysiology and
pathogenesis. Diabetes Obes Metab. 9:767–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cooper ME, Bonnet F, Oldfield M and
Jandeleit-Dahm K: Mechanisms of diabetic vasculopathy: An overview.
Am J Hypertens. 14:475–486. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neves D: Advanced glycation end-products:
A common pathway in diabetes and age-related erectile dysfunction.
Free Radic Res 47 Suppl. 1:49–69. 2013. View Article : Google Scholar
|
|
4
|
Prasad A, Bekker P and Tsimikas S:
Advanced glycation end products and diabetic cardiovascular
disease. Cardiol Rev. 20:177–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Basta G, Schmidt AM and De Caterina R:
Advanced glycation end products and vascular inflammation:
Implications for accelerated atherosclerosis in diabetes.
Cardiovasc Res. 63:582–592. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldin A, Beckman JA, Schmidt AM and
Creager MA: Advanced glycation end products: Sparking the
development of diabetic vascular injury. Circulation. 114:597–605.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Min C, Kang E, Yu SH, Shinn SH and Kim YS:
Advanced glycation end products induce apoptosis and procoagulant
activity in cultured human umbilical vein endothelial cells.
Diabetes Res Clin Pract. 46:197–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mendelsohn ME and Karas RH: The protective
effects of estrogen on the cardiovascular system. N Engl J Med.
340:1801–1811. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bagatell CJ and Bremner WJ: Androgens in
men-uses and abuses. N Engl J Med. 334:707–714. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sullivan ML, Martinez CM, Gennis P and
Gallagher EJ: The cardiac toxicity of anabolic steroids. Prog
Cardiovasc Dis. 41:1–15. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jones RD, Nettleship JE, Kapoor D, Jones
HT and Channer KS: Testosterone and atherosclerosis in aging men:
Purported association and clinical implications. Am J Cardiovasc
Drugs. 5:141–154. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi BG and McLaughlin MA: Why men's
hearts break: Cardiovascular effects of sex steroids. Endocrinol
Metab Clin North Am. 36:365–377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith AM, English KM, Malkin CJ, Jones RD,
Jones TH and Channer KS: Testosterone does not adversely affect
fibrinogen or tissue plasminogen activator (tPA) and plasminogen
activator inhibitor-1 (PAI-1) levels in 46 men with chronic stable
angina. Eur J Endocrinol. 152:285–291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adams MR, Williams JK and Kaplan JR:
Effects of androgens on coronary artery atherosclerosis and
atherosclerosis-related impairment of vascular responsiveness.
Arterioscler Thromb Vasc Biol. 15:562–570. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nathan L, Shi W, Dinh H, Mukherjee TK,
Wang X, Lusis AJ and Chaudhuri G: Testosterone inhibits early
atherogenesis by conversion to estradiol: Critical role of
aromatase. Proc Natl Acad Sci USA. 98:3589–3593. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
English KM, Mandour O, Steeds RP, Diver
MJ, Jones TH and Channer KS: Men with coronary artery disease have
lower levels of androgens than men with normal coronary angiograms.
Eur Heart J. 21:890–894. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin H, Lin J, Fu L, Mei YF, Peng G, Tan X,
Wang DM, Wang W and Li YG: Physiological testosterone stimulates
tissue plasminogen activator and tissue factor pathway inhibitor
and inhibits plasminogen activator inhibitor type 1 release in
endothelial cells. Biochem Cell Biol. 85:246–251. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin H, Wang DY, Mei YF, Qiu WB, Zhou Y,
Wang DM, Tan XR and Li YG: Mitogen-activated protein kinases
pathway is involved in physiological testosterone-induced tissue
factor pathway inhibitor expression in endothelial cells. Blood
Coagul Fibrinolysis. 21:420–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai J, Hong Y, Weng C, Tan C,
Imperato-McGinley J and Zhu YS: Androgen stimulates endothelial
cell proliferation via an androgen receptor/VEGF/cyclin A-mediated
mechanism. Am J Physiol Heart Circ Physiol. 300:H1210–H1221. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu J, Akishita M, Eto M, Ogawa S, Son BK,
Kato S, Ouchi Y and Okabe T: Androgen receptor-dependent activation
of endothelial nitric oxide synthase in vascular endothelial cells:
Role of phosphatidylinositol 3-kinase/akt pathway. Endocrinology.
151:1822–1828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu Y, Feng L, Wang S, Zhu Q, Zheng Z,
Xiang P, He B and Tang D: Calycosin protects HUVECs from advanced
glycation end products-induced macrophage infiltration. J
Ethnopharmacol. 137:359–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhuang X, Pang X, Zhang W, Wu W, Zhao J,
Yang H and Qu W: Effects of zinc and manganese on advanced
glycation end products (AGEs) formation and AGEs-mediated
endothelial cell dysfunction. Life Sci. 90:131–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rashid G, Benchetrit S, Fishman D and
Bernheim J: Effect of advanced glycation end-products on gene
expression and synthesis of TNF-alpha and endothelial nitric oxide
synthase by endothelial cells. Kidney Int. 66:1099–1106. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grimm S, Ott C, Horlacher M, Weber D, Hohn
A and Grune T: Advanced-glycation-end-product-induced formation of
immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem J.
448:127–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nedić O, Rattan SI, Grune T and Trougakos
IP: Molecular effects of advanced glycation end products on cell
signalling pathways, ageing and pathophysiology. Free Radic Res.
47:(Suppl 1). S28–S38. 2013. View Article : Google Scholar
|
|
26
|
Rodiño-Janeiro BK, González-Peteiro M,
Ucieda-Somoza R, González-Juanatey JR and Alvarez E: Glycated
albumin, a precursor of advanced glycation end-products,
up-regulates NADPH oxidase and enhances oxidative stress in human
endothelial cells: Molecular correlate of diabetic vasculopathy.
Diabetes Metab Res Rev. 26:550–558. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Orasanu G and Plutzky J: The pathologic
continuum of diabetic vascular disease. J Am Coll Cardiol.
53:(Suppl 5). S35–S42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Méndez JD, Xie J, Aguilar-Hernández M and
Méndez-Valenzuela V: Trends in advanced glycation end products
research in diabetes mellitus and its complications. Mol Cell
Biochem. 341:33–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sang HQ, Gu JF, Yuan JR, Zhang MH, Jia XB
and Feng L: The protective effect of Smilax glabra extract
on advanced glycation end products-induced endothelial dysfunction
in HUVECs via RAGE-ERK1/2-NF-κB pathway. J Ethnopharmacol.
155:785–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alexandersen P, Haarbo J, Byrjalsen I,
Lawaetz H and Christiansen C: Natural androgens inhibit male
atherosclerosis: A study in castrated, cholesterol-fed rabbits.
Circ Res. 84:813–819. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nettleship JE, Jones TH, Channer KS and
Jones RD: Physiological testosterone replacement therapy attenuates
fatty streak formation and improves high-density lipoprotein
cholesterol in the Tfm mouse: An effect that is independent of the
classic androgen receptor. Circulation. 116:2427–2434. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hulsmans M and Holvoet P: The vicious
circle between oxidative stress and inflammation in
atherosclerosis. J Cell Mol Med. 14:70–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ho E, Galougahi Karimi K, Liu CC, Bhindi R
and Figtree GA: Biological markers of oxidative stress:
Applications to cardiovascular research and practice. Redox Biol.
1:483–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Allen JD, Giordano T and Kevil CG: Nitrite
and nitric oxide metabolism in peripheral artery disease. Nitric
Oxide. 26:217–222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang JS, Guh JY, Hung WC, Yang ML, Lai
YH, Chen HC and Chuang LY: Role of the Janus kinase (JAK)/signal
transducters and activators of transcription (STAT) cascade in
advanced glycation end-product-induced cellular mitogenesis in
NRK-49F cells. Biochem J. 342:231–238. 1999. View Article : Google Scholar : PubMed/NCBI
|