Nuclear receptors regulate cell function by
controlling the expression of specific gene networks. They are
essential for the regulation of energy metabolism and immune
homeostasis (1). Receptor
interacting protein (RIP) 140 is a ligand-dependent nuclear
receptor that controls the transcription of target genes in various
tissues, including adipose, skeletal muscle, cardiac muscle, liver
and tumor tissues (1–3). RIP140 functions as a metabolic switch
that regulates numerous metabolic pathways involved in defensive
functions via interaction with transcription factors (4). As a co-repressor, RIP140 facilitates
high-fat diet-induced obesity, increases energy expenditure and
induces insulin resistance (5,6). In
addition, RIP140 affects tumorigenesis and tumor metastasis via the
E2F transcription factor and wingless-type mouse mammary tumor
virus/adenomatous polyposis coli/β-catenin signaling pathways
(7–9). As a co-activator, RIP140 activates
nuclear factor κB (NF-κB) and promotes the expression of
proinflammatory cytokines, including tumor necrosis factor (TNF)-α,
interleukin (IL)-1β and IL-6 in immune cells, particularly in
macrophages (10). Furthermore,
NF-κB-mediated degradation of the RIP140 co-activator may induce
endotoxin tolerance (ET) (11).
Type 2 diabetes and cardiovascular diseases are the result of
insulin resistance and atherosclerosis, respectively. These
metabolic disorders are macrophage-mediated chronic inflammatory
diseases (1). Sepsis is a systemic
inflammatory response syndrome (SIRS) and is a common clinical
disease. ET may significantly alleviate the inflammatory response
and reduce the mortality rates of individuals with sepsis and
septic shock (12). The present
brief review summarizes the role of RIP140 in the
macrophage-mediated inflammatory response involving insulin
resistance, atherosclerosis, sepsis and ET.
Insulin resistance and obesity are important factors
in the development of metabolic syndromes, and pose a significant
threat to human health. It has previously been demonstrated that
the inflammatory response of macrophages in adipose tissues appears
to result in insulin resistance in the skeletal muscle (13).
Insulin resistance is a hallmark of type 2 diabetes
and metabolic syndrome. A high level of free fatty acids (FFAs) in
the blood plasma has been identified as an important mediator of
obesity-associated insulin resistance in the skeletal muscle
(14). Adipocytes demonstrate the
ability to synthesize and store a large quantity of triglycerides
(TG). Adipocytes may additionally hydrolyze and release TGs as FFAs
and glycerol during fasting. These properties of adipocytes
maintain a dynamic equilibrium between FFA release into the
circulation and FFA uptake and oxidation by the peripheral tissues,
primarily in skeletal muscles. Kelley et al (15) demonstrated that elevated levels of
circulating FFAs may lead to insulin resistance in the peripheral
tissues of animals and humans.
Adipose tissue may be classified as white adipose
tissue (WAT) or brown adipose tissue (BAT). WAT is primarily
observed in adults, whereas BAT is primarily observed in infants
(16–18). Numerous studies have demonstrated
that macrophages in adipose tissue represent ~50% of the total
number of cells in high-fat diet-induced obese patients. However,
this value decreases to ~5–10% in people of a healthy weight
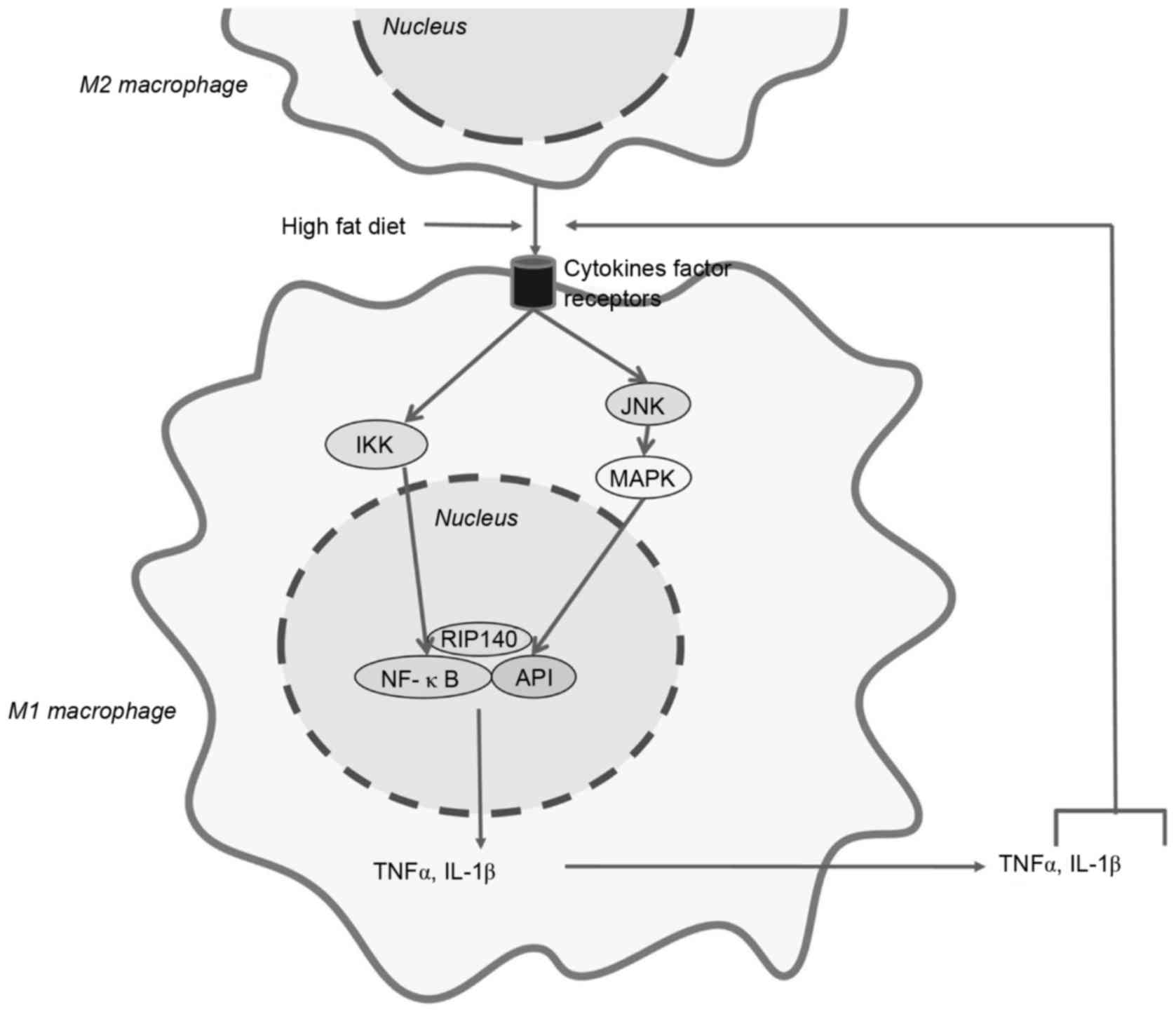
(19–21). In mice fed on a high-fat diet, the
level of RIP140 expression in macrophages is elevated, which
promotes macrophages to undergo M1-like polarization. In addition,
a high-fat diet enhances macrophage recruitment to WAT and
facilitates insulin resistance (22,23).
By contrast, knockout of RIP140 in monocytes or macrophages
decreases the level of RIP140 expression in differentiated
macrophages and promotes an anti-inflammatory phenotype via M2-like
polarization, which increases insulin sensitivity (22–24).
Macrophage-mediated chronic inflammation in adipose tissue promotes
the release of FFAs from adipocytes, which is associated with the
development of insulin resistance in skeletal muscles. Adipocytes
and macrophages secrete a significant quantity of monocyte
chemoattractant protein-1 (MCP-1)/chemokine (C-C motif) ligand-2
(CCL2), TNF-α and IL-1, which induces an inflammatory response in
adipose tissue (25). Transgenic
overexpression of MCP-1 in adipocytes enhances macrophage
infiltration in adipose tissues, which subsequently promotes the
inflammatory response and induces insulin resistance in skeletal
muscles. By contrast, knockout of the MCP-1 receptor chemotactic
cytokine receptor 2 in adipocytes reduces the inflammatory response
in adipose tissues and increases insulin sensitivity in skeletal
muscles. In normal physiology, MCP-1/CCL2, TNFα and IL-1 mediate
the inflammatory response in adipose tissues, which is important
for metabolic regulation in adipocytes. However, when adipocytes
and macrophages secrete a large quantity of these cytokines, two
significant effects on adipocyte function occur; an increase in
lipolysis and decrease in TG synthesis (26,27).
These actions result in increased levels of circulating TGs and
FFAs. The excess of circulating TG and FFAs leads to their
accumulation in skeletal muscles, which subsequently disrupts
mitochondrial oxidative phosphorylation and insulin-mediated
glucose transport, thereby facilitating insulin resistance in
skeletal muscle.
In people of a healthy weight, ~5% of total adipose
tissue cells are macrophages, and these demonstrate an
anti-inflammatory M2-like polarization state. By contrast, a large
number of M1-like macrophages accumulate in the adipose tissues of
patients with obesity (24).
Numerous studies have revealed that RIP140 is important for the
development of the M1-like polarization characteristic of
macrophages (22,23). Decreasing the level of RIP140 in
macrophages reduced the number of M1-like macrophages and increased
the number of M2-like macrophages (21). M1-like macrophages promote the
expression of pro-inflammatory cytokines, including TNFα and IL-1β,
which decrease WAT browning and enhance high-fat diet-induced
insulin resistance (11). RIP140
activates the NF-κB pathway, which is the central step for the
production of TNFα and IL-1β in macrophages. The Jun N-terminal
kinase-mitogen-activated-protein 4-kinase-4 (MAP4K4)-activator
protein-1 (AP1) and inhibitor of NF-κB (IκB)
kinase-β-NF-κB-dependent signaling pathways are additionally
important for the activation of NF-κB (Fig. 1) (28–31).
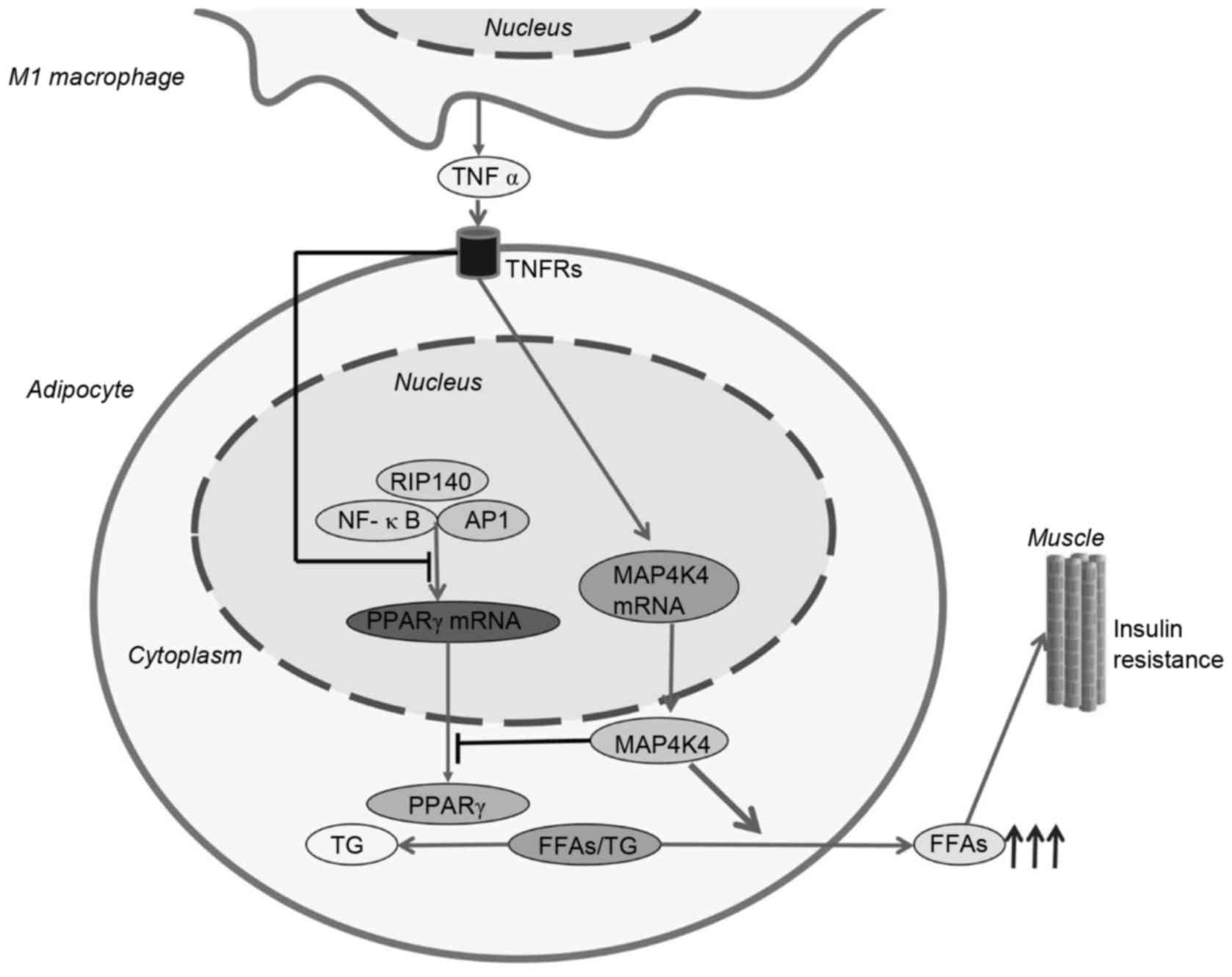
High levels of TNF-α and IL-1β interact with adipocytes to promote
the production of FFAs (32). The
specific mechanisms underlying this interaction remain to be
elucidated; however the evidence suggests that TNF-α and IL-1β may
induce peroxisome proliferator-activated receptor γ (PPARγ) at the
transcriptional and translational levels (33). PPARγ is a member of the nuclear
receptor family, is a key transcriptional regulator of the uptake
and storage of TG, and promotes adipogenesis (34). At the transcriptional level, TNF-α
inhibits the expression of PPARγ mRNA. This is due, in part, to
TNF-α-mediated activation of the RIP140, NF-κB and AP1 signaling
pathways. At the translational level, mitogen-activated protein
kinase (MAPK) is a negative regulator of PPARγ protein expression.
MAPK functions to decrease the expression of PPARγ and promote the
release of TG and FFAs. High levels of TG and FFAs in the
circulation facilitate insulin resistance in skeletal muscles
(Fig. 2) (35–37).
The specific mechanisms underlying PPARγ-mediated regulation of
fatty acid esterification, TG synthesis and hydrolysis remains to
be elucidated. TNF-α and IL-1β promote M1-like polarization of
macrophages and induce insulin resistance (Fig. 1) (38). The mechanism underlying
IL-1β-impaired insulin sensitivity in adipose tissues may be
associated with the inhibition of insulin signal transduction;
however this remains to be fully elucidated (39).
Atherosclerosis is an early-stage lesion of coronary
artery disease and myocardial infarction, which poses a serious
threat to human health. It is known that hypercholesterolemia is an
essential component for the development of various cardiovascular
diseases, particularly atherosclerosis. During
hypercholesterolemia, accumulating cholesterol leads to the
formation of an atheroma plaque. Numerous inflammatory cells, such
as macrophages, are recruited and may lead to chronic inflammation
(40). The primary function of
macrophages is to scavenge excess peripheral cholesterol. However,
macrophages differentiate into foam cells with long-term high
levels of cholesterol in blood. The emergence of foam cells
signifies the development of atherosclerosis (41). Under physiological conditions,
high-density lipoprotein (HDL) and low-density lipoprotein (LDL)
maintain the dynamic balance of cholesterol metabolism. HDL
transports excess cellular cholesterol from peripheral tissues to
the liver, which subsequently decreases the formation of
atherosclerotic plaques. By contrast, LDL transports cholesterol
from the liver to peripheral tissues and facilitates the formation
of atherosclerotic plaques (40,41).
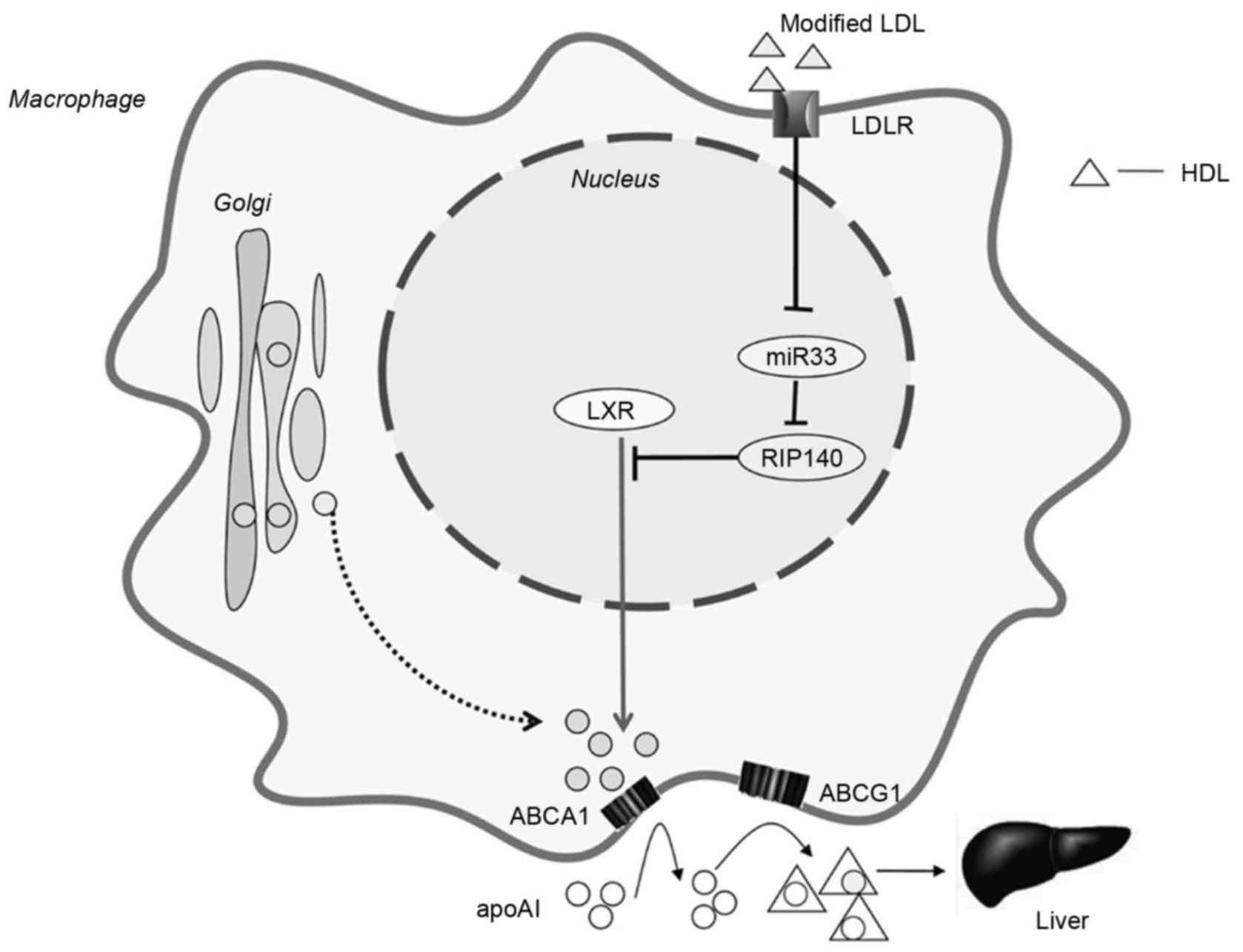
Numerous studies have demonstrated that ATP-binding membrane
cassette transporter member A1 (ABCA1) and ATP-binding cassette
subfamily G member 1 (ABCG1) interact with HDL to increase
cholesterol efflux and promote cholesterol transportation to its
lipid-depleted receptor apolipoprotein AI (apoAI), which protects
against the formation of foam cells and the development of
atherosclerosis (42,43). The specific mechanism remains to be
elucidated; however it is apparent that RIP140 contributes to foam
cell formation and atherosclerosis by regulating cholesterol
homeostasis in macrophages. RIP140 suppresses the expression of
ABCA1 and ABCG1 in macrophages and thus inhibits the efflux of
cholesterol. In an in vivo study, the short hairpin
RNA-mediated knockdown of RIP140 in peritoneal macrophages of mice
resulted in the increased expression of ABCA1 and ABCG1, which
increased cholesterol efflux (44). Conversely, overexpression of RIP140
in macrophages reduced the expression of ABCA1 and ABCG1, and
increased the accumulation of cholesterol (44,45).
Liver X receptor (LXR) is a nuclear receptor that
promotes cholesterol efflux by directly regulating the expression
of ABCA1 and ABCG1. RIP140 is a co-repressor of LXR, which inhibits
LXR-mediated ABCA1 and ABCG1 signaling pathways (46–48).
However, it has been demonstrated that activation of hepatic LXR
may induce lipogenesis and lead to hepatic steatosis. The
enhancement of peripheral LXR activity without affecting hepatic
LXR is of primary concern (47).
In addition, cholesterol-responsive microRNA (miR)-33 is a negative
regulator of RIP140 expression in macrophages, by directly binding
to a highly conserved sequence in the 3′-untranslated region of
RIP140 mRNA (49,50). A previous study demonstrated that
cholesterol upregulates RIP140 expression by repressing miR-33
expression (49). In addition,
miR-758, miR-10b, miR-144, miR-27 and miR-26 directly repress
ABCA1/ABCG1 and negatively regulate cholesterol efflux in
macrophages (51). Ultimately, the
potential of RIP140 as a target for the treatment of
atherosclerosis is evident (Fig.
3) (52).
SIRS is a very common clinical condition, with
severe sepsis and septic shock associated with a high mortality
rate. Despite the use of numerous types of antibiotics, the
mortality rate of patients with severe sepsis and septic shock
remain high at ~30% (53). During
infection, a large number of bacteria permeate the blood, thus
inducing an innate immune response. Lipopolysaccharide (LPS)
endotoxin is located in the bacterial cell wall. A number of
bacteria are killed during the innate immune response, which leads
to the release of LPS into the blood. LPS activates numerous types
of inflammatory cells, such as macrophages and monocytes, to
promote the production of inflammatory cytokines, thus resulting in
sepsis and septic shock (54,55).
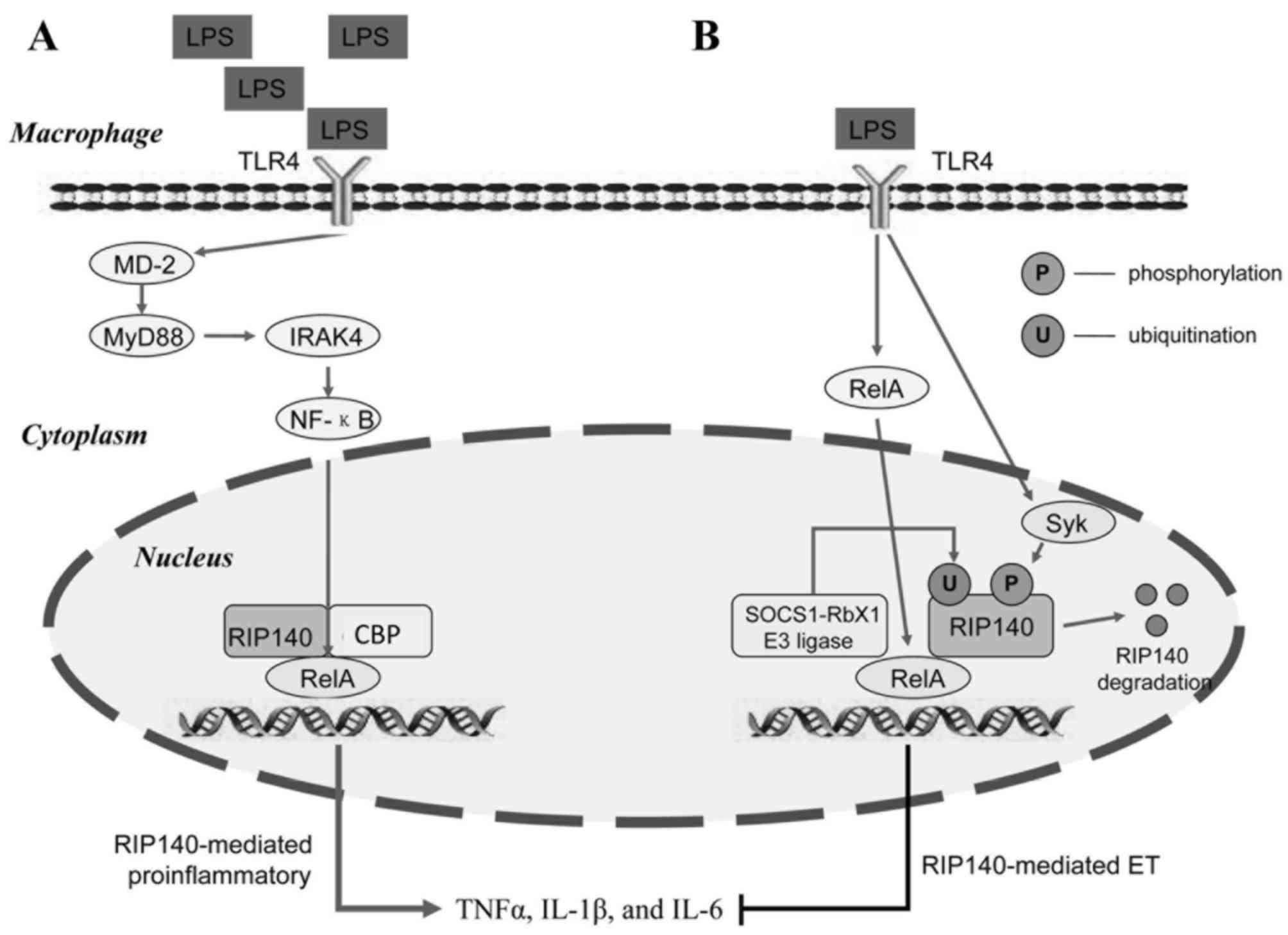
A prototypical inflammatory pathway has been established from
numerous years of research. The LPS-mediated inflammatory response
is induced by activating toll-like receptor 4 (TLR4) located on an
inflammatory cell membrane, which facilitates TLR4-mediated
activation of the downstream myeloid differentiation primary
response gene 88 (MyD88). IL-1 receptor-associated kinase (IRAK) 4
molecules on MyD88 facilitate IRAK1 phosphorylation and activation.
MyD88 and IRAK activate the NF-κB signaling pathway to facilitate
the transcription of pro-inflammatory genes, which subsequently
leads to the production of further inflammatory cytokines,
including TNFα, IL-1β and IL-6 (Fig.
4A) (56–59). Previous studies have indicated that
inflammatory cells repeatedly stimulated with low-dose LPS develop
ET (11,12). Inflammatory cytokines produced by
these inflammatory cells are subsequently reduced, which reduces
the incidence and mortality rate of sepsis and septic shock
(53). A more detailed
understanding of the mechanisms underlying ET is important for the
development of novel therapeutic treatments for sepsis and septic
shock. It has previously been demonstrated that loss of RelA
binding, histone modifications and chromatin remodeling are
essential factors for the development of ET (60). However, the specific mechanisms
underlying these alterations remain to be elucidated. Previous
studies have demonstrated that RIP140 functions as a co-activator
of the NF-κB/RelA-mediated inflammatory response by recruiting cAMP
response element binding protein-binding protein (CBP) to promote
the expression of TLR4-induced pro-inflammatory cytokines,
including TNFα, IL-1β and IL-6 (Fig.
4A) (10,11). It has previously been demonstrated
that RIP140 degradation is critical for LPS-induced ET (11). The suppressor of cytokine signaling
1 (SOCS1)-RING-box protein 1 (Rbx1) has been revealed to interact
with RelA and promote RelA degradation in cell nuclei. RIP140
interacts with RelA to co-activate SOCS1-Rbx1 transcriptional
activity. In addition, RIP140 interacts with RelA to mediate the
recruitment of SOCS1-Rbx1 E3 ligase. SOCS1 and Rbx1 may promote
RIP140 ubiquitination, which is required for RIP140 degradation
(11). In addition, LPS activates
spleen tyrosine kinase (Syk)-mediated phosphorylation of RIP140 on
Tyr364, Tyr418 and Tyr436 residues, thus facilitating its
ubiquitination and inducing ET (11). RelA-mediated SOCS1-Rbx1 E3 ligase
recruitment and Syk-mediated tyrosine phosphorylation are necessary
for LPS-induced RIP140 degradation (Fig. 4B) (11). Interferon-γ (IFN-γ) activates
macrophages to amplify the inflammatory response and promote the
expression of pro-inflammatory cytokines that abrogate ET (61). Pre-treatment of macrophages with
IFN-γ inhibits RIP140 degradation, and overexpression of
non-degradable RIP140 effectively diminishes LPS-induced ET in
vitro and in vivo (11).
Type 2 diabetes, cardiovascular diseases, sepsis and
septic shock are the most common diseases with major societal
implications (11,13,41).
These diseases are all macrophage-meditated inflammatory diseases;
Sepsis and septic shock may additionally be classified as acute
inflammatory diseases. However, chronic inflammatory diseases,
including diabetes and cardiovascular diseases may be more harmful
than acute diseases, and it is important to investigate the
underlying molecular mechanisms involved. RIP140 functions as a
nuclear receptor and co-regulator that is involved in insulin
resistance, atherosclerosis, sepsis and ET (13,41).
In mice with high-fat diet-induced obesity,
macrophages with high expression levels of RIP140 accumulate in
adipose tissues (23). Elevation
of RIP140 induces M1 polarization in macrophages and facilitates
the release of FFAs, which subsequently results in insulin
resistance of skeletal muscles (15,35–37).
In mice with hyperlipidemia, RIP140 suppresses the expression of
ABCA1 and ABCG1 in macrophages (47). Inhibition of the efflux of
cholesterol then contributes to foam cell formation and
atherosclerosis (47). In acute
inflammatory diseases, RIP140 serves as a co-activator for the
NF-κB/RelA-mediated inflammatory response by recruiting CBP to
promote expression of TLR4-induced pro-inflammatory cytokines,
including TNF-α, IL-1β and IL-6 (11). Furthermore, inflammatory cells that
are repeatedly stimulated with low-dose LPS develop ET (12). The underlying mechanism involves
degradation of RIP140 via interaction with RelA, the SOCS1-Rbx1 E3
ligase and Syk, and the subsequent reduction of the inflammatory
cytokine expression, which contributes to ET (11). Despite ongoing research regarding
the role of RIP140 in inflammatory diseases, further studies are
required to determine the underlying molecular mechanisms involved
in ET (1–4). Verifying the clinical relevance of
RIP140 as a prognostic marker may be beneficial for the diagnosis
and treatment of these diseases. In addition, intestinal
LSP-mediated excessive activation of hepatic macrophages, such as
Kupffer cells (KCs), may be important in liver ischemia-reperfusion
injury following liver transplantation (62). Investigating whether RIP140 may
reduce liver ischemia-reperfusion injury during liver
transplantation via inducing ET of KCs is a focus of current
research, which may facilitate an improved understanding of the
role of RIP140 in the inflammatory response.
|
1
|
Rosell M, Jones MC and Parker MG: Role of
nuclear receptor corepressor RIP140 in metabolic syndrome. Biochim
Biophys Acta. 1812:919–928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
White R, Morganstein D, Christian M, Seth
A, Herzog B and Parker MG: Role of RIP140 in metabolic tissues:
Connections to disease. FEBS Lett. 582:39–45. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fritah A, Christian M and Parker MG: The
metabolic coregulator RIP140: An update. Am J Physiol Endocrinol
Metab. 299:E335–E340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chung HT: RIP140, a Janus metabolic switch
involved in defense functions. Cell Mol Immunol. 10:7–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ho PC, Chuang YS, Hung CH and Wei LN:
Cytoplasmic receptor-interacting protein 140 (RIP140) interacts
with perilipin to regulate lipolysis. Cell Signal. 23:1396–1403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Powelka AM, Seth A, Virbasius JV, Kiskinis
E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD,
Parker MG and Czech MP: Suppression of oxidative metabolism and
mitochondrial biogenesis by the transcriptional corepressor RIP140
in mouse adipocytes. J Clin Invest. 116:125–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Docquier A, Harmand PO, Fritsch S,
Chanrion M, Darbon JM and Cavaillès V: The transcriptional
coregulator RIP140 represses E2F1 activity and discriminates breast
cancer subtypes. Clin Cancer Res. 16:2959–2970. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lapierre M, Bonnet S, Bascoul-Mollevi C,
Ait-Arsa I, Jalaguier S, Del Rio M, Plateroti M, Roepman P, Ychou
M, Pannequin J, et al: RIP140 increases APC expression and controls
intestinal homeostasis and tumorigenesis. J Clin Invest.
124:1899–1913. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang D, Wang Y, Dai Y, Wang J, Suo T, Pan
H and Liu H, Shen S and Liu H: Downregulation of RIP140 in
hepatocellular carcinoma promoted the growth and migration of the
cancer cells. Tumour Biol. 36:2077–2085. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zschiedrich I, Hardeland U, Krones-Herzig
A, Diaz M Berriel, Vegiopoulos A, Müggenburg J, Sombroek D, Hofmann
TG, Zawatzky R, Yu X, et al: Coactivator function of RIP140 for
NFkappaB/RelA-dependent cytokine gene expression. Blood.
112:264–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ho PC, Tsui YC, Feng X, Greaves DR and Wei
LN: NF-kB-mediated degradation of the co-activator RIP140 regulates
inflammatory response and contributes to endotoxin tolerance. Nat
Immunol. 13:379–386. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kopanakis K, Tzepi IM, Pistiki A, Carrer
DP, Netea MG, Georgitsi M, Lymperi M, Droggiti DI, Liakakos T,
Machairas A and Giamarellos-Bourboulis EJ: Pre-treatment with
low-dose endotoxin prolongs survival from experimental lethal
endotoxic shock: Benefit for lethal peritonitis by Escherichia
coli. Cytokine. 62:382–388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guilherme A, Virbasius JV, Puri V and
Czech MP: Adipocyte dysfunctions linking obesity to insulin
resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 9:367–377.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang X, Huang L and Xing D:
Photoactivation of Dok1/ERK/PPARγ signaling axis inhibits excessive
lipolysis in insulin-resistant adipocytes. Cell Signal.
27:1265–1275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kelley DE, Mokan M, Simoneau JA and
Mandarino LJ: Interaction between glucose and free fatty acid
metabolism in human skeletal muscle. J Clin Invest. 92:91–98. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barma P and Bhattacharya S, Bhattacharya
A, Kundu R, Dasgupta S, Biswas A and Bhattacharya S, Roy SS and
Bhattacharya S: Lipid induced overexpression of NF-kappaB in
skeletal muscle cells is linked to insulin resistance. Biochim
Biophys Acta. 1792:190–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Unger RH: Lipotoxic diseases. Annu Rev
Med. 53:319–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Santomauro AT, Boden G, Silva ME, Rocha
DM, Santos RF, Ursich MJ, Strassmann PG and Wajchenberg BL:
Overnight lowering of free fatty acids with Acipimox improves
insulin resistance and glucose tolerance in obese diabetic and
nondiabetic subjects. Diabetes. 48:1836–1841. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Savage DB, Petersen KF and Shulman GI:
Disordered lipid metabolism and the pathogenesis of insulin
resistance. Physiol Rev. 87:507–520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Christianson JL, Nicoloro S, Straubhaar J
and Czech MP: Stearoyl-CoA desaturase 2 is required for peroxisome
proliferator-activated receptor gamma expression and adipogenesis
in cultured 3T3-L1 cells. J Biol Chem. 283:2906–2916. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weisberg SP, McCann D, Desai M, Rosenbaum
M, Leibel RL and Ferrante AW Jr: Obesity is associated with
macrophage accumulation in adipose tissue. J Clin Invest.
112:1796–1808. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu PS, Lin YW, Burton FH and Wei LN:
M1-M2 balancing act in white adipose tissue browning-a new role for
RIP140. Adipocyte. 4:146–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu PS, Lin YW, Lee B, McCrady-Spitzer SK,
Levine JA and Wei LN: Reducing RIP140 expression in macrophage
alters ATM infiltration, facilitates white adipose tissue browning,
and prevents high-fat diet-induced insulin resistance. Diabetes.
63:4021–4031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cancello R, Henegar C, Viguerie N, Taleb
S, Poitou C, Rouault C, Coupaye M, Pelloux V, Hugol D, Bouillot JL,
et al: Reduction of macrophage infiltration and chemoattractant
gene expression changes in white adipose tissue of morbidly obese
subjects after surgery-induced weight loss. Diabetes. 54:2277–2786.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sartipy P and Loskutoff DJ: Monocyte
chemoattractant protein 1 in obesity and insulin resistance. Proc
Natl Acad Sci USA. 100:7265–7270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rull A, Camps J, Alonso-Villaverde C and
Joven J: Insulin resistance, inflammation, and obesity: Role of
monocyte chemoattractant protein-1 (or CCL2) in the regulation of
metabolism. Mediators Inflamm. 2010:pii: 3265802010. View Article : Google Scholar
|
|
27
|
Uchida Y, Takeshita K, Yamamoto K, Kikuchi
R, Nakayama T, Nomura M, Cheng XW, Egashira K, Matsushita T,
Nakamura H and Murohara T: Stress augments insulin resistance and
prothrombotic state: Role of visceral adipose-derived monocyte
chemoattractant protein-1. Diabetes. 61:1552–1561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Solinas G, Vilcu C, Neels JG,
Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris
A, Scadeng M, Olefsky JM and Karin M: JNK1 in hematopoietically
derived cells contributes to diet-induced inflammation and insulin
resistance without affecting obesity. Cell Metab. 6:386–397. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang X, Guilherme A, Chakladar A, Powelka
AM, Konda S, Virbasius JV, Nicoloro SM, Straubhaar J and Czech MP:
An RNA interference-based screen identifies MAP4K4/NIK as a
negative regulator of PPAR gamma, adipogenesis, and
insulin-responsive hexose transport. Proc Natl Acad Sci USA.
103:2087–2092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shulman GI: Cellular mechanisms of insulin
resistance. J Clin Invest. 106:171–176. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tesz GJ, Guilherme A, Guntur KV, Hubbard
AC, Tang X, Chawla A and Czech MP: Tumor necrosis factor alpha
(TNFalpha) stimulates Map4k4 expression through TNFalpha receptor 1
signaling to c-Jun and activating transcription factor 2. J Biol
Chem. 282:19302–19312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-alpha:
Direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Odegaard JI, Ricardo-Gonzalez RR, Goforth
MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D,
Brombacher F, Ferrante AW and Chawla A: Macrophage-specific
PPARgamma controls alternative activation and improves insulin
resistance. Nature. 447:1116–1120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamaguchi Y, Cavallero S, Patterson M,
Shen H, Xu J, Kumar SR and Sucov HM: Adipogenesis and epicardial
adipose tissue: A novel fate of the epicardium induced by
mesenchymal transformation and PPARγ activation. Proc Natl Acad Sci
USA. 112:2070–2075. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Imai T, Takakuwa R, Marchand S, Dentz E,
Bornert JM, Messaddeq N, Wendling O, Mark M, Desvergne B, Wahli W,
et al: Peroxisome proliferator-activated receptor gamma is required
in mature white and brown adipocytes for their survival in the
mouse. Proc Natl Acad Sci USA. 101:4543–4547. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Loft A, Forss I, Siersbæk MS, Schmidt SF,
Larsen AS, Madsen JG, Pisani DF, Nielsen R, Aagaard MM, Mathison A,
et al: Browning of human adipocytes requires KLF11 and
reprogramming of PPARγ superenhancers. Genes Dev. 29:7–22. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siersbæk MS, Loft A, Aagaard MM, Nielsen
R, Schmidt SF, Petrovic N, Nedergaard J and Mandrup S: Genome-wide
profiling of peroxisome proliferator-activated receptor γ in
primary epididymal, inguinal, and brown adipocytes reveals
depot-selective binding correlated with gene expression. Mol Cell
Biol. 32:3452–3463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ait-Lounis A and Laraba-Djebari F:
TNF-alpha modulates adipose macrophage polarization to M1 phenotype
in response to scorpion venom. Inflamm Res. 64:929–936. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bing C: Is interleukin-1β a culprit in
macrophage-adipocyte cross talk in obesity? Adipocyte. 4:149–152.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McLaren JE, Michael DR, Ashlin TG and
Ramji DP: Cytokines, macrophage lipid metabolism and foam cells:
Implications for cardiovascular disease therapy. Prog Lipid Res.
50:331–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qiu Y, Yanase T, Hu H, Tanaka T, Nishi Y,
Liu M, Sueishi K, Sawamura T and Nawata H: Dihydrotestosterone
suppresses foam cell formation and attenuates atherosclerosis
development. Endocrinology. 151:3307–3316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yvan-Charvet L, Ranalletta M, Wang N, Han
S, Terasaka N, Li R, Welch C and Tall AR: Combined deficiency of
ABCA1 and ABCG1 promotes foam cell accumulation and accelerates
atherosclerosis in mice. J Clin Invest. 117:3900–3908.
2007.PubMed/NCBI
|
|
43
|
Wei H, Tarling EJ, McMillen TS, Tang C and
LeBoeuf RC: ABCG1 regulates mouse adipose tissue macrophage
cholesterol levels and the ratio of M1 to M2 cells during obesity
and caloric restriction. J Lipid Res. 56:2337–2347. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin YW, Liu PS, Adhikari N, Hall JL and
Wei LN: RIP140 contributes to foam cell formation and
atherosclerosis by regulating cholesterol homeostasis in
macrophages. J Mol Cell Cardiol. 79:287–294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dawson MI and Xia Z: The retinoid X
receptors and their ligands. Biochim Biophys Acta. 1821:21–56.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Calkin AC and Tontonoz P: Liver X receptor
signaling pathways and atherosclerosis. Arterioscler Thromb Vasc
Biol. 30:1513–1518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
He Y, Zhang L, Li Z, Gao H, Yue Z, Liu Z,
Liu X, Feng X and Liu P: RIP140 triggers foam-cell formation by
repressing ABCA1/G1 expression and cholesterol efflux via liver X
receptor. FEBS Lett. 589:455–460. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Calkin AC and Tontonoz P: Transcriptional
integration of metabolism by the nuclear sterol-activated receptors
lXR and FXR. Nat Rev Mol Cell Biol. 13:213–224. 2012.PubMed/NCBI
|
|
49
|
Ho PC, Chang KC, Chuang YS and Wei LN:
Cholesterol regulation of receptor-interacting protein 140 via
microRNA-33 in inflammatory cytokine production. FASEB J.
25:1758–1766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rayner KJ, Sheedy FJ, Esau CC, Hussain FN,
Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y,
et al: Antagonism of mir-33 in mice promotes reverse cholesterol
transport and regression of atherosclerosis. J Clin Invest.
121:2921–2931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dávalos A and Fernández-Hernando C: From
evolution to revolution: miRNAs as pharmacological targets for
modulating cholesterol efflux and reverse cholesterol transport.
Pharmacol Res. 75:60–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Karasawa T and Takahashi M: RIP140 as a
novel therapeutic target in the treatment of atherosclerosis. J Mol
Cell Cardiol. 81:136–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jawad I, Lukšić I and Rafnsson SB:
Assessing available information on the burden of sepsis: Global
estimates of incidence, prevalence and mortality. J Glob Health.
2:0104042012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Charchaflieh J, Wei J, Labaze G, Hou YJ,
Babarsh B, Stutz H, Lee H, Worah S and Zhang M: The role of
complement system in septic shock. Clin Dev Immunol.
2012:4073242012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang X and Quinn PJ: Lipopolysaccharide:
Biosynthetic pathway and structure modification. Prog Lipid Res.
49:97–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xiong Y, Pennini M, Vogel SN and Medvedev
AE: IRAK4 kinase activity is not required for induction of
endotoxin tolerance but contributes to TLR2-mediated tolerance. J
Leukoc Biol. 94:291–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Laird MH, Rhee SH, Perkins DJ, Medvedev
AE, Piao W, Fenton MJ and Vogel SN: TLR4/MyD88/PI3K interactions
regulate TLR4 signaling. J Leukoc Biol. 85:966–977. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nahid MA, Satoh M and Chan EK: MicroRNA in
TLR signaling and endotoxin tolerance. Cell Mol Immunol. 8:388–403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liew FY, Xu D, Brint EK and O'Neill LA:
Negative regulation of Toll-like receptor-mediated immune
responses. Nat Rev Immunol. 5:446–458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Park SH, Park-Min KH, Chen J, Hu X and
Ivashkiv LB: Tumor necrosis factor induces GSK3 kinase-mediated
cross-tolerance to endotoxin in macrophages. Nat Immunol.
12:607–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen J and Ivashkiv LB: IFN-γ abrogates
endotoxin tolerance by facilitating Toll-like receptor induced
chromatin remodeling. Proc Natl Acad Sci USA. 107:19438–19443.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen Y, Liu Z, Liang S, Luan X, Long F,
Chen J, Peng Y, Yan L and Gong J: Role of Kupffer cells in the
induction of tolerance of orthotopic liver transplantation in rats.
Liver Transpl. 14:823–836. 2008. View Article : Google Scholar : PubMed/NCBI
|