Introduction
In addition to the ageing population and serious
environmental pollution, bladder cancer (BCa) is one of the most
common malignancies in China (1).
Although surgery remains the standard therapy for the treatment of
BCa, an increasing number of patients have a preference for bladder
preservation strategies to improve their quality of life (2). In 2012, James et al (3) completed a multicenter randomized
phase III trial to compare the efficacy of radiotherapy alone or
concomitant chemoradiotherapy for patients with muscle-invasive
BCa. The 5-year overall survival rates for chemoradiation and
radiotherapy were 48 and 35%, respectively. In addition, Zehnder
et al (4)reported that the
5-year recurrence-free survival rates of patients with
pT2pN0–2 and pT3pN0–2
BCa undergoing radical cystectomy and extended lymph node
dissection were 57, vs. 67% and 32, vs. 34%, respectively. Although
no studies have directly compared the outcome of bladder
preservation therapy with that of standard surgery in BCa
treatment, the data from previous clinical studies suggest that
radiotherapy or chemoradiotherapy may be an alternative to surgery,
particularly in less medically fit patients (5).
Human disabled homolog 2 interaction protein
(DAB2IP), a putative tumor suppressor gene, belongs to the Ras
GTPase-activating protein family (6). It is often downregulated in BCa with
aggressive phenotypes (7) and
confers BCa cell resistance to ionizing radiation (IR) (8) and antineoplastic drugs (9). Therefore, it may serve as a promising
biomarker of prognosis for patients with BCa treated with
radiotherapy or chemoradiotherapy. In previous investigations
(8), it was found that
ataxia-telangiectasia mutated (ATM), a key signal protein
initiating DNA damage repair upon IR (10), was upregulated at the mRNA and
protein levels in DAB2IP-deficient BCa cells. In addition,
inhibiting the expression of ATM or its activation markedly
enhanced the sensitivity of DAB2IP-deficient BCa cells to IR,
suggesting that ATM-targeted drug screening may be an effective
approach to improve the response of patients with DAB2IP-deficient
BCa to radiotherapy. In order to elucidate the mechanism underlying
the ATM-loss-induced enhancement of radiosensitivity of small
interfering (si) RNA-transfected DAB2IP cells, the effect of γ-rays
on the activation of nuclear factor-κB (NF-κB) and the
mitogen-activated protein kinase (MAPK) signaling pathway were
investigated in the present study.
Materials and methods
Cell culture
The 5637 human bladder urothelial cancer cell line,
purchased from Shanghai Cell Bank of China (Shanghai, China), was
cultured in Dulbecco's modified Eagle's medium (high glucose,
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 8% fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin and 100
µg/ml streptomycin at 37°C in a humidified atmosphere with 5%
carbon dioxide.
RNA interference
The siRNA oligonucleotides against human DAB2IP,
ATM, catalytic subunit of DNA-dependent protein kinase (DNA-PKcs)
and control siRNA have been described previously (8). In brief, transient inhibition of the
target genes was performed on 2×105 cells per ml by transfection
with 20 nM siRNA using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The resulting 5637 cells were termed siControl, siDAB2IP,
siDAB2IP+siATM and siDAB2IP+siDNA-PKcs, respectively.
Cell irradiation
The cells were irradiated at room temperature in
ambient air using a 137Cs source (γ-ray; MDS Nordion, Toronto, ON,
Canada) with a central dose rate of 0.77 Gy/min and a volume of
radiation cavity of 7.5 L.
Colony formation assay
A total of 2×105 log-phase 5637 cells were seeded
into 35 mm culture dishes (Thermo Fisher Scientific, Inc.) and
subjected to increasing doses of γ-rays (0, 2 and 5 Gy). At 4 h
post-irradiation, the cells were diluted serially to appropriate
concentrations (100, 200 and 800 cells per 3 ml) and seeded into 60
mm dishes in triplicate. Following 14 days of incubation at 37°C,
the colonies were rinsed twice with phosphate-buffered saline (PBS;
Beyotime Institute of Biotechnology, Haimen, China), fixed for 15
min using methyl alcohol (Sinopharm Chemical Reagent Co. Ltd.,
Shanghai, China), stained with 0.1% crystal violet solution (Sangon
Biotech Co., Ltd., Shanghai, China). The visible colonies (>50
cells) were counted and the surviving fraction was calculated.
Western blot analysis
The cell lysates were extracted with radio
immunoprecipitation assay lysis buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) and mixed with 10 mM PMSF (cat. no.
ST506; Beyotime Institute of Biotechnology) 2 h post-IR. The
supernatant was collected following centrifugation at 12,000 × g
for 10 mins at 4°C. The protein concentration was quantified using
a Micro BCA Protein Assay kit (cat. no. SK3061; Sangon Biotech Co.,
Ltd.) and equal quantities of total protein (40 µg) were subjected
to 10% SDS-PAGE followed by transfer onto PVDF membranes (0.45 µm;
Merck Millipore, Darmstadt, Germany). The membranes were blocked
with 5% bovine serum albumin (AR2440; Sangon Biotech Co., Ltd.) for
1 h at room temperature and incubated with primary antibodies at
4°C overnight. The primary antibodies used were as follows: DAB2IP
rabbit polyclonal antibody (1:2,000; cat. no. A302-440A; Bethyl
Laboratories, Inc., USA), ATM rabbit monoclonal antibody (1:1,000;
cat. no. 1549-1; Epitomics, Burlingame, CA, USA), DNA-PKcs rabbit
monoclonal antibody (1:1,000; cat. no. 1579-1; Epitomics),
phosphorylated (p)-p38 mouse monoclonal antibody (1:2,000; cat. no.
9216; Cell Signaling Technology, Inc., Danvers, MA, USA), p38
rabbit polyclonal antibody (1:1,000; cat. no. 9212; Cell Signaling
Technology, Inc.), p-c-Jun N-terminal kinase (JNK) rabbit
polyclonal antibody (1:1,000; cat. no. 9251; Cell Signaling
Technology, Inc.), JNK rabbit polyclonal antibody (1:1,000; cat.
no. sc-571; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
p-extracellular signal-regulated kinase (ERK) rabbit monoclonal
antibody (1:1,000, cat. no. 4370; Cell Signaling Technology, Inc.)
and ERK rabbit monoclonal antibody (1:1,000; cat. no. ER31218;
HuaAn Biotechnology, Inc., Hangzhou, China). Anti-GAPDH was
purchased from Xianzhi Biotechnical Co., Ltd. (Hangzhou, Zhejiang,
China). The membranes were washed three times and then incubated
with anti-mouse horseradish peroxidase-conjugated secondary
antibodies (1:1,000; cat. no. A0208; Beyotime Institute of
Biotechnology) or anti-rabbit (1:1,000; cat. no. A0216; Beyotime
Institute of Biotechnology) for 1 h at room temperature. Finally,
the protein bands were visualized using chemiluminescence (BeyoECL
Plus; cat. no. P0018; Beyotime Institute of Biotechnology) and
detected using the ChemiDoc XRS system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Immunofluorescence assay
The cells were diluted to the appropriate
concentration, seeded onto cover slips and exposed to γ-rays of 2
Gy. At 30 min, 2 h, 8 h or 24 h post-IR, the cells were rinsed
three times with pre-cooled PBS and fixed with fixing solution
(cat. no. P0098; Beyotime Institute of Biotechnology, Inc.) for 15
min. Subsequently, 0.5%Triton X-100 was used to permeate the
membrane at room temperature (RT) for 30 min, followed by blocking
with goat serum (cat. no. P0102; Beyotime Institute of
Biotechnology) for 30 min. The cells were then incubated with
primary antibody γ-H2AX (1:100; cat. no. 2212-1; Epitomics) or
NF-κB p65 (1:50; cat. no. sc-372; Santa Cruz Biotechnology, Inc.)
at RT for 1 h. Alexa Fluor 488-conjugated goat anti-rabbit IgG
(1:300; cat. no. A0423; Beyotime Institute of Biotechnology) was
used as the secondary antibody and incubated with samples at RT for
1 h, protected from light. The cells were mounted in Vectashield®
mounting medium containing 4′,6-diamidino-2-phenylindole and
examined using a fluorescence microscope (Olympus Corporation,
Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± standard error
of the mean of at least three independent experiments. The results
were analyzed for significance using Student's t test (unpaired).
P<0.05 was considered to indicate a statistically significant
difference. Statistical analyses were performed using SPSS 18.0
statistics software (SPSS, Inc., Chicago, IL, USA).
Results
Decreased expression of ATM enhances
DAB2IP-deficient cell radiosensitivity
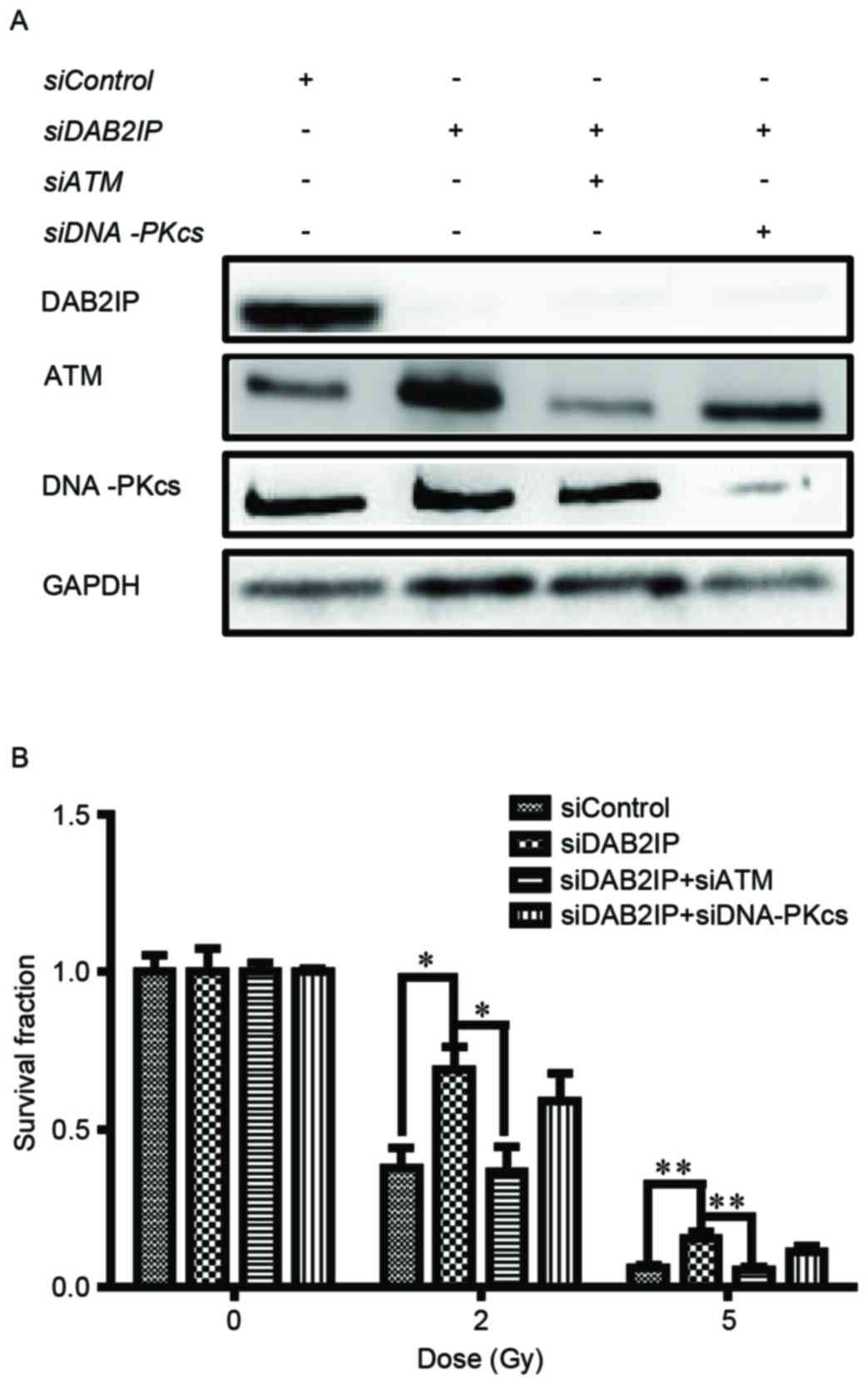
The endogenous expression of DAB2IP, ATM and
DNA-PKcs in the 5637 cells were respectively knocked down by siRNA
transfection and their levels were detected using western blot
analysis (Fig. 1A). The cells were
then exposed to γ-irradiation at increasing doses (0, 2 and 5 Gy).
IR caused a dose-dependent reduction in clonogenic survival of the
siControl cells, siDAB2IP cells, siDAB2IP+siATM cells and
siDAB2IP+siDNAPKcs cells (Fig.
1B). As expected, the siDAB2IP cells exhibited higher
radioresistance, compared with the siControl cells. The surviving
fractions at 2 Gy (SF2) for the siDAB2IP and siControl
cells were 0.69±0.06 and 0.38±0.07 (P=0.0022), respectively. When
the expression of ATM was inhibited in the siDAB2IP cells the
SF2 value was reduced to 0.37±0.08, which was
significantly lower, compared with that in the siDAB2IP cells
(P=0.0026). This radiosensitization effect was not observed for the
siDAB2IP cells treated with DNA-PKcs-knockdown
(SF2=0.59±0.10, vs. siDAB2IP cells; P=0.1058).
Loss of ATM delays siDAB2IP cell DNA
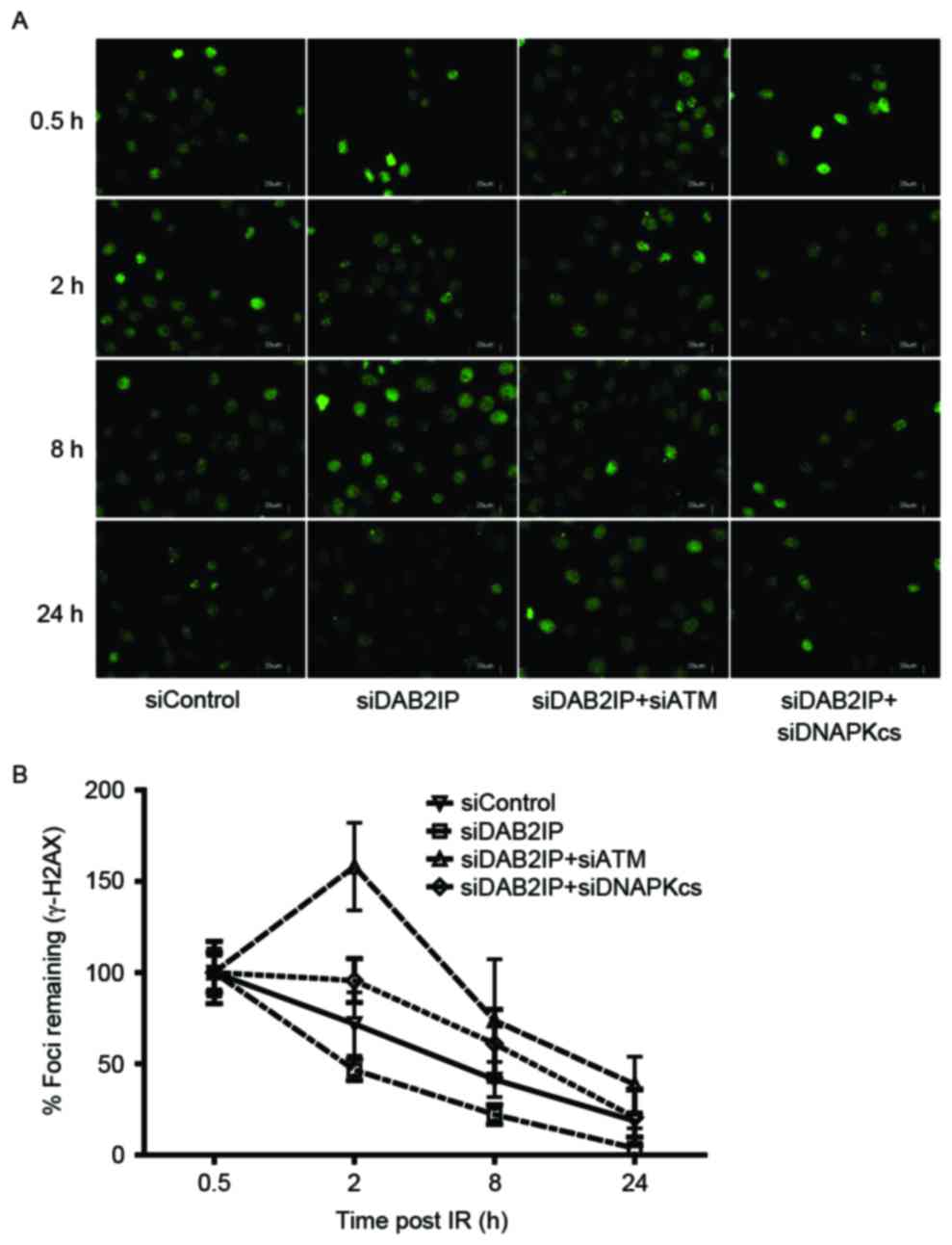
double-strand break (DSB) repair kinetics
Enhanced DSB repair is an important mechanism by
which DAB2IP-deficient cells become resistant to IR (11). The rapid phosphorylation of H2AX
and foci formation at damage sites occurs early in DSB repair. In
the present study, the cells treated with the indicated siRNAs were
subjected to a total dose of 2 Gy. The DSB repair kinetics were
determined over a 24 h period by counting the γH2AX foci (green;
Fig. 2A). The results revealed
that ~80% repair was completed at 8 h post-IR in the siDAB2IP
cells, whereas the siContol cells retained almost 50% of the foci
at this time. Compared with the DNA-PKcs, inhibition delayed the
DSB repair rate in DAB2IP-deficient cells, and the loss of ATM
caused siDAB2IP cells to exhibit unique DSB repair kinetics.
Firstly, the peak of γH2AX foci formation was observed 2 h
following radiation in the siDAB2IP+siATM cells, whereas this peak
occurred immediately following IR in the other groups. Secondly,
>40% of the DSB remained unrepaired in cells lacking DAB2IP and
ATM at 24 h post-IR. This value was double that in the siControl or
siDAB2IP+siDNA-PKcs cells (~20%), and ~10 times higher than that in
the siDAB2IP cells (~3%; Fig.
2B).
Loss of ATM compromises the activation
of NF-κB in siDAB2IP cells
To elucidate the mechanism underlying the ATM
deficiency-enhanced siDAB2IP cell radiosensitivity, the effect of
γ-rays on the activation of NF-κB was investigated using an
immunofluorescence assay (Fig.
3A). The translocation of p65 (green) into the nucleus
following radiation was monitored (Fig. 3B) and the number of p65-posive
nuclei were counted (12).
Compared with the siControl cells, the activation of NF-κB was
significantly increased in the cells lacking DAB2IP. IR induced the
increased movement of cytoplasmic p65 into the nuclei in
DABIP-deficient and DAB2IP-proficient cells. By contrast, knocking
down the endogenous expression of ATM in the siDAB2IP cells
resulted in dose-independent impairment of the response of NF-κB to
IR. However, these changes were not observed in cells deficient in
DNA-PKcs and DAB2IP, suggesting that ATM was involved in the
activation of NF-κB.
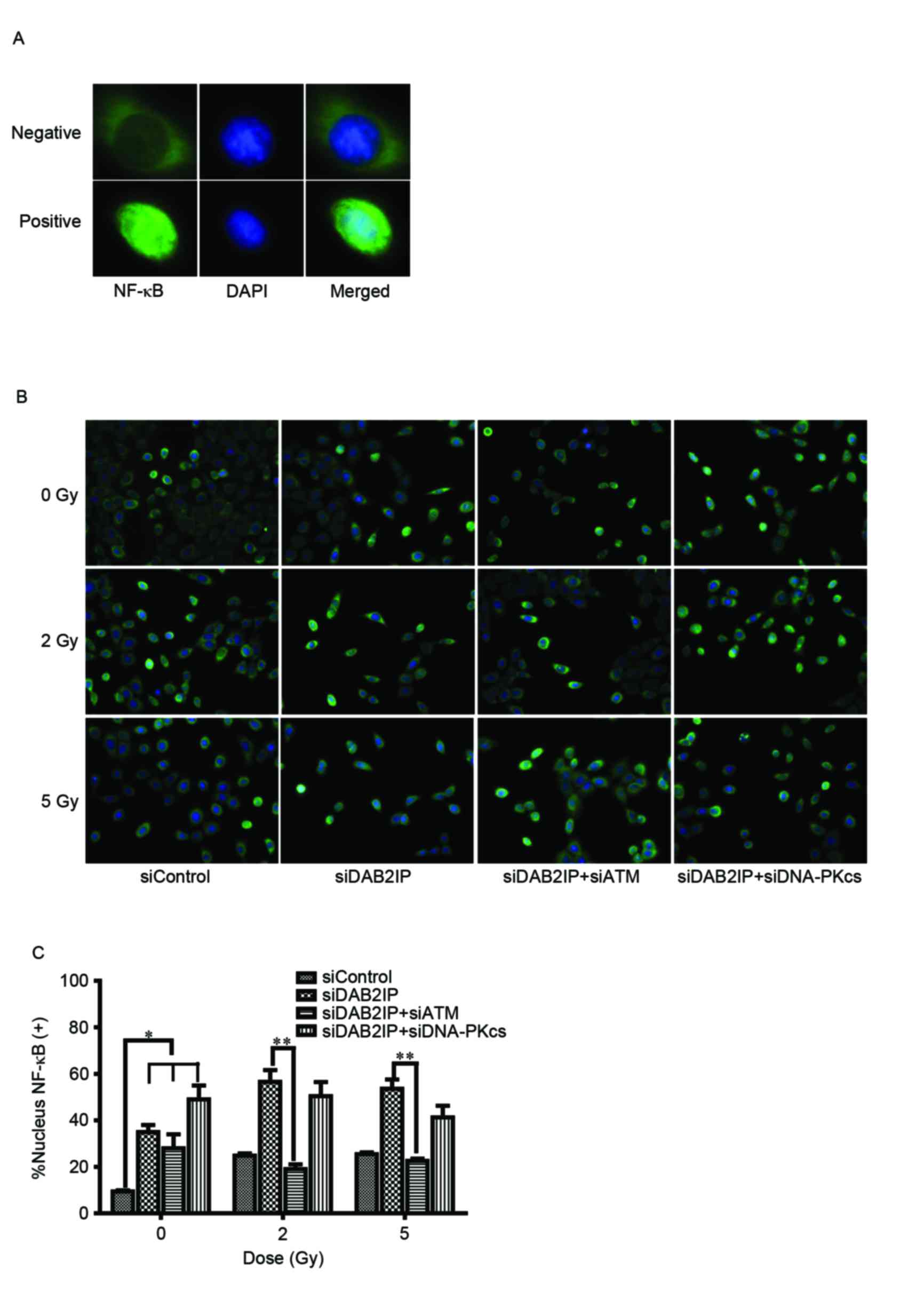 | Figure 3.Loss of ATM delays DSB repair kinetics
of siDAB2IP cells. (A) Distribution of NF-κB (green) was determined
using an immunofluorescence assay (magnification ×100). DAPI (blue)
was utilized to mark the location of nuclei. Above, NF-κB
distributed in the cytoplasm only was determined as negative.
Below, NF-κB translocated into the nucleus was determined as
positive. (B) All sublines were irradiated with 2 Gy and
immunostained for NF-κB at 2 h post-IR (magnification ×40). (C)
Percentage of nuclear NF-κB (+) cells against time were plotted
(average 100 nuclei). The results are presented as the mean ±
standard error of the mean of triplicate experiments. *P<0.05,
compared with the siControl cells; **P<0.05, compared with the
siDAB2IP cells. ATM, ataxia-telangiectasia mutated; DSB,
double-strand break; DAB2IP, disabled homolog 2 interactive
protein; DNA-PKcs, catalytic subunit of DNA-dependent protein
kinase; si, small interfering RNA; NF-κB, nuclear factor-κB; DAPI,
4′,6-diamidino-2-phenylindole. |
Loss of ATM affects the
phosphorylation of p38MAPK and JNK in siDAB2IP cells subjected to
γ-rays
It is known that the mitogen-activated protein
kinase (MAPK) signaling pathway is involved in a variety of
fundamental cellular processes, including the stress response,
apoptosis and survival. In the present study, the cells treated
with the indicated siRNAs were exposed to IR of 2 or 5 Gy. The
phosphorylation and the total protein levels of p38, JNK and ERK
were detected using western blot analysis (Fig. 4A) and the ratios were calculated
using QuantityOne software, version 4.6.2 (Bio-Rad Laboratories,
Inc.) (Fig. 4B). Neither RNA
interference nor IR altered the total protein expression levels of
p38, JNK or ERK. However, the levels of p-MAPK were altered
markedly in the cells in response to all types of stress. Compared
with the siControl cells, knocking down DAB2IP alone caused
elevated expression levels of p-p38, p-JNK and p-ERK. When exposed
to IR, a simultaneous increase in the expression levels of p-p38
and p-ERK, and a decrease in the expression of p-JNK were observed
in the siDAB2IP cells. In the siControl cells, only the
phosphorylation of JNK was enhanced upon IR. ATM and DAB2IP
deficiency in the cells led to differences in MAPK activation
compared with the siDAB2IP cells, in which IR enhanced the
phosphorylation of JNK and decreased the activation of p38.
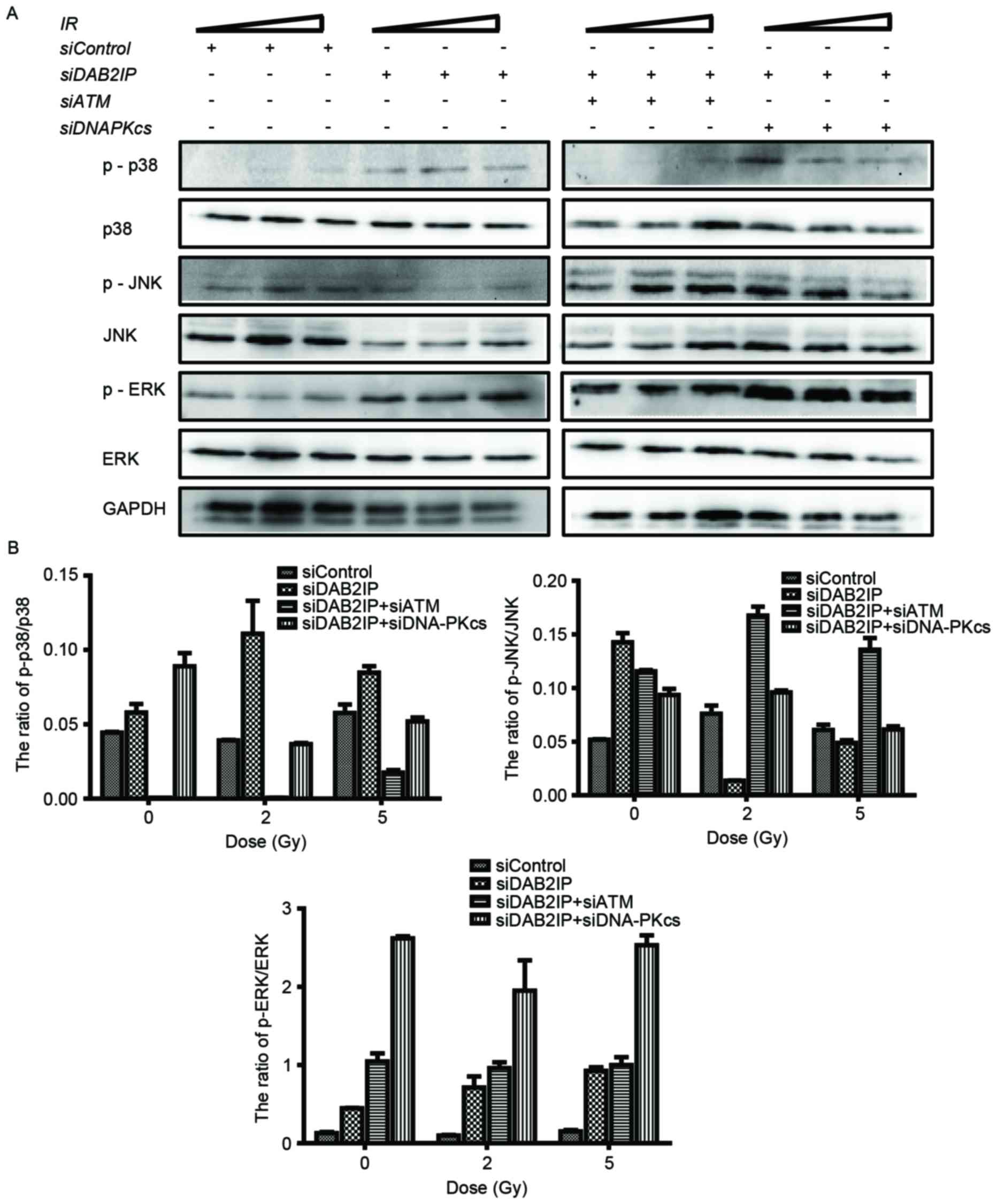 | Figure 4.Loss of ATM affects the
phosphorylation of MAPK in irradiated siDAB2IP cells. (A) All
sublines were subjected to increasing doses of IR (0, 2 and 5 Gy).
The phosphorylation and the total protein levels of p38, JNK and
ERK were detected using western blot analtsis GADPH was loaded as
an internal control. (B) Ratios of p-MAPK/MAPK were determined
using QuantityOne software and plotted against the radiation dose.
The results are presented as the mean ± standard error of the mean
of triplicate experiments. IR, irradiation; ATM,
ataxia-telangiectasia mutated; DSB, double-strand break; DAB2IP,
disabled homolog 2 interactive protein; DNA-PKcs, catalytic subunit
of DNA-dependent protein kinase; si, small interfering RNA; JNK,
c-Jun N-terminal kinase; ERK, extracellular signal-regulated
kinase; p-, phosphorylated. |
Discussion
Our previous study demonstrated that BCa cells
deficient in DAB2IP exhibited increased clonogenic survival in
response to IR, compared with cells expressing endogenous levels of
DAB2IP, and was associated with elevated expression and activation
of ATM, increased S phase cell distribution and faster DSB repair
kinetics (8). Therefore, the
present study aimed to determine whether knocking down ATM enhanced
the radiosensitivity of DAB2IP-deficient cells and examine the
possible underlying mechanism. Firstly, it was observed that the
downregulation of ATM decreased the survival fraction of the
irradiated siDAB2IP cells, however, this enhanced sensitivity to IR
was not detected in siDAB2IP cells with DNA-PKcs deficiency
(Fig. 1). It has been reported
that ATM and DNA-PKcs, which belong to the phosphatidyl inositol
3-kinase related kinase superfamily, are involved in DNA damage
repair following IR (13). The
data obtained in the present study showed that ATM, but not
DNA-PKcs, may be the major protein mediating radioresistance in
DAB2IP-deficient BCa cells. Secondly, the present study monitored
the dynamic course of DSB repair following IR in cells with
different phenotypes. A unique time-lag of γH2AX foci formation and
high levels of unpaired DSB were detected in the cells treated with
siDAB2IP+siATM (Fig. 2). Similar
results were shown previously in KU55933-treated DAB2IP-deficient
5637 cells in response to IR (8).
KU55933 specifically inhibited the phosphorylation of ATM (14), whereas siRNA transfection decreased
the total expression of ATM, the results of the present study
supported these findings that the activation of ATM is important in
mediating cell response to DSB repair.
To clarify the mechanism by which ATM affected the
radiosensitivity of DAB2IP-deficient cells, activation of the NF-κB
and MAPK signaling pathways were identified using
immunofluorescence and western blot analyses, respectively. NF-κB
is usually maintained in an inactive state in the cytoplasm and
enters the nucleus in response to various stimuli, including IR
exposure (15). The activation of
NF-κB is recognized as a key feature in protecting cells from
apoptosis and is associated with reinforcing radioresistance in the
majority of cell types (16). In
the preγΔβδσπκχsent study, knocking down DAB2IP resulted in the
significant nuclear translocation of NF-κB. Neither siATM nor
siDNA-PKcs resulted in such activation of NF-κB (Fig. 3). Although radiation exposure led
to the activation of NF-κB in the DAB2IP-positive and
DAB2IP-negative cells, ATM deficiency markedly suppressed the
induction of NF-κB by IR in the siDAB2IP cells. By contrast, the
downregulation of DNA-PKcs did not affect the activation of NF-κB
in the IR-treated DAB2IP-deficient cells. These results suggested
that ATM, but not DNA-PKcs, was involved in the activation of NF-κB
by IR. Of note, the activation of NF-κB was similar in the siDAB2IP
and siDAB2IP+siATM cells, and no differences in the percentage of
cells positive for nuclear NF-κB were found between the IR-treated
and unirradiated cells with DAB2IP and ATM deficiency. Of note, the
increase of IR dose did not alter the above trend. These results
suggested that siDAB2IP exhibits a robust DNA damage/repair system,
in which ATM occupies a pivotal position. Following the induction
of DSB in the nucleus upon IR, ATM is exported to the neoplasm and
triggers the activation of NF-κB (17). In unirradiated cells, the increased
activation of NK-κB observed in the DAB2IP-deficient cells may be
associated with the protein-protein interaction between p53 and
DAB2IP (18). It was hypothesized
that mutated p53 in 5637 cells (19) bound to and inhibited the tumor
suppressor, DAB2IP, in the cytoplasm. By contrast, in
DAB2IP-negative cells, an increase in mutated p53 release and
accumulation in the neoplasm induced the activation of NF-κB and
reduced the activation of JNK (Fig.
4).
The present study also revealed that the elevated
activation of p38 and ERK, and decreased phosphorylation of JNK
occurred simultaneously in the IR-treated siDAB2IP cells, but not
in the siControl cells exposed to IR. The downregulation of ATM
inhibited the expression of p-p38 and stimulated the activation of
JNK, but did not affect the activation of ERK in the
DAB2IP-negative cells in response to IR. These findings indicated
that DAB2IP deficiency induced the radioresistance of the BCa
cells, and this may be due to ATM-dependent enhancement of DSB
repair kinetics, sensitization of NF-κB and MARK signaling cascade
transfer following IR.
Acknowledgements
This study was supported by National Nature Science
Foundation of China (grant no. 31270896), the Shanghai Nature
Science Foundation (grant no. 11ZR1402100), the Scientific Research
Foundation for the Returned Overseas Chinese Scholars, State
Education Ministry (grant no. 44-8) and the Zhuoxue Project of
Fudan University to Dr Zhaolu Kong.
References
|
1
|
Han SJ, Zhang SW, Chen WQ and Li CL:
Analysis of the status and trends of bladder cancer incidence in
China. Oncology Progress. 11:89–95. 2013.
|
|
2
|
Eapen L: A review of the 2014 English
Language publications pertinent to the treatment of invasive
bladder cancer by radiotherapy. Curr Opin Support Palliat Care.
9:245–248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
James ND, Hussain SA, Hall E, Jenkins P,
Tremlett J, Rawlings C, Crundwell M, Sizer B, Sreenivasan T,
Hendron C, et al: Radiotherapy with or without chemotherapy in
muscle-invasive bladder cancer. N Engl J Med. 366:1477–1488. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zehnder P, Studer UE, Skinner EC, Dorin
RP, Cai J, Roth B, Miranda G, Birkhäuser F, Stein J, Burkhard FC,
et al: Super extended versus extended pelvic lymph node dissection
in patients undergoing radical cystectomy for bladder cancer: A
comparative study. J Urol. 186:1261–1268. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kotwal S, Choudhury A, Johnston C, Paul
AB, Whelan P and Kiltie AE: Similar treatment outcomes for radical
cystectomy and radical radiotherapy in invasive bladder cancer
treated at a united kingdom specialist treatment center. Int J
Radiat Oncol Biol Phys. 70:456–463. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen H, Pong R, Wang Z and Hsieh JT:
Differential regulation of the human gene DAB2IP in normal and
malignant prostatic epithelia: Cloning and characterization.
Genomics. 79:573–581. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shen YJ, Kong ZL, Wan FN, Wang HK, Bian
XJ, Gan HL, Wang CF and Ye DW: Downregulation of DAB2IP results in
cell proliferation and invasion and contributes to unfavorable
outcomes in bladder cancer. Cancer Sci. 105:704–712. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang T, Shen Y, Chen Y, Hsieh JT and Kong
Z: The ATM inhibitor KU55933 sensitizes radioresistant bladder
cancer cells with DAB2IP gene defect. Int J Radiat Biol.
91:368–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu K, Wang B, Chen Y, Zhou J, Huang J, Hui
K, Zeng J, Zhu J, Zhang K, Li L, et al: DAB2IP regulates the
chemoresistance to pirarubicin and tumor recurrence of non-muscle
invasive bladder cancer through STAT3/Twist1/P-glycoprotein
signaling. Cell Signal. 27:2515–2523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guleria A and Chandna S: ATM kinase: Much
more than a DNA damage responsive protein. DNA Repair (Amst).
39:1–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kong Z, Xie D, Boike T, Raghavan P, Burma
S, Chen DJ, Habib AA, Chakraborty A, Hsieh JT and Saha D:
Downregulation of Human DAB2IP gene expression in prostate cancer
cells results in resistance to ionizing radiation. Cancer Res.
70:2829–2839. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Otterson MF, Nie L, Schmidt JL, Link BJ,
Jovanovic N, Lyros O and Rafiee P: EUK-207 protects human
intestinal microvascular endothelial cells (HIMEC) against
irradiation-induced apoptosis through the Bcl2 pathway. Life Sci.
91:771–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shrivastav M, Miller CA, de Haro LP,
Durant ST, Chen BP, Chen DJ and Nickoloff JA: DNA-PKcs and ATM
co-regulate DNA double-strand break repair. DNA Repair (Amst).
8:920–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Juvekar A, Burga LN, Hu H, Lunsford EP,
Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis
CA, et al: Combining a PI3K inhibitor with a PARP inhibitor
provides an effective therapy for BRCA1-related breast cancer.
Cancer Discov. 2:1048–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dent P, Yacoub A, Contessa J, Caron R,
Amorino G, Valerie K, Hagan MP, Grant S and Schmidt-Ullrich R:
Stress and radiation-induced activation of multiple intracellular
signaling pathways. Radiat Res. 159:283–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai M, Ma X, Li X, Mei Q, Li X, Wu Z and
Han W: The Accomplices of NF-κB Lead to Radioresistance. Curr
Protein Pept Sci. 16:279–294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hadian K and Krappmann D: Signals from the
nucleus: Activation of NF-kappaB by cytosolic ATM in the DNA damage
response. Sci Signal. 4:pe22011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Di Minin G, Bellazzo A, Dal Ferro M,
Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R,
Sommaggio R, et al: Mutant p53 reprograms TNF signaling in cancer
cells through interaction with the tumor suppressor DAB2IP. Mol
Cell. 56:617–629. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu HB, Yang K, Xie YQ, Lin YW, Mao QQ and
Xie LP: Silencing of mutant p53 by siRNA induces cell cycle arrest
and apoptosis in human bladder cancer cells. World J Surg Oncol.
11:222013. View Article : Google Scholar : PubMed/NCBI
|