Introduction
Breast cancer is one of the most common malignancies
among females worldwide (1).
Estrogens have been implicated in the etiology of breast cancer.
Epidemiological and clinical evidence has indicated that factors
associated with elevated estrogen levels throughout the lifetime of
a female, including the early onset of menstruation, late
menopause, use of oral contraceptives, late first full-term
pregnancy, and hormone replacement therapy, are associated with an
increase in breast cancer risk among pre- and postmenopausal women
(2). Estrogens are generally
considered to cause proliferation of breast cancer cells via the
estrogen receptor (ER), and serve as a transcription factor to
regulate the expression of target genes encoding proteins with
important biological functions (3). The ER-mediated signaling pathway may
have an important role in the development of cancer; however, they
do not serve a crucial role in cancer initiation (4). Compared with ER-mediated processes,
substantial evidence suggests that the oxidative metabolism of
estrogens serves a major role in the initiation of breast cancer
(5,6). Specific estrogen metabolites,
predominantly catechol estrogens-3,4-quinones (CE-3,4-Q), have the
potential to initiate the cancer process by binding to DNA and
forming depurinating adducts,
4-OHE1(E2)-1-N3Ade and
4-OHE1(E2)-1-N7Gua (6,7).
These depurinating DNA adducts are rapidly lost from DNA by
cleavage of the glycosyl bond, leaving apurinic sites in DNA that
may generate mutations that initiate cancer (8). In addition, redox cycling of quinone
and semiquinone metabolites results in the generation of free
radicals and reactive oxygen species (ROS). Excessive ROS not only
exerts genotoxicity by indirectly increasing genomic instability,
but also stimulates progression of mammary carcinogenicity by
transducing redox-associated signal pathways (9,10).
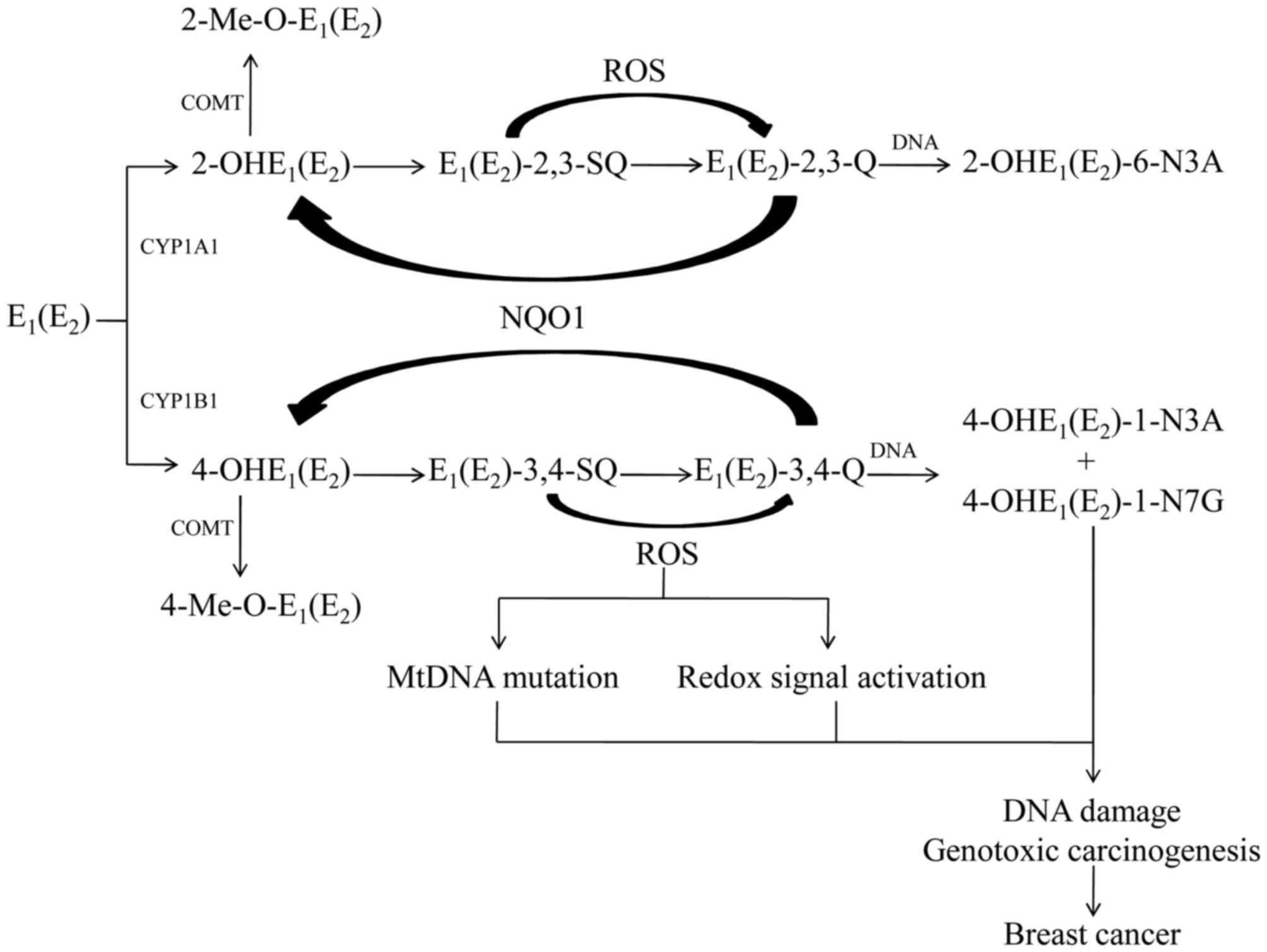
Estrogen metabolism and elimination can be
characterized by two steps. Phase is the conversion of estrogen
into CEs by cytochrome P450 (CYP) 450 enzymes. The major P450
enzymes involved in estrogens metabolize include CYP1A1 and CYP1B1.
Phase II is the inactivation and detoxification pathways of CEs,
including O-methylation by catechol-O-methyltransferase (COMT). The
present review discusses the process of estrogen metabolism, the
role of estrogen metabolites and ROS in breast carcinogenesis, and
the effect of metabolism enzyme polymorphisms on generation of
procarcinogens and breast cancer susceptibility.
Estrogen metabolism
Endogenous estrogen mainly refers to estrone
(E1) and 17β-estradiol (E2). E1
and E2 are interconverted by 17β-hydroxy steroid
dehydrogenase (17β-HSD; Fig. 1)
(11). Endogenous estrogens are
metabolized by two main pathways: Formation of the 2-OH- and
4-OH-estrogens, which are known as CEs, and to a lesser extent,
16a-OHE1(E2) (11). CYP3A5, CYP3A7and CYP1A1 have
catalytic ability for 16a-OHE1(E2) (11). In extrahepatic tissues, estrogens
are transformed into 2-OHE1(E2) by CYP1A1,
whereas 4-OHE1(E2) is catalyzed primarily by
CYP1B1 (11). Since CYP1B1 has
been demonstrated to be expressed in healthy human breast ductal
tissue and over-expressed in invasive ductal carcinomas,
estrogen-quinone depurinating adducts levels are greater in women
with breast cancer compared with healthy women (4,12–14).
In extrahepatic tissues, the most common pathway of
conjugation of CEs is O-methylation inactivation via COMT, which
prevents their conversion to estrogen semiquinones and quinines
(15). COMT catalyzes
2-OHE1(E2) at the 2-OH and 3-OH positions,
and 4-OHE1(E2) at the 4-OH position, with the
methyl group derived from S-adenosylmethionine (16). Inhibition of COMT increases the
amount of oxidative DNA damage and 4-OH quinine depurinating adduct
levels, which means that COMT is responsible for preventing the
oxidative metabolism of CEs to genotoxic quinone metabolites
(17,18). Previous studies have suggested that
2-methoxyestradiol, the major O-methylation metabolite of
2-OHE1(E2), has anticancer activities by
growth inhibitory effects (19).
Unless detoxified, 2-OHE1(E2)
and 4-OHE1(E2) are further oxidized to the
corresponding quinones, E1(E2)-2,3-Q and
E1(E2)-3,4-Q. Of the two estrogen quinones,
E1(E2)-3,4-Q is believed to be a critical
metabolite which reacts with DNA to form depurinating adducts.
Quinones also undergoes a two-electron reduction to form
corresponding hydroquinones, which are transformed by
NAD(P)H-Quinone oxidoreductase 1 (NQO1) (20). NQO1 transforms CE-Q back to CEs,
thus making quinone metabolites unavailable for reaction with DNA
and oxidative stress (20).
Role of estrogen and its metabolites in
carcinogenesis
Chemical carcinogens covalently bind to DNA to
generate two types of adducts: stable ones and depurinating ones.
E1(E2)-3,4-Q produces much higher levels of
depurinating adducts and smaller amounts of stable adducts
(7). Depurinating estrogen-DNA
adducts serve an important role in cancer initiation. The
depurinating adducts are rapidly lost from DNA by cleavage of the
glycosyl bond, and then produce apurinic sites that may lead to
cancer (21). Mounting experiments
on estrogen metabolism, formation of DNA adducts, carcinogenicity,
mutagenicity and cell transformation has identified that estrogen
metabolites, especially CE-3,4-Q, react with DNA to form
predominantly depurinating adducts,
4-OHE1(E2) −1-N3Ade and
4-OHE1(E2)-1-N7Gua, leading to the
accumulation of mutations and potentially cell transformation
(6–8,22).
2-OHE1(E2) is transformed to
E1(E2)-2,3-Q;E1(E2)-2,3-Q
is much less reactive with DNA than E1(E2)
−3,4-Q, because different mechanisms of adduction are responsible
for different reactivity. E1(E2)-3,4-Q reacts
via a proton-assisted 1,4-Michael addition; however, reaction of
E1(E2)-2,3-Q with Ade results in the
generation of 2-OHE1(E2)-6-N3Ade by
1,6-Michael addition (23,24). The greater carcinogenic activity of
4-OHE1(E2) is associated with a higher amount
of depurinating DNA adducts formed by
E1(E2)-3,4-Q, compared with
E1(E2)-2,3-Q (25). According to previous studies,
2-OHE1(E2) methylation by COMT may have an
inhibitory effect on cell proliferation (19). This may be another reason for the
reduced genotoxic of 2-OHE1(E2).
Accumulating evidence for the initiation of cancer
by estrogen-DNA adducts has been identified by using human breast
epithelial cell lines such as MCF-10F, which is an immortalized,
non-transformed ER-a-negative cell line. Treatment of these cells
with E2 or 4-OHE2 produces depurinating
estrogen-DNA adducts (26–28). These adducts induce colony
formation in soft agar, the expression of which are indicative of
neoplastic transformation ability. (29–31).
The cells are transformed by estrogens even in the presence of the
anti-estrogen tamoxifen or ICI-182,780 (31). The results further indicate that
transformation occurs via the genotoxic effects of the estrogen
metabolites. The 2-OHE2 metabolite induces these
alterations to a much smaller extent. Implantation of
estrogen-transformed MCF-10F cells, selected by their invasiveness,
into severely compromised immune-deficient mice, produces tumors
(30). Female ERKO/Wnt-1 mice were
ovariectomized at 15 days old and implanted with E2.
Breast tumors developed in a dose-dependent manner (32). Tumors were induced even following
implantation of E2 plus the anti-estrogen ICI-182,780
(33). These results support the
hypothesis that estrogen metabolism is a crucial event in the
initiation of estrogen-induced cancer.
The mutagenicity of
E1(E2)-3,4-Q was first studied in female
SENCAR mice by determining the H-ras mutations induced, and the
estrogen-DNA adducts formed (34).
Equal amounts of the depurinating
4-OHE1(E2)-1-N3Ade and
4-OHE1(E2)-1-N7Gua adducts were identified in
the skin, representing >99% of the total adducts formed
(34). Mutations were observed in
the H-ras oncogene within 6–12 h after treatment. The rapid
appearance of mutations indicated that they arose by error-prone
repair of the apurinic sites generated by the depurinating
estrogen-DNA adducts. In a second study, female ACI rats, which are
susceptible to estrogen-induced mammary tumors, were treated with
E1(E2)-3,4-Q by intramammillary injection.
Depurinating N3Ade and N7Gua adducts as well as H-ras mutations
were detected in mammary skin tissue. These results demonstrate the
mutagenic activity of these estrogen metabolites (35). With multiple treatments of the
mammalian cells with 4-OHE1(E2), a
dose-dependent, statistically significant increase in mutant
fraction was observed (36). The
reactive quinone formed from 4-OHE1(E2),
E1(E2)-3,4-Q, was similarly mutagenic.
However, no mutagenicity was detected when the cells were treated
with 2-OHE1(E2) (36).
To date, three studies have been conducted in women
at normal or high risk for breast cancer. The risk of developing
breast cancer is measured as the ratio of estrogen-DNA adducts to
their respective estrogen metabolites and conjugates. High levels
of estrogen-DNA adducts have been seen in analyses of urine and
serum from women that are at high risk of breast cancer (14,37,38).
Observation of higher levels of estrogen-DNA adducts in women at
high risk for breast cancer would suggest that formation of these
adducts is a causative factor in the etiology of breast cancer
rather than a consequence of the disease (4).
Role of ROS in estrogen carcinogenesis
Redox cycling via reduction of CE-Q to semiquinones,
catalyzed by CYP reductase, and subsequent oxidation back to CE-Q
by O2, forms super-anion radicals and then
H2O2 (39).
Estrogen-mediated high ROS accumulation serves a key role in
driving carcinogenesis (40).
Excessive ROS serves as an important effector to increase genomic
instability and activate the redox-associated signaling pathway.
Physiologically available concentrations of estrogens or estrogen
metabolites directly acting on the mitochondria of mammary
epithelial cells produces ROS, which subsequently enhances the
phosphorylation of kinases to activate redox-sensitive
transcription factors (41,42).
Therefore, ROS serves an important role in estrogen-induced
cancer.
2-OHE1(E2) and
4-OHE1(E2) are highly redox active and
generate ROS in breast epithelial cells (9). The prolonged exposure to estrogen
aggravates mutations in mitochondrial DNA (mtDNA) and mitochondrial
protein damage by inducing ROS overproduction. Previous studies
have identified that instability of mtDNA induces cancer cell
metastasis and triggers cancer malignant transformation, whereas
ROS scavengers suppress the metastatic potential in mice via
alleviation of mtDNA mutation (43). Furthermore, mutations in mtDNA
altering expression and function of the mitochondrial respiratory
chain was observed in breast cancer cells, blocking of estrogens
attenuated the respiratory and metabolic responses and superoxide
accumulation (44,45). These results indicated that
ROS-triggered mtDNA mutations may contribute to cancer malignant
transformation. Furthermore, overproduction of ROS induced by
alteration of mitochondrial metabolism is also involved in
estrogen-mediated carcinogenesis via induction of oxidative DNA
damage (40).
A recent study identified that ROS induced by
4-OHE1(E2) causes malignant transformation of
MCF-10A cells, and co-treatment with
4-OHE1(E2) and biological or chemical ROS
scavengers prevents the tumorigenic conversion of MCF-10A cells
(41). It appears that
oxidant-mediated activation of redox-sensitive phosphatidylinositol
3-kinase/protein kinase B (AKT) pathways serves a pivotal role in
tumor malignant transformation of healthy breast epithelial cells
by estrogens (41). In addition to
AKT, the nuclear factor-κB (NF-κB) family is another important
redox transcription factor activated by ROS that has been observed
during neoplastic transformation of mammary epithelial cells
(46). ROS overproduced by CEs
accelerates the nuclear translocation of NF-κB by induction of IκB
kinase (IKK) α and -β activities (47). At the same time, inhibition of
NF-κB activation by antioxidants has demonstrated a positive
associated between NF-κB-associated neoplastic transformation and
ROS overproduction (47,48). Excess ROS generated by repeated
exposure to 4-OHE1(E2) causes malignancy of
human mammary epithelial cells in nude mice (41).
In conclusion, estrogen induces the overproduction
of ROS, which subsequently initiates multiple biological functions,
including mtDNA mutation, alteration of mitochondrial metabolism,
and activation of the redox-associatedsignaling pathway, thereby
accelerating cell proliferation involved in tumor progression.
Association of metabolism enzyme
polymorphisms in breast cancer
Levels of 4-OHE1(E2) in breast
cancer are increased compared with healthy breast tissue (14). As CYP1B1 is the key enzyme for the
formation of 4-OHE1(E2), expression and
genetic variations in CYP1B1 may influence breast cancer
progression by increasing concentration of
4-OHE1(E2). To date, >300 polymorphisms
have been found in CYP1B1 (49).
The most common polymorphisms of CYP1B1, including Arg48Gly,
Ala119Ser, Val432Leu and Asn453Ser, lead to alterations in estrogen
metabolism and may influence the risk of breast cancer (49). An in vitro study
demonstrated that the 4-hydroxylase activities of estradiol by
Ala119Ser and Asn453Ser variants of CYP1B1 are 2–4-fold higher
compared with wild-types (49).
The Val432Leu variant increases CYP1B1 catalytic ability, with a
subsequent elevation in 4-OHE1(E2) formation
(50). The Asn453Ser polymorphism
is associated with decreased levels of CYP1B1 cellular protein,
which is associated with a reduced risk of breast cancer in
postmenopausal women (51).
However, the effect of CYP1B1 polymorphisms in breast cancer
etiology remains controversial. It has been reported that there are
no associations between Arg48Gly, Ala119Ser, Val432Leu and
Asn453Ser and breast cancer risk in Polish population, but another
study identified that Arg48Gly, Ala119Ser and Val432Leu variants
were associated with increased breast cancer risk in the Polish
population (52,53). A case-control study reported that
Leu432Val and Val432Val genotypes significantly increased breast
cancer risk (50). Jiao et
al (54) also identified that
CYP1B1 432 Val variants appear to be a factor for susceptibility to
breast cancer.
In contrast to 4-OHE1(E2),
2-OHE1(E2) is weakly carcinogenic or has
protective activity. An increase in CYP1A1 activity directs CEs
toward 2-OHE1(E2) and away from the genotoxic
4-OHE1(E2) (11,19).
T3801C, T3205C, A2455 G and C2453A variants in CYP1A1 have been
studied in regard to their potential implication in breast cancer
risk. The two former variants are located in the 3′-noncoding
region, whereas the latter two lead to amino acid substitutions in
exon 7 (Ile462Val and Thr461Asp, respectively), which increase
CYP1A1 activity (55). Chen et
al (56) demonstrated that the
A2455 G G/G genotype is associated with increased breast cancer
risk in East-Asians. Additionally, Caucasian subjects carrying the
A2455 G allele also exhibited an elevated breast cancer risk
(57). T3801C, T3205C and C2453A
variants were not associated with breast cancer risk (57).
COMT is a phase II protective enzyme in that
methylation of the catechol metabolites blocks oxidative metabolism
to reactive quinones, and thus is protective against formation of
the depurinating adducts and ROS. A widely studied single
nucleotide polymorphism (SNP) in exon 4 results in the amino acid
substitution of Val with Met, termed Val158Met (16). Compared with wild-types, the Met158
variant demonstrates thermo instability; thus, COMT activity is
reduced in cells expressing Met158 (58). Previous studies have identified
that the Val158Met variant affects protein stability, and altered
conformation renders it more susceptible to recognition by the
cellular protein degradation processes, thus reducing cell activity
(58,59). Meta-analyses have examined the
influence of COMT on breast cancer incidence in women. The results
suggested that a Val158Met polymorphism in the COMT gene may be a
risk factor for breast cancer in the Chinese population (60,61).
However, Li et al (62)
suggested that the COMT Val158Met polymorphism is not a risk factor
for breast cancer in the Asian population.
However, numerous studies have been performed to
investigate the association of these polymorphisms with
susceptibility to breast cancer. However, the results of these
studies remain conflicting. Together, these results demonstrate
that without some measure of enzyme expression or activity, it is
difficult to predict and interpret results from these types of
SNP-association studies.
Conclusion
Increasing studies have identified that excessive
exposure to estrogens is associated with increased breast cancer
risk. Results from in vivo and in vitro studies have
indicated that oxidative metabolism of estrogens serves a major
role in the initiation of breast cancer. The oxidative metabolism
of estrogens to reactive quinones causes both formation of
depurinating adducts and production of ROS, which are associated
with breast cancer progression. The regulation of metabolism
enzymes, which are responsible for estrogen metabolism, are
critical for the homeostasis of estrogen. SNPs in enzymes may
influence the risk of breast cancer, but the results remain
conflicting. Future investigations into the role of estrogen
metabolism, phase I and phase II involvement in estrogen
metabolism, and their SNPs are required to measure specific
biomarkers of metabolites. This may involve determination of the
levels of the specific adenine and guanine DNA adducts and markers
of oxidative DNA damage, detectable in urine and plasma. Through
this approach, the role of SNPs in the formation and inactivation
of enzymes, including CYP1A1, CYP1B1 and COMT, maybe
determined.
Acknowledgements
This review was supported by the National Scientific
Foundation of China (no. 81603201), and the Funds for outstanding
young scholars in Chongqing Medical University (no.
CYYQ201401).
References
|
1
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics. CA Cancer J Clin. 61:409–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Santen RJ, Boyd NF, Chlebowski RT,
Cummings S, Cuzick J, Dowsett M, Easton D, Forbes JF, Key T,
Hankinson SE, et al: Critical assessment of new risk factors for
breast cancer: Considerations for development of an improved risk
prediction model. Endocr Relat Cancer. 14:169–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hofseth LJ, Raafat AM, Osuch JR, Pathak
DR, Slomski CA and Haslam SZ: Hormone replacement therapy with
estrogen or estrogen plus medroxyprogesterone acetate is associated
with increased epithelial proliferation in the normal
postmenopausal breast. J Clin Endocrinol Metab. 84:4559–4565. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cavalieri E and Rogan E: The molecular
etiology and prevention of estrogen-initiated cancers: Ockham's
Razor: Pluralitas non est ponenda sine necessitate. Plurality
should not be posited without necessity. Mol Aspects Med. 36:1–55.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Devanesan P, Santen RJ, Bocchinfuso WP,
Korach KS, Rogan EG and Cavalieri E: Catechol estrogen metabolites
and conjugates in mammary tumors and hyperplastic tissue from
estrogen receptor-alpha knock-out (ERKO)/Wnt-1 mice: Implications
for initiation of mammary tumors. Carcinogenesis. 22:1573–1576.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cavalieri EL, Stack DE, Devanesan PD,
Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD,
Gross ML, Gooden JK, et al: Molecular origin of cancer: Catechol
estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl
Acad Sci USA. 94:10937–10942. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li KM, Todorovic R, Devanesan P,
Higginbotham S, Köfeler H, Ramanathan R, Gross ML, Rogan EG and
Cavalieri EL: Metabolism and DNA binding studies of
4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female
ACI rat mammary gland in vivo. Carcinogenesis. 25:289–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cavalieri EL and Rogan EG: Depurinating
estrogen-DNA adducts in the etiology and prevention of breast and
other human cancers. Future Oncol. 6:75–91. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fussell KC, Udasin RG, Smith PJ, Gallo MA
and Laskin JD: Catechol metabolites of endogenous estrogens induce
redox cycling and generate reactive oxygen species in breast
epithelial cells. Carcinogenesis. 32:1285–1293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Valko M, Rhodes CJ, Moncol J, Izakovic M
and Mazur M: Free radicals, metals and antioxidants in oxidative
stress-induced cancer. Chem Biol Interact. 160:1–40. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee AJ, Cai MX, Thomas PE, Conney AH and
Zhu BT: Characterization of the oxidative metabolites of
17beta-estradiol and estrone formed by 15 selectively expressed
human cytochrome p450 isoforms. Endocrinology. 144:3382–3398. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jefcoate CR, Liehr JG, Santen RJ, Sutter
TR, Yager JD, Yue W, Santner SJ, Tekmal R, Demers L, Pauley R, et
al: Tissue-specific synthesis and oxidative metabolism of
estrogens. J Natl Cancer Inst Monogr. 95–112. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rahman M, Lax SF, Sutter CH, Tran QT,
Stevens GL, Emmert GL, Russo J, Santen RJ and Sutter TR: CYP1B1 is
not a major determinant of the disposition of aromatase inhibitors
in epithelial cells of invasive ductal carcinoma. Drug Metab
Dispos. 36:963–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pruthi S, Yang L, Sandhu NP, Ingle JN,
Beseler CL, Suman VJ, Cavalieri EL and Rogan EG: Evaluation of
serum estrogen-DNA adducts as potential biomarkers for breast
cancer risk. J Steroid Biochem Mol Biol. 132:73–79. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salama SA, Kamel M, Awad M, Nasser AH,
Al-Hendy A, Botting S and Arrastia C: Catecholestrogens induce
oxidative stress and malignant transformation in human endometrial
glandular cells: Protective effect of catechol-O-methyltransferase.
Int J Cancer. 123:1246–1254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yager JD: Catechol-O-methyltransferase:
Characteristics, polymorphisms and role in breast cancer. Drug
Discov Today Dis Mech. 9:e41–e46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lavigne JA, Goodman JE, Fonong T, Odwin S,
He P, Roberts DW and Yager JD: The effects of
catechol-O-methyltransferase inhibition on estrogen metabolite and
oxidative DNA damage levels in estradiol-treated MCF-7 cells.
Cancer Res. 61:7488–7494. 2001.PubMed/NCBI
|
|
18
|
Zahid M, Saeed M, Lu F, Gaikwad N, Rogan E
and Cavalieri E: Inhibition of catechol-O-methyltransferase
increases estrogen-DNA adduct formation. Free Radic Biol Med.
43:1534–1540. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu BT and Conney AH: Is
2-methoxyestradiol an endogenous estrogen metabolite that inhibits
mammary carcinogenesis? Cancer Res. 58:2269–2277. 1998.PubMed/NCBI
|
|
20
|
Gaikwad NW, Rogan EG and Cavalieri EL:
Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen
ortho-quinones. Free Radic Biol Med. 43:1289–1298. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cavalieri E, Rogan E and Chakravarti D:
The role of endogenous catechol quinones in the initiation of
cancer and neurodegenerative diseases. Methods Enzymol.
382:293–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cavalieri EL and Rogan EG: A unifying
mechanism in the initiation of cancer and other diseases by
catechol quinones. Ann N Y Acad Sci. 1028:247–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stack DE, Li G, Hill A and Hoffman N:
Mechanistic insights into the Michael addition of deoxyguanosine to
catechol estrogen-3,4-quinones. Chem Res Toxicol. 21:1415–1425.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bolton JL and Shen L: p-Quinone methides
are the major decomposition products of catechol estrogen
o-quinones. Carcinogenesis. 17:925–929. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zahid M, Kohli E, Saeed M, Rogan E and
Cavalieri E: The greater reactivity of estradiol-3,4-quinone vs
estradiol-2,3-quinone with DNA in the formation of depurinating
adducts: Implications for tumor-initiating activity. Chem Res
Toxicol. 19:164–172. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu F, Zahid M, Saeed M, Cavalieri EL and
Rogan EG: Estrogen metabolism and formation of estrogen-DNA adducts
in estradiol-treated MCF-10F cells. The effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin induction and
catechol-O-methyltransferase inhibition. J Steroid Biochem Mol
Biol. 105:150–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu F, Zahid M, Wang C, Saeed M, Cavalieri
EL and Rogan EG: Resveratrol prevents estrogen-DNA adduct formation
and neoplastic transformation in MCF-10F cells. Cancer Prev Res.
1:135–145. 2008. View Article : Google Scholar
|
|
28
|
Saeed M, Rogan E, Fernandez SV, Sheriff F,
Russo J and Cavalieri E: Formation of depurinating N3Adenine and
N7Guanine adducts by MCF-10F cells cultured in the presence of
4-hydroxyestradiol. Int J Cancer. 120:1821–1824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Russo J and Russo IH: Genotoxicity of
steroidal estrogens. Trends Endocrinol Metab. 15:211–214. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Russo J, Lareef M Hasan, Balogh G, Guo S
and Russo IH: Estrogen and its metabolites are carcinogenic agents
in human breast epithelial cells. J Steroid Biochem Mol Biol.
87:1–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lareef MH, Garber J, Russo PA, Russo IH,
Heulings R and Russo J: The estrogen antagonist ICI-182-780 does
not inhibit the transformation phenotypes induced by
17-beta-estradiol and 4-OH estradiol in human breast epithelial
cells. Int J Oncol. 26:423–429. 2005.PubMed/NCBI
|
|
32
|
Yue W, Santen RJ, Wang JP, Li Y, Verderame
MF, Bocchinfuso WP, Korach KS, Devanesan P, Todorovic R, Rogan EG
and Cavalieri EL: Genotoxic metabolites of estradiol in breast:
Potential mechanism of estradiol induced carcinogenesis. J Steroid
Biochem Mol Biol. 86:477–486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santen R, Cavalieri E, Rogan E, Russo J,
Guttenplan J, Ingle J and Yue W: Estrogen mediation of breast tumor
formation involves estrogen receptor-dependent, as well as
independent, genotoxic effects. Ann NY Acad Sci. 1155:132–140.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chakravarti D, Mailander PC, Li KM,
Higginbotham S, Zhang HL, Gross ML, Meza JL, Cavalieri EL and Rogan
EG: Evidence that a burst of DNA depurination in SENCAR mouse skin
induces error-prone repair and forms mutations in the H-ras gene.
Oncogene. 20:7945–7953. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mailander PC, Meza JL, Higginbotham S and
Chakravarti D: Induction of A.T to G.C mutations by erroneous
repair of depurinated DNA following estrogen treatment of the
mammary gland of ACI rats. J Steroid Biochem Mol Biol. 101:204–215.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao Z, Kosinska W, Khmelnitsky M,
Cavalieri EL, Rogan EG, Chakravarti D, Sacks PG and Guttenplan JB:
Mutagenic activity of 4-hydroxyestradiol, but not
2-hydroxyestradiol, in BB rat2 embryonic cells and the mutational
spectrum of 4-hydroxyestradiol. Chem Res Toxicol. 19:475–479. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gaikwad NW, Yang L, Muti P, Meza JL,
Pruthi S, Ingle JN, Rogan EG and Cavalieri EL: The molecular
etiology of breast cancer: Evidence from biomarkers of risk. Int J
Cancer. 122:1949–1957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gaikwad NW, Yang L, Pruthi S, Ingle JN,
Sandhu N, Rogan EG and Cavalieri EL: Urine biomarkers of risk in
the molecular etiology of breast cancer. Breast Cancer. 3:1–8.
2009.PubMed/NCBI
|
|
39
|
Cavalieri E, Chakravarti D, Guttenplan J,
Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R
and Sutter T: Catechol estrogen quinones as initiators of breast
and other human cancers: Implications for biomarkers of
susceptibility and cancer prevention. Biochim Biophys Acta.
1766:63–78. 2006.PubMed/NCBI
|
|
40
|
Tian H, Gao Z, Wang G, Li H and Zheng J:
Estrogen potentiates reactive oxygen species (ROS) tolerance to
initiate carcinogenesis and promote cancer malignant
transformation. Tumour Biol. 37:141–150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Okoh VO, Felty Q, Parkash J, Poppiti R and
Roy D: Reactive oxygen species via redox signaling to PI3K/AKT
pathway contribute to the malignant growth of 4-hydroxy
estradiol-transformed mammary epithelial cells. PLoS One.
8:e542062013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Felty Q, Xiong WC, Sun D, Sarkar S, Singh
KP, Parkash J and Roy D: Estrogen-induced mitochondrial reactive
oxygen species as signal-transducing messengers. Biochemistry.
44:6900–6909. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ishikawa K, Takenaga K, Akimoto M,
Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y and Hayashi
J: ROS-generating mitochondrial DNA mutations can regulate tumor
cell metastasis. Science. 320:661–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Doan VD, Gagnon S and Joseph V: Prenatal
blockade of estradiol synthesis impairs respiratory and metabolic
responses to hypoxia in newborn and adult rats. Am J Physiol Regul
Integr Comp Physiol. 287:R612–R618. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tan DJ, Bai RK and Wong LJ: Comprehensive
scanning of somatic mitochondrial DNA mutations in breast cancer.
Cancer Res. 62:972–976. 2002.PubMed/NCBI
|
|
46
|
Kim DW, Sovak MA, Zanieski G, Nonet G,
Romieu-Mourez R, Lau AW, Hafer LJ, Yaswen P, Stampfer M, Rogers AE,
et al: Activation of NF-kappaB/Rel occurs early during neoplastic
transformation of mammary cells. Carcinogenesis. 21:871–879. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Park SA, Na HK, Kim EH, Cha YN and Surh
YJ: 4-hydroxyestradiol induces anchorage-independent growth of
human mammary epithelial cells via activation of IkappaB kinase:
Potential role of reactive oxygen species. Cancer Res.
69:2416–2424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Benhar M, Engelberg D and Levitzki A: ROS,
stress-activated kinases and stress signaling in cancer. EMBO Rep.
3:420–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hanna IH, Dawling S, Roodi N, Guengerich
FP and Parl FF: Cytochrome P450 1B1 (CYP1B1) pharmacogenetics:
Association of polymorphisms with functional differences in
estrogen hydroxylation activity. Cancer Res. 60:3440–3444.
2000.PubMed/NCBI
|
|
50
|
Delort L, Satih S, Kwiatkowski F, Bignon
YJ and Bernard-Gallon DJ: Evaluation of breast cancer risk in a
multigenic model including low penetrance genes involved in
xenobiotic and estrogen metabolisms. Nutr Cancer. 62:243–251. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bandiera S, Weidlich S, Harth V, Broede P,
Ko Y and Friedberg T: Proteasomal degradation of human CYP1B1:
Effect of the Asn453Ser polymorphism on the post-translational
regulation of CYP1B1 expression. Mol Pharmacol. 67:435–443. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matyjasik J, Cybulski C, Masojć B,
Jakubowska A, Serrano-Fernandez P, Górski B, Debniak T, Huzarski T,
Byrski T, Gronwald J, et al: CYP1B1 and predisposition to breast
cancer in Poland. Breast Cancer Res Treat. 106:383–388. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gaudet MM, Chanock S, Lissowska J, Berndt
SI, Yang XR, Peplonska B, Brinton LA, Welch R, Yeager M,
Bardin-Mikolajczak A, et al: Genetic variation of Cytochrome P450
1B1 (CYP1B1) and risk of breast cancer among Polish women.
Pharmacogenet Genomics. 16:547–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jiao H, Liu C, Guo W, Peng L, Chen Y and
Martin FL: Association of CYP1B1 Polymorphisms with Breast Cancer:
A Case-Control Study in the Han Population in Ningxia Hui
Autonomous Region, P. R. China. Biomark Insights. 5:21–27.
2010.PubMed/NCBI
|
|
55
|
Masson LF, Sharp L, Cotton SC and Little
J: Cytochrome P-450 1A1 gene polymorphisms and risk of breast
cancer: A HuGE review. Am J Epidemiol. 161:901–915. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen C, Huang Y, Li Y, Mao Y and Xie Y:
Cytochrome P450 1A1 (CYP1A1) T3801C and A2455G polymorphisms in
breast cancer risk: A meta-analysis. J Hum Genet. 52:423–435. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sergentanis TN and Economopoulos KP: Four
polymorphisms in cytochrome P450 1A1 (CYP1A1) gene and breast
cancer risk: A meta-analysis. Breast Cancer Res Treat. 122:459–469.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shield AJ, Thomae BA, Eckloff BW, Wieben
ED and Weinshilboum RM: Human catechol O-methyltransferase genetic
variation: Gene resequencing and functional characterization of
variant allozymes. Mol Psychiatry. 9:151–160. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Doyle AE and Yager JD:
Catechol-O-methyltransferase: Effects of the val108met polymorphism
on protein turnover in human cells. Biochim Biophys Acta.
1780:27–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tian C, Liu L, Yang X, Wu H and Ouyang Q:
The Val158Met polymorphism in the COMT gene is associated with
increased cancer risks in Chinese population. Tumour Biol.
35:3003–3008. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wan GX, Cao YW, Li WQ, Li YC and Li F: The
catechol-O-methyltransferase Val158Met polymorphism contributes to
the risk of breast cancer in the chinese population: an updated
meta-analysis. J Breast Cancer. 17:149–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li K, Li W and Zou H:
Catechol-O-methyltransferase Val158Met polymorphism and breast
cancer risk in Asian population. Tumour Biol. 35:2343–2350. 2014.
View Article : Google Scholar : PubMed/NCBI
|