Introduction
According to GLOBOCAN, lung cancer is among the most
common types of cancer worldwide (1). Non-small cell lung cancer (NSCLC)
comprises 85% of all types of lung cancer. NSCLC is relatively
insensitive to chemotherapy compared with small cell lung cancer
(2,3). Despite recent advances in the
chemotherapy of NSCLC, the therapeutic efficacy of currently
available agents remains unsatisfactory and the majority of
patients with advanced NSCLC are refractory to medication (4,5).
Therefore, the development of novel therapeutic strategies
characterized by higher efficacy is imperative for the treatment of
patients with NSCLC.
Gefitinib (ZD1839, Iressa) is a chemotherapeutic
agent that was approved by the US Food and Drug Administration in
2003 for the treatment of patients with local or metastatic NSCLC,
following failure of treatment with platinum- and docetaxel-based
chemotherapeutic schemes (6).
Gefitinib is an effective and well-tolerated agent; however, some
patients with NSCLC are insensitive to gefitinib chemotherapy
(7). Dihydroartemisinin (DHA) is
the active metabolite of all artemisinin compounds and is widely
used as an antimalarial therapeutic agent. In addition, previous
studies have reported that DHA exhibited pronounced anticancer
effects in breast, colorectal, cervical and lung cancer (8–11),
whereas in combination with chemotherapeutic agents it exerted
synergistic effects in the treatment of various types of cancer
(12,13).
The NCI-H1975 human NSCLC cell line was established
in July 1988, and is characterized by much lower sensitivity to
gefitinib than other human NSCLC lines, such as A431 and H3255
(14). The present study aimed to
ascertain whether the combination of gefitinib with DHA have a
higher therapeutic efficacy than currently used gefitinib
monotherapy. Therefore, the present study investigated the
combination of gefitinib with DHA to improve the chemotherapeutic
sensitivity of NCI-H1975 cell, and explored the molecular
mechanisms underlying actions of the drugs.
Materials and methods
Cell culture
The NCI-H1975 human lung adenocarcinoma cell line
was purchased from American Type Culture Collection (Manassas, VA,
USA) and cultured in RPMI-1640 medium (catalog no. SH30809.01;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA), supplemented
with 10% fetal bovine serum (FBS; HyClone; GE Healthcare Life
Sciences). NCI-H1975 cells were maintained at 37°C in a humidified
5% CO2 atmosphere.
Cell Counting kit 8 (CCK8) assay
Cellular viability was evaluated using CCK8
(Beyotime Institute of Biotechnology, Haimen, China), according to
the manufacturer's protocol. Briefly, NCI-H1975 cells were seeded
at 5×103 into 96-well plates containing RPMI 1640
medium, supplemented with 10% FBS and incubated for 24 h. When
NCI-H1975 cells reached 80% confluence they were treated with
gefitinib (catalog no. HY-50895; ApexBio; MedChemExpress, New
Jersey, USA) or DHA (catalog no.S2290; Selleck Chemicals, Houston,
TX, USA) for 24 h. Subsequently, viable cells were detected using
CCK8. The absorbance of each sample at 450 nm was measured using a
microplate reader (Tecan Group Ltd., Salzburg, Austria).
Terminal deoxynucleotidyl transferase
dUTP nick-end labeling (TUNEL) assay
NCI-H1975 cells were incubated with DHA and
gefitinib for 24 h to induce cell apoptosis. Cellular apoptosis was
assessed via evaluating DNA fragmentation in NCI-H1975 cells, using
a Cell Death Detection kit (catalog no. 11684817910; Roche
Diagnostics, Basel, Switzerland), as previously described (15). Briefly, cells were fixed for 24 h
with 4% paraformaldehyde at room temperature, washed with PBS and
permeabilized for 30 min with 1% Triton X-100 at 4°C. Subsequently,
cells were treated with the terminal deoxynucleotidyl
transferase-labeled nucleotide mix and maintained at 37°C for 1 h
in the dark. Slides were rinsed and counterstained (TE 2000-U;
Nikon Corporation, Tokyo, Japan) for 15 min with 10 mg/ml
4,6-diamidino-2-phenylindole (DAPI) at 37°C.
Cell cycle analysis
The effects of gefitinib and DHA on cell cycle
distribution were assessed using flow cytometric analysis of the
DNA content of NCI-H1975 cells, following staining with propidium
iodide (PI). Briefly, NCI-H1975 cells were seeded in 6-well plates
and allowed to attach overnight at 37°C. Fresh complete RPMI-1640
medium was then added, containing the desired concentrations of
gefitinib (10 µM) or DHA (10 µM), and cells were incubated for 24 h
at 37°C. Cells were then washed with PBS and fixed in 70% ethanol
overnight at 4°C. Subsequently, cells were treated at room
temperature with 80 mg/ml RNaseA and 50 mg/ml PI for 30 min, and
analyzed (ModFitLT version 2.0; Verity Software House Inc.,
Topsham, ME, USA) using a Coulter® Epics®
XL™ Flow Cytometer (Beckman Coulter, Inc., Brea, CA,
USA).
Cell migration and invasion
analysis
To assess the effect of chemotherapeutics on the
migratory and invasive capabilities of NCI-H1975 cells, cells were
incubated with gefitinib (10 µM) or DHA (10 µM) at 37°C for 24 h.
NCI-H1975 cells were harvested, resuspended in RPMI-1640 medium and
seeded at 5×105 into the upper chambers of Transwell
inserts with 8 µm pore size (EMD Millipore, Billerica, MA, USA).
The lower chambers contained culture medium supplemented with 10%
FBS as a chemoattractant. Following incubation at 37°C in 5%
CO2 for 24 h, non-migrated cells on the top of the
membrane were removed with cotton swabs. Cells that had migrated to
the lower membrane were fixed with 95% ethanol, stained for 1 h
with 0.2% crystal violet (Sigma-Aldrich; Merck KGaA) and counted
under a light microscope. Each experiment was performed in
triplicate. Matrigel-coated 24-well Boyden chambers (EMD Millipore)
were used in the cell invasion assay. Cells were seeded in the
upper chamber in serum-free medium. The lower chambers contained
culture medium supplemented with 10% FBS as a chemoattractant.
Following incubation at 37°C in 5% CO2 for 24 h,
non-invasion cells on the top of the membrane were removed with
cotton swabs. After 24 h, cells that had invaded to the lower
membrane were fixed, stained with 0.2% crystal violet
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 1 h, and
counted (Image-Pro Plus 6.0, Media Cybernetics, Inc., Rockville,
MD, USA) under a light microscope.
Western blot analysis
Total protein was extracted from NCI-H1975 cells.
NCI-H1975 cells were digested with lysis buffer (catalog no.
P0013B; Beyotime Institute of Biotechnology). Briefly, the
NCI-H1975 cells were harvested and centrifuged at 12,000 × g at 4°C
for 15 min and the supernatant was collected for the western blot
experiment. Total protein concentration in the supernatant was
determined using a Bicinchoninic Acid assay (BCA; Beyotime
Institute of Biotechnology). Briefly, the BCA working solution was
prepared at a ratio of 50:1 with BCA reagent A and BCA reagent B,
respectively, and mixed thoroughly. Following this, 200 µl of BCA
working solution was added to each of the 96-well plates containing
protein samples, which were incubated at 37°C for 30 min. The
absorbance was measured at a wavelength of 562 nm by a microplate
reader, and the protein concentration of the sample was calculated
from the standard curve. A total of 60–80 µg extracted protein
samples were separated by 10–15% SDS-PAGE and transferred onto
nitrocellulose membranes. The membranes were blocked with 5%
non-fat milk at room temperature for 1 h. Subsequently, the
membranes were incubated at 4°C overnight with the following
primary antibodies: Anti-mechanistic target of rapamycin (mTOR;
1:1,000; catalog no. 9964; Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-phosphorylated (p)-mTOR (1:1,000),
anti-cyclin B1, anti-B-cell lymphoma 2 (Bcl-2; 1:1,000; catalog no.
2872; Cell Signaling Technology, Inc.) anti-Bcl-2-associated X
protein (Bax; 1:1,000; catalog no. 2774; Cell Signaling Technology,
Inc.) anti-signal transducer and activator of transcription (STAT)
3 (1:1,000; catalog no. ab119352; Abcam, Cambridge, MA, USA),
anti-p-STAT3 (1:1,000; catalog no. ab30647; Abcam),
anti-cyclin-dependent kinase (Cdk) 1 (1:1,000; catalog no. ab18;
Abcam) and anti-β-actin (1:4,000; KangChen Bio-tech, Inc.,
Shanghai, China). Membranes were washed three times for 10 min in
PBS containing 0.5% Tween-20, and incubated with Alexa
Fluor™-conjugated secondary antibody (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 1 h at room temperature.
IRDye®800CW Goat anti-Rabbit (catalog nos. 926-32211,
C60607-15; LI-COR Biosciences, Lincoln, NE, USA).
IRDye®800CW Goat anti-Mouse (catalog nos. 926-32210,
C60405-05; LI-COR Biosciences). The bands were visualized using the
Odyssey Imaging system (LI-COR Biosciences) and semi-quantified
using the Odyssey software version 3.0. Relative protein expression
was normalized to β-actin, which was used as an internal
control.
Statistical analysis
The statistical significance of the difference
between groups was assessed by one-way analysis of variance,
followed by Dunnett's test for multiple comparisons. Data are
expressed as the mean ± standard error of the mean. P<0.05 was
considered to indicate a statistically significant difference.
Results
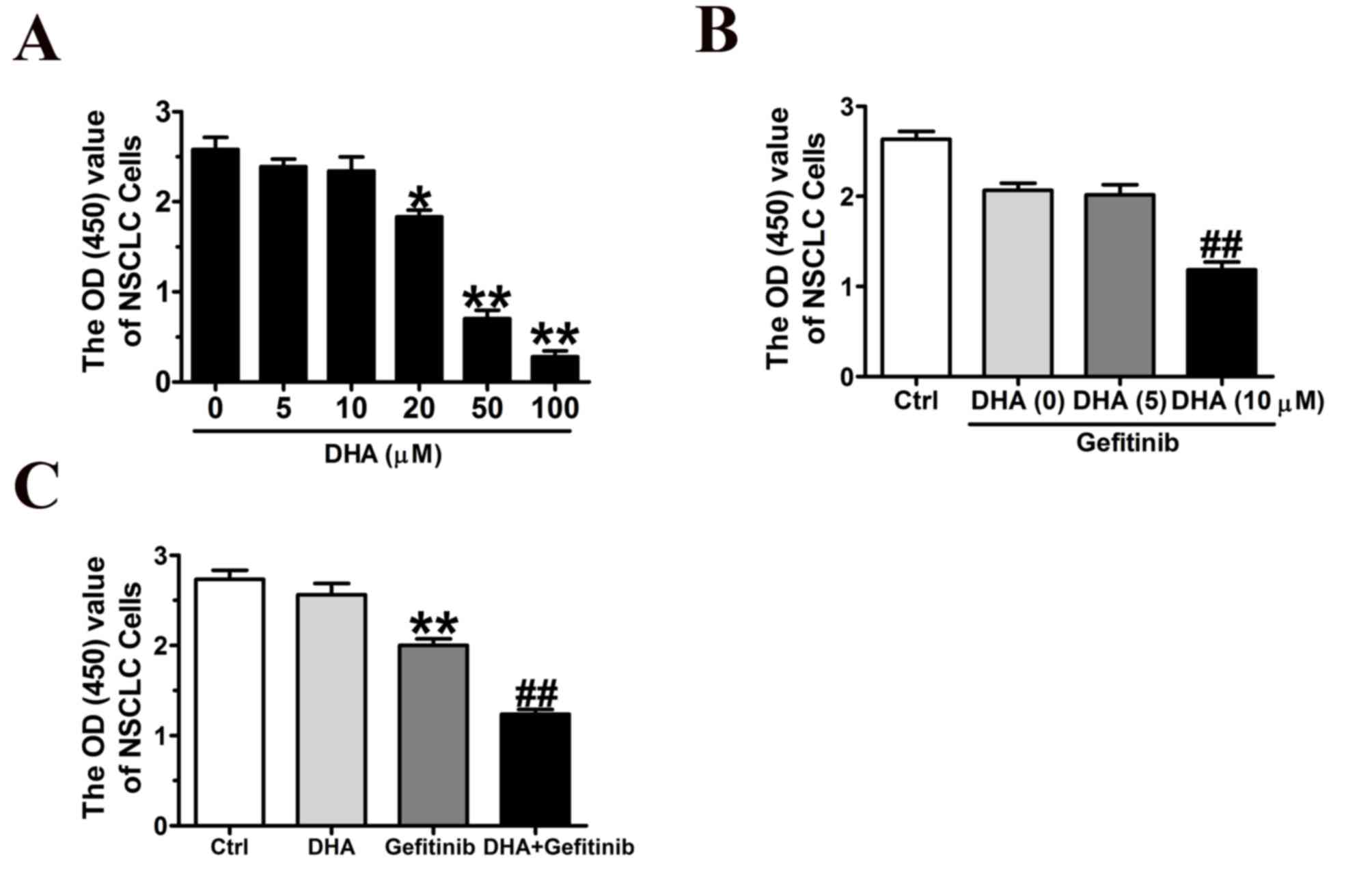
DHA inhibits NCI-H1975 cellular
proliferation
To investigate whether DHA may inhibit the growth of
lung cancer cells, CCK8 assay was performed. Results demonstrated
that DHA significantly inhibited NCI-H1975 cellular viability in a
dose-dependent manner (Fig. 1A).
The combination of DHA (10 µM) and gefitinib (10 µM) exhibited
significantly enhanced inhibitory effects on NCI-H1975 cells
compared with gefitinib alone (Fig.
1B); however, a lower dose of DHA (5 µM), did not appear to
potentiate the effects of gefitinib (Fig. 1B).
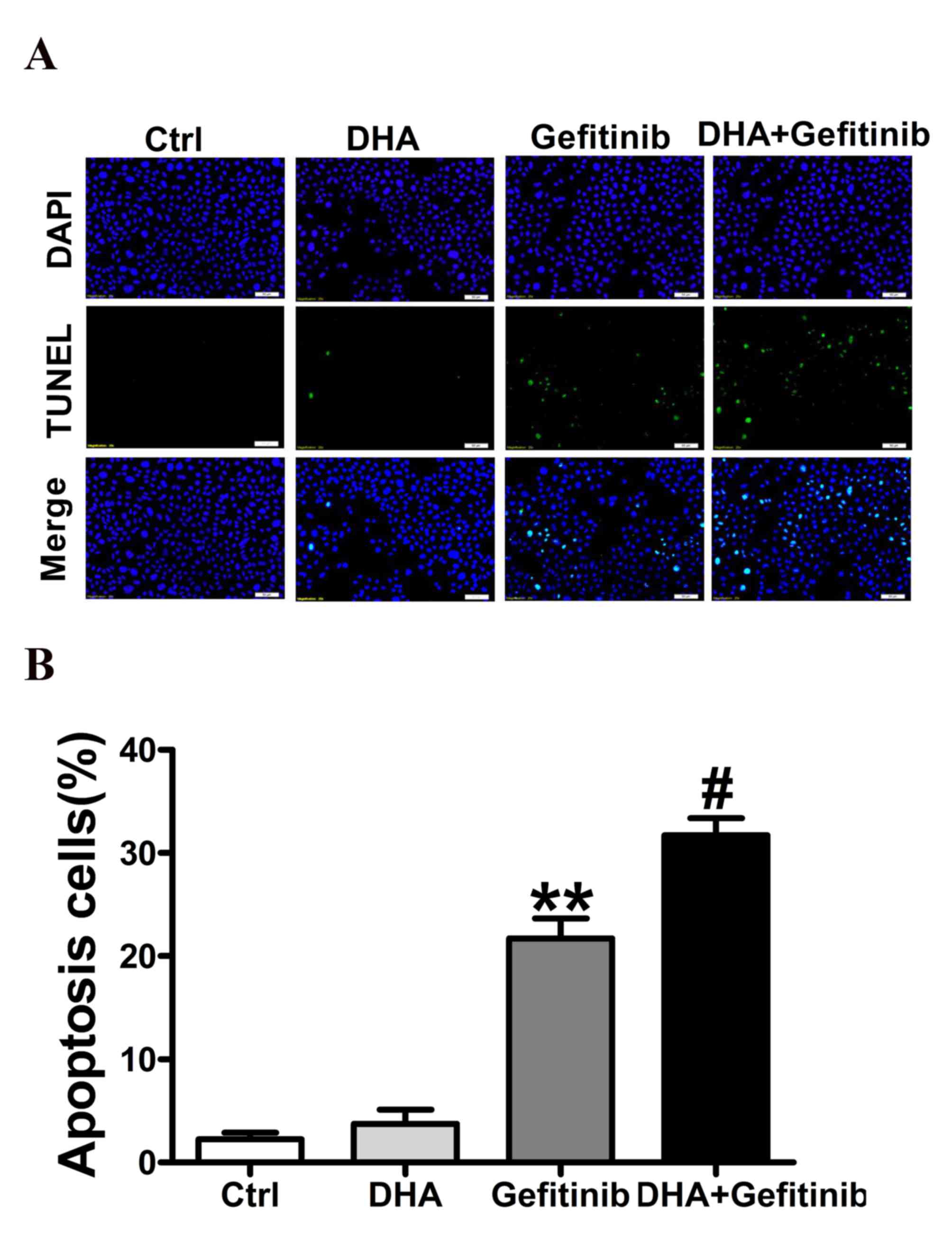
DHA potentiates the gefitinib-induced
apoptosis of NCI-H1975 cells
In accordance with previous studies (16–19),
Co-treatment of DHA with gefitinib significantly increased cancer
cell apoptosis. Treatment with a combination of DHA (10 µM) and
gefitinib resulted in significantly higher cancer cell apoptotic
rate compared with treatment with gefitinib alone. DHA administered
alone demonstrated no marked effect on cell apoptosis (Fig. 2B). Representative images were
indicated in Fig. 2A. These
results suggested that DHA may enhance the proapoptotic effects of
gefitinib, and therefore may have potential as an agent for
adjuvant therapy in NSCLC.
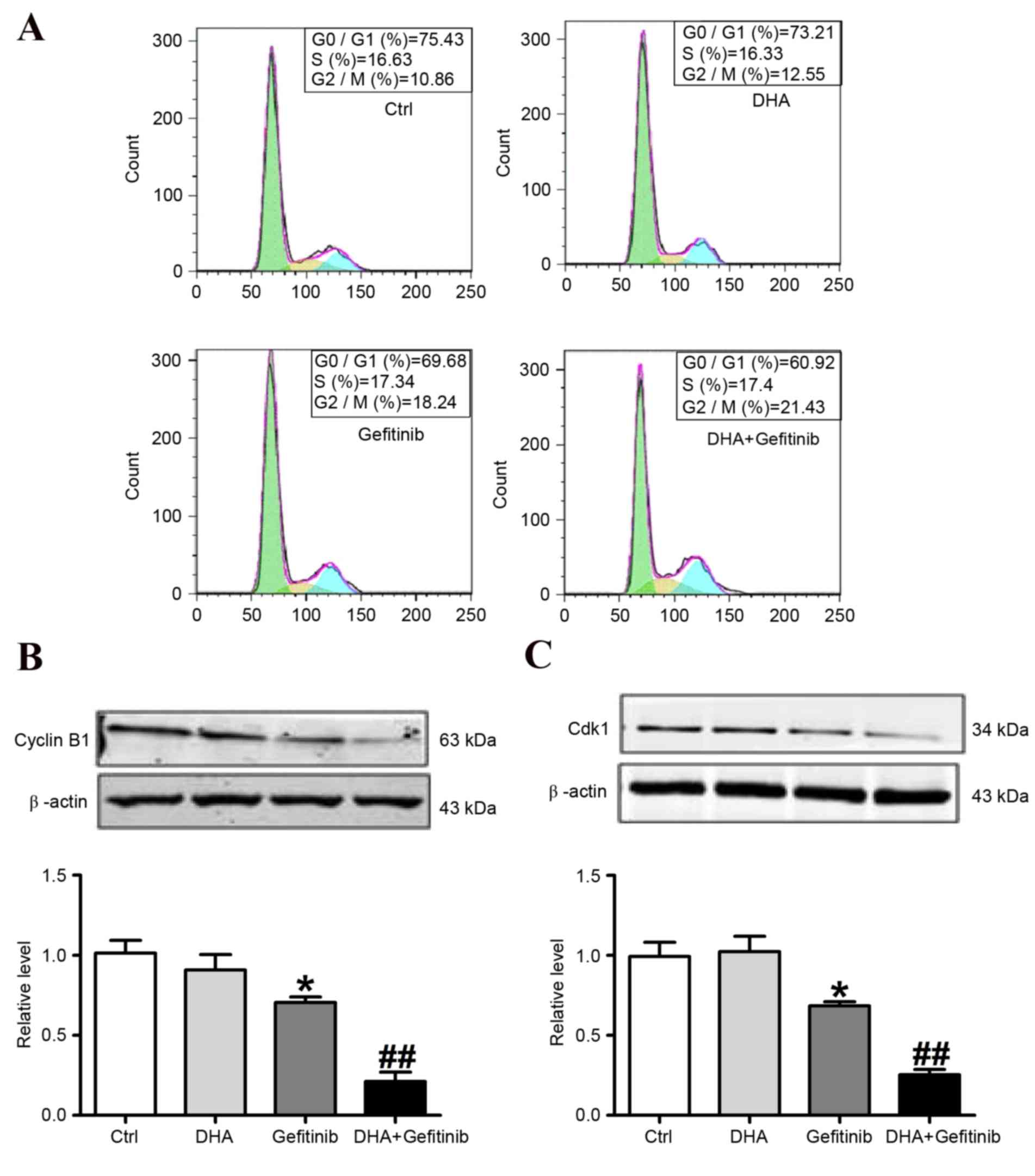
DHA potentiates the gefitinib-induced
downregulation of cyclin B1 and Cdk1 expression in NCI-H1975
cells
Cell cycle progression of NCI-H1975 cells is
controlled by the sequential activation of Cdks, whose activity
depends on their association with regulatory cyclins. The formation
of a Cdk1-cyclin B1 complex is crucial for the initiation of
mitosis in several organisms (20,21).
The analysis of cell cycle distribution using flow cytometry
revealed a G2/M arrest in NCI-H1975 cells treated with
gefitinib or a combination of gefitinib and DHA (Fig. 3A and Table I). To investigate the underlying
mechanisms, Cdk1 and cyclin B1 expression was assessed using
western blot analysis (Fig. 3B).
Treatment of NCI-H1975 cells with gefitinib for 24 h resulted in a
significant increase in the percentage of cells in G2/M
phase, accompanied by a decrease in the percentage of cells in
G0/G1 phase. These effects were potentiated
following DHA and gefitinib co-treatment (Fig. 3A). Furthermore, western blot
analysis demonstrated that treatment of NCI-H1975 cells with
gefitinib (10 µM) caused a significant downregulation in cyclin B1
and Cdk1 protein expression levels. Notably, DHA co-administration
significantly potentiated the effects of gefitinib on cyclin B1 and
Cdk1 downregulation (Fig. 3B and
C). These results suggested that the addition of DHA to
gefitinib chemotherapy may potentiate the downregulation of cyclin
B1 and Cdk1 protein levels and prevent Cdk1-cyclin B1 complex
formation, and thus promote G2/M phase arrest in cancer
cells.
 | Table I.Effects of DHA and gefitinib on cell
cycle distribution of NCI-H1975 cells. |
Table I.
Effects of DHA and gefitinib on cell
cycle distribution of NCI-H1975 cells.
|
| Proportion of cells
(%) |
|---|
|
|
|
|---|
| Group | G0/G1 phase | S phase | G2/M phase |
|---|
| Ctrl | 75.54±1.12 | 15.01±2.01 | 10.96±1.27 |
| DHA | 73.02±3.15 | 15.58±1.61 | 11.75±1.86 |
| Gefitinib |
67.21±1.07a | 15.94±2.46 |
18.98±1.42a |
| DHA+Gefitinib |
60.92±1.27b | 16.33±1.85 |
21.73±1.03b |
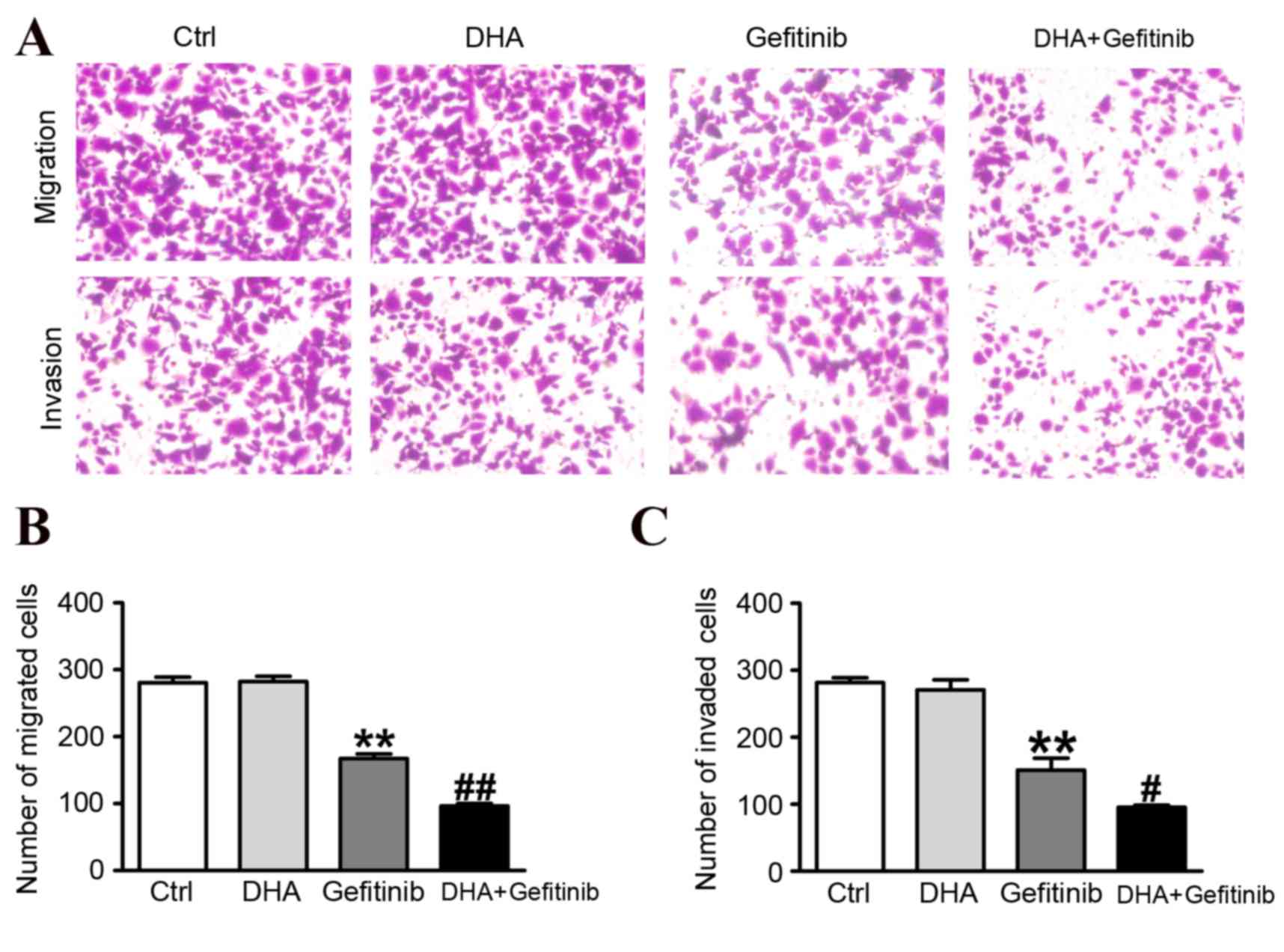
DHA enhances gefitinib-induced
inhibition of migration and invasion of NCI-H1975 cells
Representative results of the migration and invasion
assays are presented in Fig. 4A.
The migratory capabilities of NCI-H1975 cells were significantly
reduced following treatment with gefitinib, and the addition of DHA
potentiated the inhibitory actions of gefitinib (Fig. 4B). The effects of DHA
co-administration were further investigated on the invasive
capabilities of gefitinib-treated cancer cells. The number of
NCI-H1975 cells that invaded the lower chamber was significantly
reduced following treatment with a combination of DHA and gefitinib
compared with treatment with gefitinib alone (Fig. 4C). These results demonstrated that
DHA significantly enhanced the inhibitory effects of gefitinib on
cancer cell migration and invasion.
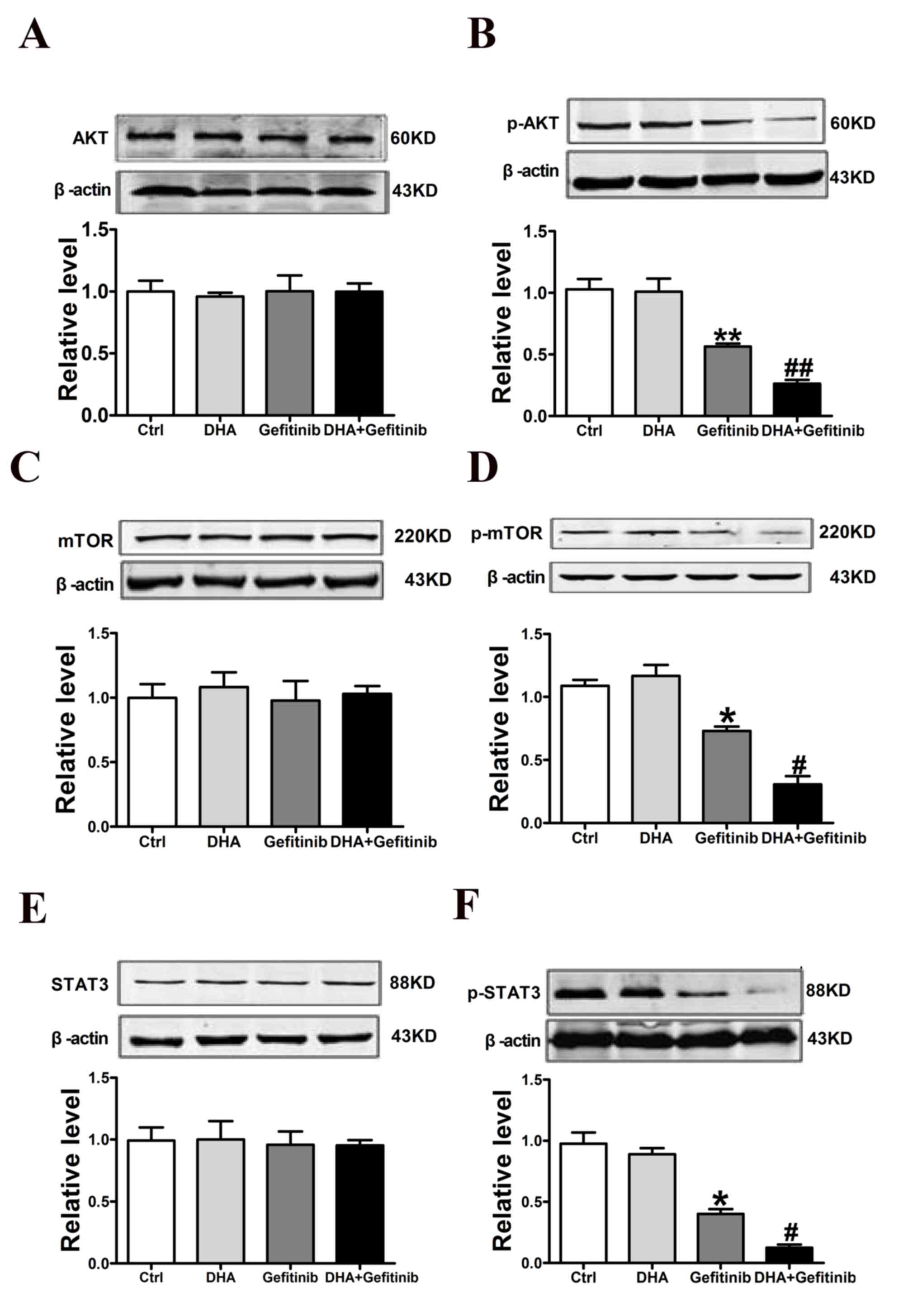
DHA enhances gefitinib-induced
downregulation of p-Akt, p-mTOR and p-STAT3 in NCI-H1975 cells
Western blot analysis revealed that p-Akt protein
expression levels were significantly downregulated following DHA
and gefitinib co-administration. The β-actin was recognized as
internal control; However, the levels of total Akt remained
unaltered (Fig. 5A and B). In
addition, DHA significantly enhanced the gefitinib-induced
downregulation of p-mTOR, whereas total mTOR levels remained
unaffected across treatment groups (Fig. 5C and D). Similarly, p- but not
total STAT3 protein expression levels were significantly
downregulated following combined treatment with DHA and gefitinib
(Fig. 5E and F). These results
suggested that DHA may enhance the inhibitory actions of gefitinib
on cellular migration and invasion possibly through the regulation
of p-AKT/p-mTOR/p-STAT3 pathways.
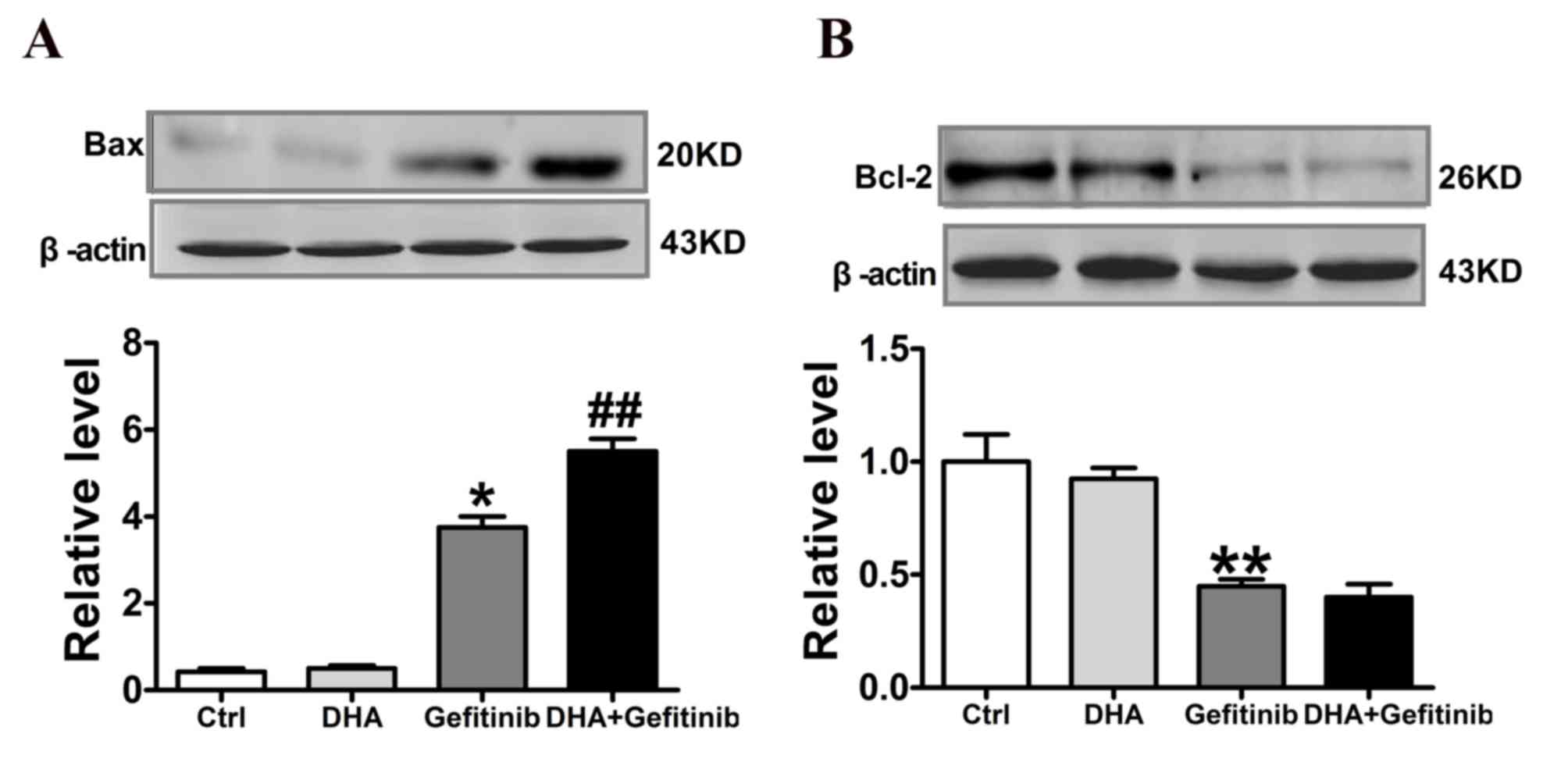
DHA enhances gefitinib-induced
upregulation of Bax and downregulation of Bcl-2 in NCI-H1975
cells
Western blot analysis revealed that Bax protein
expression levels were significantly upregulated following
treatment with gefitinib. Notably, Bax protein levels were
significantly increased following DHA co-administration compared
with cells treated with gefitinib alone (Fig. 6A). Conversely, Bcl-2 protein
expression levels were significantly downregulated following
treatment with gefitinib. Treatment with DHA and Gefitinib combined
did not produce any further significant reduction in Bcl-2 levels,
as presented in Fig. 6B.
Discussion
NSCLC is among the most common types of cancer and
one of the leading causes of cancer-associated mortality. Drug
resistance during cancer chemotherapy is a major concern in cancer
therapeutics (22). Therefore,
strategies aimed at increasing the efficacy of chemotherapy may
help improve disease prognosis and improve the patients' quality of
life. Natural products have garnered attention as a source for the
development of novel anticancer drugs. Numerous natural products
have exhibited anticancer potential and may prove useful, either as
single agents or in combination with existing antineoplastic drugs,
in the treatment of various types of cancer (23–26).
DHA is derived from artemisinin, a natural product
isolated from Artemisia apiacea. It has previously been reported
that DHA exhibited antitumor effects in several types of cancer
(27). In accordance with previous
studies, the present results demonstrated that DHA exhibited
anticancer effects in NCI-H1975 cells. DHA appeared to suppress
NCI-H1975 cellular proliferation in a concentration manner. A
prominent role has previously been reported for mTOR in cancer.
Activation of mTOR signaling pathways has been demonstrated to
contribute to the initiation and progression of tumorigenesis
(28,29). Conversely, the inhibition of mTOR
by rapamycin has been reported to enhance the chemosensitivity of
cancer cells (30), and in NSCLC
mTOR inhibition induced apoptosis and autophagy (31). In addition, it has been reported
that DHA suppressed p-mTOR activity in ovarian cancer and
rhabdomyosarcoma cells (32).
Therefore, the present study evaluated the effects of DHA on mTOR.
The result indicated that DHA inhibited the activity of p-mTOR. The
western blot results revealed that administration of DHA alone had
no effect on mTOR or p-mTOR, appearing only to potentiate the
actions of gefitinib.
It has previously been demonstrated that DHA may
enhance the efficacy of chemotherapy in the treatment of cancer
(33). Therefore, the effects of
the combination of DHA with the antineoplastic drug gefitinib were
investigated in NCI-H1975 cells. Flow cytometric analysis
demonstrated that the combination of DHA and gefitinib caused cell
cycle arrest at the G0/G1 and G2/M
phase. The percentage of cells in the G0/G1 phase was decreased by
DHA+gefitinib. which was accompanied by a downregulation in Cdk1
and cyclin B1 protein expression levels. It has previously been
demonstrated that Bcl-2 is a type of apoptosis-suppressing gene,
which promotes cell survival by inhibiting adapters needed for
activation of the proteases (caspases) (34). Bcl-2 elevation may enhance various
types of cell survival and promote cancer progression and this
activity has been observed in colon, prostate and lung cancers
(35–37). Bax encodes a dominant-inhibitor of
the Bcl-2 protein (38) and its
upregulation may promote cell apoptosis in multiple types of cancer
(38–42). Furthermore, it was revealed to
induce apoptosis in NCI-H1975 cells, as well as a decrease in Bcl-2
and increase in Bax protein expression levels. The expression ratio
of Bcl-2/Bax was also decreased (Fig.
6A and B). Notably, although gefitinib induced cell cycle
arrest and apoptosis, in combination with DHA its effects were
significantly potentiated. These results suggested that DHA may act
synergistically with gefitinib in cancer cells.
To explore the molecular mechanisms underlying the
anticancer effects of combined treatment with DHA and gefitinib,
the protein levels of Akt, mTOR and STAT3 were evaluated. The
Akt/mTOR/STAT3 signaling pathway is critical in the regulation of
cancer cell growth, migration and apoptosis (43,44).
The present results revealed that p-Akt, p-mTOR and p-STAT3 were
significantly downregulated in NCI-H1975 cells that received a
combination of DHA and gefitinib; however, total protein levels of
Akt, mTOR and STAT3 remained unaltered across treatment groups.
These results suggested that the synergistic effects of DHA and
gefitinib may be exerted through the Akt/mTOR/STAT3 pathway.
However, further experiments are required to elucidate the complex
mechanisms underlying the anticancer effects of DHA and
gefitinib.
In conclusion, the present study demonstrated that
DHA enhanced the inhibitory effects of gefitinib on cancer cell
proliferation. The combination of DHA with gefitinib significantly
inhibited the growth and promoted the apoptosis of NCI-H1975 NSCLC
cells. The mechanisms underlying the anticancer effects presently
observed may involve regulation of the Akt/mTOR/STAT3 signaling
pathway.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song L, Xiong H, Li J, Liao W, Wang L, Wu
J and Li M: Sphingosine kinase-1 enhances resistance to apoptosis
through activation of PI3K/Akt/NF-κB pathway in human non-small
cell lung cancer. Clin Cancer Res. 17:1839–1849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chemotherapy in non-small cell lung
cancer: A meta-analysis using updated data on individual patients
from 52 randomised clinical trials. Non-small Cell Lung Cancer
Collaborative Group. BMJ. 311:899–909. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwamoto Y, Mitsudomi T, Sakai K, Yamanaka
T, Yoshioka H, Takahama M, Yoshimura M, Yoshino I, Takeda M,
Sugawara S, et al: Randomized phase II study of adjuvant
chemotherapy with long-term S-1 versus cisplatin+S-1 in completely
resected stage II–IIIA non-small cell lung cancer. Clin Cancer Res.
21:5245–5252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scagliotti GV, Fossati R, Torri V, Crinò
L, Giaccone G, Silvano G, Martelli M, Clerici M, Cognetti F, Tonato
M, et al: Adjuvant Lung Project Italy/European Organisation for
Research Treatment of Cancer-Lung Cancer Cooperative Group
Investigators: Randomized study of adjuvant chemotherapy for
completely resected stage I, II, or IIIA non-small-cell lung
cancer. J Natl Cancer Inst. 95:1453–1461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cohen MH, Williams GA, Sridhara R, Chen G,
McGuinn WD Jr, Morse D, Abraham S, Rahman A, Liang C, Lostritto R,
et al: United States food and drug administration drug approval
summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res.
10:1212–1218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu CJ, Zhou L and Cai Y:
Dihydroartemisinin induces apoptosis of cervical cancer cells via
upregulation of RKIP and downregulation of bcl-2. Cancer Biol Ther.
15:279–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong Q, Chen L, Lu Q, Sharma S, Li L,
Morimoto S and Wang G: Quercetin attenuates doxorubicin
cardiotoxicity by modulating Bmi-1 expression. Br J Pharmacol.
171:4440–4454. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang S, Ma Y, Jiang J, Dai Z, Gao X, Yin
X, Xi W1 and Min W: Inhibition of urokinase-type plasminogen
activator expression by dihydroartemisinin in breast cancer cells.
Oncol Lett. 7:1375–1380. 2014.PubMed/NCBI
|
|
11
|
Zhou HJ, Zhang JL, Li A, Wang Z and Lou
XE: Dihydro-artemisinin improves the efficiency of
chemotherapeutics in lung carcinomas in vivo and inhibits murine
Lewis lung carcinoma cell line growth in vitro. Cancer Chemother
Pharmacol. 66:21–29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mi YJ, Geng GJ, Zou ZZ, Gao J, Luo XY, Liu
Y2, Li N, Li CL, Chen YQ, Yu XY2 and Jiang J: Dihydroartemisinin
inhibits glucose uptake and cooperates with glycolysis inhibitor to
induce apoptosis in non-small cell lung carcinoma cells. PLoS One.
10:e01204262015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Wei H, Zhao B, Li M, Lv W, Lv L,
Song B and Lv S: The reverse effect of X-ray irradiation on
acquired gefitinib resistance in non-small cell lung cancer cell
line NCI-H1975 in vitro. J Mol Histol. 45:641–652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan Y, Chen M, Meng J, Yu L, Tu Y, Wan L,
Fang K and Zhu W: Arsenic trioxide and resveratrol show synergistic
anti-leukemia activity and neutralized cardiotoxicity. PLoS One.
9:e1058902014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao X, Zhong H, Wang R, Liu D, Waxman S,
Zhao L and Jing Y: Dihydroartemisinin and its derivative induce
apoptosis in acute myeloid leukemia through Noxa-mediated pathway
requiring iron and endoperoxide moiety. Oncotarget. 6:5582–5596.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao C, Gao W and Chen T: Synergistic
induction of apoptosis in A549 cells by dihydroartemisinin and
gemcitabine. Apoptosis. 19:668–681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H and Kinoshita
I: Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yusuf SW, Kim P and Durand JB: Erlotinib
or gefitinib for non-small-cell lung cancer. N Engl J Med.
364:2367–2368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsui TA, Murata H, Sakabe T, Sowa Y,
Horie N, Nakanishi R, Sakai T and Kubo T: Sulforaphane induces cell
cycle arrest and apoptosis in murine osteosarcoma cells in vitro
and inhibits tumor growth in vivo. Oncol Rep. 18:1263–1268.
2007.PubMed/NCBI
|
|
21
|
Jakubikova J, Bao Y and Sedlak J:
Isothiocyanates induce cell cycle arrest, apoptosis and
mitochondrial potential depolarization in HL-60 and
multidrug-resistant cell lines. Anticancer Res. 25:3375–3386.
2005.PubMed/NCBI
|
|
22
|
Chang A: Chemotherapy, chemoresistance and
the changing treatment landscape for NSCLC. Lung Cancer. 71:3–10.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simmons TL, Andrianasolo E, McPhail K,
Flatt P and Gerwick WH: Marine natural products as anticancer
drugs. Mol Cancer Ther. 4:333–342. 2005.PubMed/NCBI
|
|
24
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clinical Translational Oncol. 9:767–776. 2007.
View Article : Google Scholar
|
|
25
|
Altmann KH and Gertsch J: Anticancer drugs
from nature-natural products as a unique source of new
microtubule-stabilizing agents. Nat Prod Rep. 24:327–357. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Newman DJ and Cragg GM: Natural products
as sources of new drugs over the last 25 years. J Nat Prod.
70:461–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun H, Meng X, Han J, Zhang Z, Wang B, Bai
X and Zhang X: Anti-cancer activity of DHA on gastric cancer-an in
vitro and in vivo study. Tumour Biol. 34:3791–3800. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Follo MY, Manzoli L, Poli A, McCubrey JA
and Cocco L: PLC and PI3K/Akt/mTOR signalling in disease and
cancer. Adv Biol Regul. 57:10–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cornu M, Albert V and Hall MN: mTOR in
aging, metabolism, and cancer. Curr Opin Genet Dev. 23:53–62. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Faried LS, Faried A, Kanuma T, Nakazato T,
Tamura T, Kuwano H and Minegishi T: Inhibition of the mammalian
target of rapamycin (mTOR) by rapamycin increases chemosensitivity
of CaSki cells to paclitaxel. Eur J Cancer. 42:934–947. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li YC, He SM, He ZX, Li M, Yang Y, Pang
JX, Zhang X, Chow K, Zhou Q, Duan W, et al: Plumbagin induces
apoptotic and autophagic cell death through inhibition of the
PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells.
Cancer Lett. 344:239–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng X, Li L, Jiang H, Jiang K, Jin Y and
Zheng J: Dihydro-artemisinin potentiates the anticancer effect of
cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer
cells: involvement of apoptosis and autophagy. Biochem Biophys Res
Commun. 444:376–381. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu GS, Lu JJ, Guo JJ, Huang MQ, Gan L,
Chen XP and Wang YT: Synergistic anti-cancer activity of the
combination of dihydroartemisinin and doxorubicin in breast cancer
cells. Pharmacol Rep. 65:453–459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sheng H, Shao J, Morrow JD, Beauchamp RD
and DuBois RN: Modulation of apoptosis and Bcl-2 expression by
prostaglandin E2 in human colon cancer cells. Cancer Res.
58:362–366. 1998.PubMed/NCBI
|
|
36
|
McDonnell TJ, Troncoso P, Brisbay SM,
Logothetis C, Chung LW, Hsieh JT, Tu SM and Campbell ML: Expression
of the protooncogene bcl-2 in the prostate and its association with
emergence of androgen-independent prostate cancer. Cancer Res.
52:6940–6944. 1992.PubMed/NCBI
|
|
37
|
Katz HR: bcl-2 protein in non-small-cell
lung carcinoma. N Engl J Med. 330:2211994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miyashita T, Krajewski S, Krajewska M,
Wang HG, Lin HK, Liebermann DA, Hoffman B and Reed JC: Tumor
suppressor p53 is a regulator of bcl-2 and bax gene expression in
vitro and in vivo. Oncogene. 9:1799–1805. 1994.PubMed/NCBI
|
|
39
|
Zhang L, Yu J, Park BH, Kinzler KW and
Vogelstein B: Role of BAX in the apoptotic response to anticancer
agents. Science. 290:989–992. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choudhuri T, Pal S, Agwarwal ML, Das T and
Sa G: Curcumin induces apoptosis in human breast cancer cells
through p53-dependent Bax induction. FEBS Lett. 512:334–340. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brambilla E, Negoescu A, Gazzeri S,
Lantuejoul S, Moro D, Brambilla C and Coll JL: Apoptosis-related
factors p53, Bcl2, and Bax in neuroendocrine lung tumors. Am J
Pathol. 149:1941–1952. 1996.PubMed/NCBI
|
|
42
|
Gupta S, Afaq F and Mukhtar H: Involvement
of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle
arrest and apoptosis by apigenin in human prostate carcinoma cells.
Oncogene. 21:3727–3738. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sakamoto KM, Grant S, Saleiro D, Crispino
JD, Hijiya N, Giles F, Platanias L and Eklund EA: Targeting novel
signaling pathways for resistant acute myeloid leukemia. Mol Genet
Metab. 114:397–402. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He SQ, Gao M, Fu YF and Zhang YN:
Glycyrrhizic acid inhibits leukemia cell growth and migration via
blocking AKT/mTOR/STAT3 signaling. Int J Clin Exp Pathol.
8:5175–5181. 2015.PubMed/NCBI
|