Introduction
For thousands of years, ginseng has been used as a
nutrient and medicine, and as a component in cosmetics and
beverages, to improve quality of life and maintain health in humans
(1–3). In Asia in particular, it is
considered to be an invaluable herb (4,5).
Ginseng can be divided into two types, wild and cultivated,
according to their different growing conditions (6). The annual growth cycle of ginseng
begins with seeding, followed by leaf expansion, flowering, green
fruiting, red fruiting and wilitng (7).
The leaf expansion stage is a critical period in the
early growth and differentiation of the plant. During the leaf
expansion period, plant nutrients are transported from storage in
the roots to the aerial regions, as required for growth. During
this period, the plant organs exhibit significant morphological and
physiological changes (8). These
changes include the rapid growth of the stems and leaves, during
which the plants increase in height more rapidly than they grow in
width, and the rates of ginsenoside Rb1, Rc and Rd
synthesis during this stage are higher, compared with those in
other stages of growth (7,9). In addition, plant pests and other
diseases, which severely inhibit ginseng growth, frequently take
hold during this period (10,11).
The molecular mechanisms associated with these features in ginseng
remain to be fully elucidated (12,13).
Therefore, the present study aimed to perform transcriptome
analysis on ginseng leaves (GL) and stems (GS) during this
important growth stage.
During the last decade, next-generation sequencing
technology has improved the efficiency and speed of gene discovery
(14). The development of a novel,
high-throughput DNA sequencing method has provided a technique
enabling the mapping and quantifying of transcriptomes, and this
technology has led to a change in genomics and genetics, which has
provided cheaper and faster sequencing information (15–17).
In the present study, using the HiSeq™ 2000
Sequencing System platform and the paired-end sequencing method,
two de novo transcriptome databases were constructed from
cDNA libraries generated from Panax ginseng stems (GS) and
leaves (GL). The most frequent transcripts, growth-related,
stress-related, pathogenesis-related and redox-associated proteins,
and ginsenoside biosynthesis enzymes, expressed at low levels, of
interest were identified. These datasets provide useful information
on the transcript profile, which can assist in the understanding of
tissue-specific biological functions and of the transcriptional
regulatory expression mechanism.
Materials and methods
Plant materials and preparation
The plant materials used in the present study were
originally collected from the Fusong ginseng planting base (Jilin,
China). The stems and leaves were collected from 5-year-old ginseng
plants during the leaf expansion period. All samples were washed
with distilled water, cut into small sections with a thickness of
<1 cm, and then immediately stored in liquid nitrogen for
further processing.
RNA isolation and construction of cDNA
libraries
Total RNA was isolated from the GS and GL samples
separately using a modified TRIzol method according to the
manufacturer's protocol. To evaluate the RNA integrity, 10 mg of
RNA was fractionated on a 1% agarose gel, stained with ethidium
bromide, and visualized using UV light. The presence of intact 28S
and 18S rRNA bands was used as the criterion for RNA integrity
(18). The quality of the RNA
samples was confirmed using an Agilent 2100 bioanalyzer (Agilent
Technologies, Inc., Santa Clara, CA, USA), with a minimum RNA
integrated number value of eight (19). The samples for the transcriptome
analysis were prepared using the Illumina kit (Beijing Genomics
Institute, Shenzhen, China) according to the manufacturer's
protocol. The mRNA was purified from 10 µg of total RNA using oligo
(dT) magnetic beads (Beijing Genomics Institute). Following
purification, the mRNA was fragmented into small sections using
divalent cations at 94°C. Using these short fragments as templates,
reverse transcriptase and random primers were used to synthesize
first-strand cDNA. Second-strand cDNA was synthesized using DNA
polymerase I (Takara Biotechnology Co., Ltd., Dalian, China) and
RNase H (Takara Biotechnology Co, Ltd.), respectively. The cDNA
fragments underwent an end-repair process and were ligated to
adapters. These products were purified and enriched using
polymerase chain reaction (PCR) to produce the final cDNA library.
The PCR amplification was performed in a 50 µl reaction mixture
containing 10 µl reverse transcription product and 40 µl PCR
reaction solution (0.5 µl of each primer, 0.25 µl TaKaRa Ex
Taq® HS, 10 µl 5× PCR buffer and 28.75 µl ddH2O).
Cycling conditions were as follows: An initial predenaturation step
at 94°C for 2 min; followed by 30 consecutive cycles of
denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec and
extension at 72°C for 1 min.
Gene sequencing, de novo assembly and
functional annotation
The cDNA library was sequenced using the Illumina
sequencing platform (HiSeq 2000). The average size of library
inserts was 200 bp. Image deconvolution and quality value
calculations were performed using Illumina GA pipeline 1.3. The raw
reads were cleaned by removing adaptor sequences, empty reads and
low-quality sequences (20).
Trinity version 2.0 software (http://www.trinitysoftware.nl/) was used for de novo
assembly.
Different sequences were used for the Basic Local
Alignment Search Tool (BLAST) search and annotation against the
plant protein database of NR (ftp://ftp.ncbi.nih.gov/blast/db/FASTA) with a
significance threshold of E-value ≤10−5 and Swiss-Prot
(http://www.uniprot.org/). Functional annotations
using Gene Ontology (GO) terms were performed using Blast2go
version 3.0 software (https://www.blast2go.com/) with an E-value cut-off of
10−5. After getting GO annotation for every Unigene, all
Unigenes were classified based on GO function by Web Gene Ontology
Annotation Plot (WEGO) online software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) to
understand the distribution of gene function of the species.
Annotation with Clusters of Orthologous Groups (COG) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways was performed
using BLASTx against the COG (http://www.ncbi.nlm.nih.gov/COG/) and KEGG (http://www.genome.jp/kegg/kegg1.html)
databases (21–25).
To obtain distinct gene sequences, the unigenes were
clustered using TIGR Gene Indices clustering (TGICL) software
version 2.1 (https://sourceforge.net/projects/tgicl). For gene
expression analysis, the number of expressed reads were counted and
normalized using RPKM values based on the following formula:
RPKM=109 C/(N × L), where C is the number of mappable
reads uniquely aligned to a unigene, N is the total number of
mappable reads uniquely aligned to all unigenes, and L is the sum
of the unigene in base pairs (26).
Reverse transcription-quantitative PRC
(RT-qPCR)
To confirm the relative expression of the target
genes in the GS and GL transcriptome data, RT-qPCR was performed
using the Mx3000p Real-Time PCR detection system with a One Step
SYBR PrimeScript PLUS RT-PCR kit (Takara Biotechnology Co., Ltd.)
(27). PrimerPremier 5.0 (Premier
Biosoft International, Palo Alto, CA, USA) was applied to determine
the primer sequences (28). The
PCR amplification was performed in a 25 µl mixture containing 2 µl
of the reverse transcription product and 23 µl of the PCR reaction
solution (0.5 µl of each primer, 12.5 µl of SYBR® Primix
Ex TaqTM (2*), 0.5 µl of ROX Reference Dye II (50*) and 9 µl of
ddH2O). The reaction was performed using the following reaction
cycles: initial denaturation at 95°C for 30 sec; 40 consecutive
cycles of denaturation at 95°C for 5 sec, annealing at 54°C for 15
sec, and extension at 72°C for 30 sec. Tyrosine hydroxylase (TH)
and WNK lysine deficient protein kinase 1 genes were used as
internal standards. The primer sequences used for RT-qPCR are
listed in Tables I and II. The thermal cycle conditions for PCR
were as follows: 42°C for 5 min, 95°C for 10 sec, and then 40
cycles of 95°C for 5 sec followed by 60°C for 30 sec. The relative
expression levels were calculated using the 2−ΔΔCq
method (29).
 | Table I.Primers sequences used for reverse
transcription-quantitative polymerase chain reaction analysis
selected from the Panax ginseng stem database. |
Table I.
Primers sequences used for reverse
transcription-quantitative polymerase chain reaction analysis
selected from the Panax ginseng stem database.
| No. | Gene | RPKM | Sequence
(5′-3′) |
|---|
| 1 | GBR5-like
protein | 32,147.61 | F:
ATTAGTTCAGAGGTCGCAGC |
|
|
|
| R:
ATCCGCTCCTCCCATCAAC |
| 2 | Specific abundant
protein 3 | 11,366.95 | F:
GTTGCTCTGGTGGTGCTTCT |
|
|
|
| R:
TGTAAACACTTGCCCTGCCG |
| 3 | Protease
inhibitor/seed storage/LTP family protein |
4,117.47 | F:
GCGTTGCCTATGTGCTGTTA |
|
|
|
| R:
CTTGTAACCAACTGGGCGAT |
| 4 | Major latex-like
protein |
3,731.75 | F:
GAAAAGTTGGCTCCGTCGTC |
|
|
|
| R:
TCACCAAGTTGTCTTCACCC |
| 5 | Cyclophilin |
2,136.74 | F:
GGCAGGATTGTGATGGAGC |
|
|
|
| R:
TTGAGGGATGACTCGGTGG |
| 6 | Plasma membrane
intrinsic protein 2–1 |
400.39 | F:
GCCAGGAAAGTGTCGCTAAT |
|
|
|
| R:
TCTCAGCCCCTAATCCAGTG |
| Table II.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction selected from
the Panax ginseng leaf database. |
Table II.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction selected from
the Panax ginseng leaf database.
| No. | Gene | RPKM | Sequence
(5′-3′) |
|---|
| 1 | Chlorophyll a/b
binding protein of | 99,115.65 | F:
CCCTCTCCTCCCCATCATTC |
|
| LHCII type I
precursor |
| R:
CGGGCTTTTTTCCTGTTTTC |
| 2 | Chloroplast
light-harvesting chlorophyll | 23,716.69 | F:
CTGACCCCGAGACATTTGCT |
|
| a/b-binding
protein |
| R:
ACTTGACACCATTGCGAGCC |
| 3 | Chloroplast
ferredoxin I | 14,780.64 | F:
ATGGGTCAGGCTCTGTTCG |
|
|
|
| R:
TCTTTCTCCCCTTCTGGTGT |
| 4 | Specific abundant
protein 3 | 37,15.27 | F:
TCTGACTCTGGCAACCGATG |
|
|
|
| R:
CAGGAAGAACCTTGACAGCG |
| 5 | Catalase-1
precursor | 3,442.73 | F:
CAGGCAGGAGACAGATACCG |
|
|
|
| R:
CGTTGAGGCGAGACGCTATT |
| 6 | Cytokine binding
protein CBP57 | 2,291.8387 | F:
ATCAAACCCCAAACCGACAG |
|
|
|
| R:
GCTGGGCAATCACTTGGTTC |
Results
Transcriptome sequencing and
assembly
Total RNA was extracted from GS and GL during the
leaf-expansion period, followed by reverse transcription into cDNA.
Using the Illumina sequencing platform, >38,000,000 and
118,000,000 sequencing reads, respectively, were generated with an
average length of 90 bp. The data sets were deposited in the NCBI
Array-Express repository with the accession number E-MTAB-937.
Following a stringent quality check and data cleaning, 39,000,000
high-quality reads were obtained in the GS and GL libraries. Based
on the high-quality reads, a total of 168,300 and 120,241 contigs
were assembled, with an average length of 305 and 267 bp,
respectively, in GS and GL. The size distribution of these contigs
is shown in Table III. The reads
were then mapped back to contigs. Using paired-end reads, it was
possible to detect contigs from the same transcript, and the
distances between these contigs. Following clustering of these
unigenes using TGICL software, the contigs generated 98,808 and
73,162 unigenes in GS and GL, respectively, with mean lengths of
572 and 413 bp, respectively. In addition, 57,371 and 50,523
sequences were obtained, respectively, using an E-value cut-off of
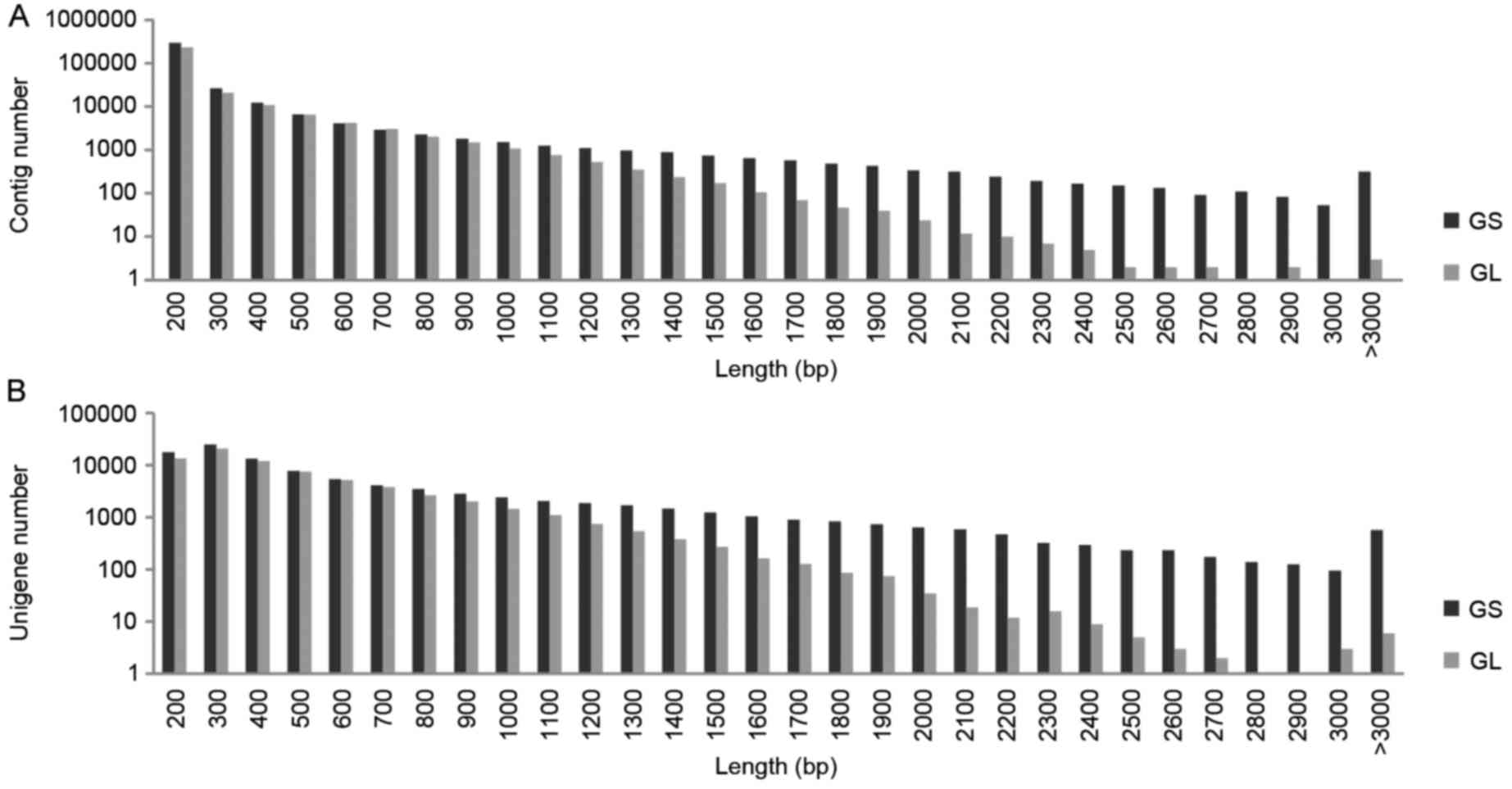
10−5. As shown in Fig. 1A
and B, the longer gene length resulted in higher quantities of
contigs and unigenes in GS, compared with in GL.
| Table III.Overview of the sequencing and
assembly in the GS and GL libraries. |
Table III.
Overview of the sequencing and
assembly in the GS and GL libraries.
| Feature | Statistic in
GS | Statistic in
GL |
|---|
| Total base pairs
(bp) | 3,451,590,540 | 3,479,796,000 |
| Total number of
reads | 38,351,006 | 38,664,400 |
| Average read length
(bp) | 90 | 90 |
| Total number of
contigs | 168,300 | 120,241 |
| Mean length of
contigs (bp) | 305 | 267 |
| Total number of
unigenes | 98,808 | 73,162 |
| Mean length of
unigenes (bp) | 572 | 413 |
Functional annotation by searching
against public databases
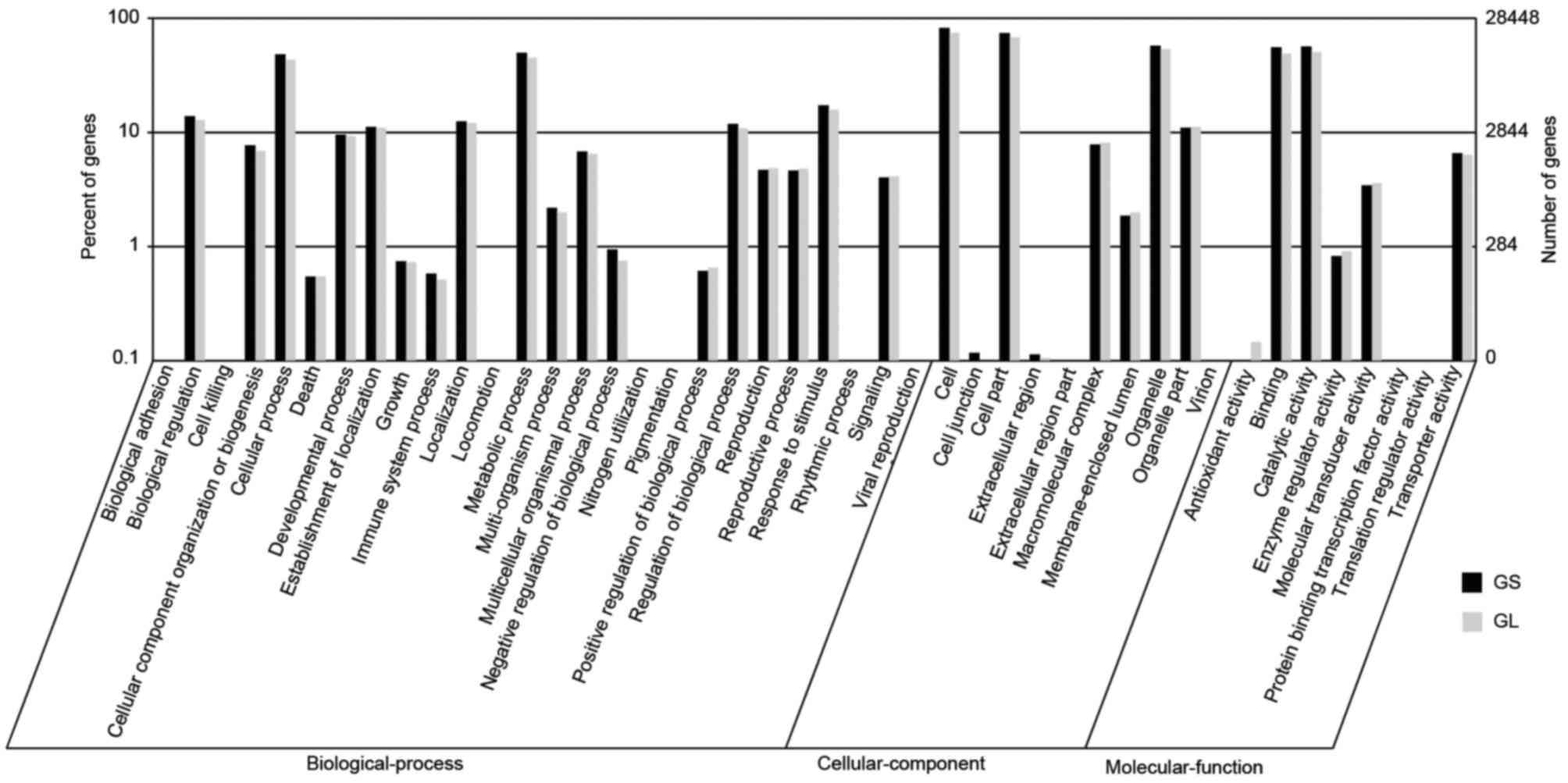
The GO terms were used to classify the functions of
the predicted GS and GL unigenes. Based on the sequence homology,
the annotated unigenes were analyzed with Blast2GO for GO
classification. From the GS library, 135,355 sequences were
categorized into 44 functional groups using WEGO tool, whereas
126,504 sequences were categorized from the GL library (Fig. 2) (30). The three major categories of
biological process, cellular component, and molecular function,
were assigned to 50,621/46,912, 55,437/52,700, and 29,297/26,892 GO
terms in the GS/GL libraries, respectively. There were high
percentages of genes from the categories ‘cell’ (19,229 GS/17,983
GL), ‘cell part’ (17,365/16,421), ‘organelle’ (13,584/12,982),
‘catalytic activity’ (13,343/12,240), ‘binding’ (13,155/11,907),
‘metabolic process’ (11,795/10,892), and ‘cellular process’
(11,497/10,427) in GS and GL. However, the categories ‘cell
killing’ (2), ‘locomotion’
(2), ‘nitrogen utilization’
(2), and ‘rhythmic process’
(2) contained the fewest GS genes,
and the categories ‘cell killing’ (2), ‘translation regulator activity’
(2), and ‘nitrogen utilization’
(1) contained the fewest GL
genes.
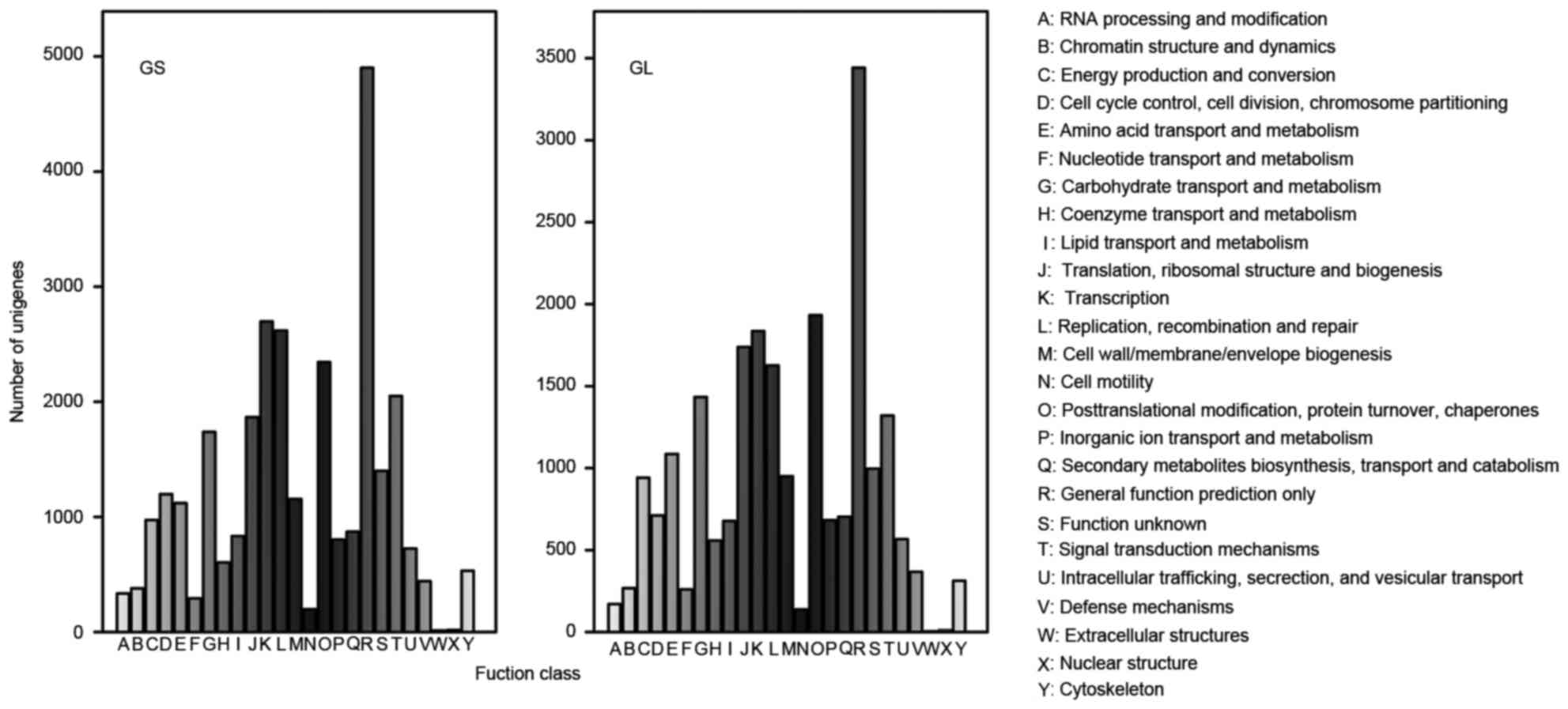
COG is an orthologous gene classification database,
where each COG protein is assumed to come from protein ancestors.
Overall, 30,105 and 22,726 sequences were clustered into 25 COG
classifications in GS and GL, respectively.
Among the GS categories, the cluster for ‘General
function prediction only’ (4,901; 16.28%) associated with basic
physiological and metabolic functions represented the largest
group, followed by ‘Transcription’ (2,697; 8.96%), ‘Replication,
recombination and repair’ (2,619; 8.70%), ‘Post-translational
modification, protein turnover, chaperones’ (2,345; 7.79%), and
‘Signal transduction mechanisms’ (2,050, 6.81%), whereas only a few
unigenes were assigned to ‘Extra cellular structures’ (13, 0.04%)
and ‘Nuclear structure’ (18; 0.06%), as shown in Fig. 3.
Among the GL categories, the cluster for ‘General
function prediction only’ (3,442), ‘Post-translational
modification, protein turnover, chaperones’ (1,933), and
‘Transcription’ (1,836) were the three largest groups, representing
15.15, 8.51, and 8.08%, respectively. The groups with the fewest
unigenes were ‘Extracellular structures’ (3; 0.01%) and ‘Nuclear
structure’ (10; 0.04%), which were the same as for GS (Fig. 3).
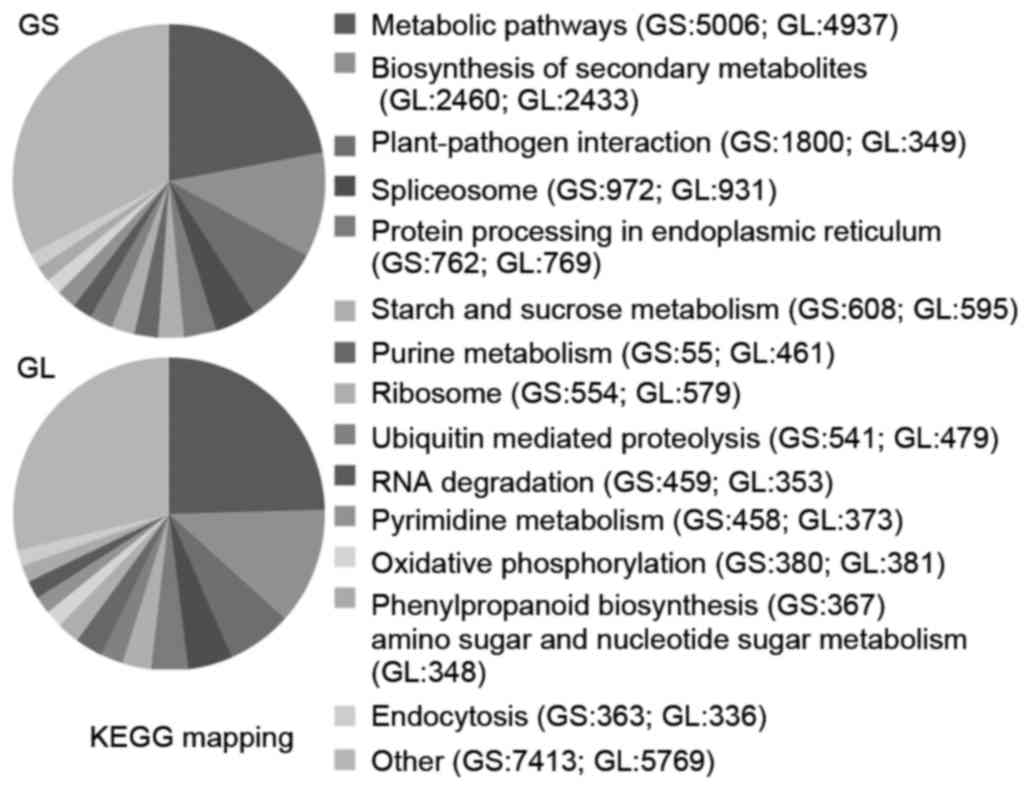
Ginseng transcriptome pathway analysis was performed
using KEGG mapping. In total, 22,697 and 20,093 sequences were
identified with pathway annotations in GS and GL, respectively, and
these were functionally assigned to 121 KEGG pathways.
In the GS and GL aerial tissues, the ‘metabolic
pathways’ had the highest contribution (22.06 and 24.57%,
respectively), followed by ‘Biosynthesis of secondary metabolites’
(10.84 and 12.11%) and ‘Plant-pathogen interaction’ (7.93 and
6.71%), as show in Fig. 4. These
annotations of gene or protein names and descriptions, GO terms,
putative conserved domains, and potential metabolic pathways
provide valuable resources for investigating the specific
processes, functions and pathways involved in GS and GL
development.
Transcriptional data analysis and
summary
The genes with the highest levels of expression in
GS encoded a GBR5-like protein, followed by At5g54075, allergen,
chlorophyll a/b binding protein of light-harvesting complex II
(LHCII) type I precursor, specific abundant protein 3, a
hypothetical protein, GBR5, peroxidase, phloem protein 2–2, and
pathogenesis-related protein 10 (Table IV). The phytochrome-associated
genes were expressed at a high level in GL data, and the gene with
the highest expression was chlorophyll a/b binding protein,
followed by the LHCII protein Lhcb1, GBR5-like protein, a
hypothetical protein, chloroplast light-harvesting chlorophyll
a/b-binding protein, At5g54075, chloroplast ferredoxin I,
cytochrome P450 like-TBP, photosystem II protein I, and RNA
polymerase α subunit (Table V). A
number of functional genes were obtained from both transcriptome
databases, including growth-associated proteins (GBR5-like protein,
GBR5, cyclophilin, cytokinin-repressed protein, and auxin response
factor 3), stress-related proteins (specific abundant and
drought-induced proteins), pathogenesis-related proteins
(pathogenesis-related protein 10, Avr9/Cf-9 rapidly elicited
protein, Erwinia-induced protein, and fungal elicitor-induced
protein), and redox-related proteins (peroxidase and catalase-1
precursor), as shown in Table
VI.
| Table IV.Top 10 most frequent transcripts in
the Panax ginseng stem transcriptome library. |
Table IV.
Top 10 most frequent transcripts in
the Panax ginseng stem transcriptome library.
| No. | Gene | Species | Accession no. | RPKM |
|---|
| 1 | GBR5-like
protein | Panax ginseng | gb|ABD73293.1 | 32,147.61 |
| 2 | At5g54075 | Arabidopsis
thaliana | gb|AAT46037.1 | 16,817.52 |
| 3 | Allergen | Kirola | sp|P85524.1 | 12,958.45 |
| 4 | Chlorophyll a/b
binding protein of LHCII type I precursor | Panax
ginseng | gb|AAB87573.1 | 11,366.95 |
| 5 | Specific abundant
protein 3 | Panax
ginseng | gb|AAX40471.1 | 10,665.18 |
| 6 | Hypothetical
protein | Vitis
vinifera |
ref|XP_002280773.1 | 8,902.68 |
| 7 | GBR5 | Panax
ginseng | gb|AAP55852.1 | 8,511.19 |
| 8 | Peroxidase | Populus
trichocarp | gb|ACN97180.1 | 7,838.47 |
| 9 | Phloem protein
2–2 | Apium
graveolens var. Dulce | gb|AAM62133.1 | 7,704.61 |
| 10 |
Pathogenesis-related protein 10 | Panax
ginseng | gb|ACY36943.1 | 5,465.76 |
| Table V.The top 10 most frequent transcripts
in Panax ginseng leaf transcriptome library. |
Table V.
The top 10 most frequent transcripts
in Panax ginseng leaf transcriptome library.
| No. | Gene | Species | Accession no. | RPKM |
|---|
| 1 | Chlorophyll a/b
binding protein of LHCII type I precursor | Panax
ginseng | gb|AAB87573.1 | 99,115.65 |
| 2 | Light-harvesting
complex II protein Lhcb1 | Populus
trichocarpa |
ref|XP_002316737.1 | 30,255.99 |
| 3 | GBR5-like
protein | Panax
ginseng | gb|ABD73293.1 | 29,122.78 |
| 4 | Hypothetical
protein VITISV_001840 | Vitis
vinifera | emb|CAN65763.1 | 25,185.48 |
| 5 | Chloroplast
light-harvesting chlorophyll a/b-binding protein | Artemisia
annua | gb|ABQ32304.1 | 23,716.69 |
| 6 | At5g54075 | Arabidopsis
thaliana | gb|AAT46037.1 | 16,026.90 |
| 7 | Chloroplast
ferredoxin I | Camellia
sinensis | gb|AEI83424.1 | 14,780.64 |
| 8 | Cytochrome P450
like_TBP | Nicotiana
tabacum | dbj|BAA10929.1 | 14,505.00 |
| 9 | Photosystem II
protein I | Davidia
involucrata | gb|ADM92705.1 | 13,675.17 |
| 10 | RNA polymerase α
subunit | Panax
ginseng |
ref|YP_086997.1 | 10,301.30 |
| Table VI.Expressed transcripts of associated
protein in GS and GL library. |
Table VI.
Expressed transcripts of associated
protein in GS and GL library.
|
| GS | GL |
|---|
|
|
|
|
|---|
| Category | Associated protein
gene | RPKM | Associated protein
gene | RPKM |
|---|
|
Growth-associated | GBR5-like
protein | 32,147.61 | GBR5-like
protein | 29,122.78 |
| proteins | GBR5 | 85,11.19 | GBR5 | 7,096.61 |
|
| Cyclophilin | 21,36.74 | Cytokinin binding
protein | 2,291.83 |
|
| Cytokinin-repressed
protein | 21,2.24 | Auxin response
factor 3 | 64.14 |
|
| Auxin response
factor 3 | 51.93 | Cyclophilin | 64.06 |
| Stress-related | Specific abundant
protein | 10,665.18 | Specific abundant
protein | 3,715.27 |
| genes | Metallothionein-1
like protein | 3,629.83 | Drought-induced
protein | 3,011.41 |
|
| Drought-induced
protein | 1,710.70 | Metallothionein-1
like protein class I | 930.61 |
|
| Cold-inducible
protein | 984.20 | Cold-inducible
protein | 623.07 |
|
| Dehydrin 3 | 854.14 | Dehydrin 2 | 388.63 |
|
| Dehydrin 2 | 843.81 | Dehydration-induced
protein | 48.83 |
|
| Dehydration-induced
protein | 127 | Dehydrin 1 | 18.33 |
|
Disease-related | Pathogensis-related
protein 10 | 5,465.76 | Pathogensis-related
protein 10 | 823.96 |
| proteins | Avr9/Cf-9 rapidly
elicited protein | 589.12 | Defensin-like
protein 1 | 73.29 |
|
| Defensin-like
protein 1 | 586.17 | Erwinia induced
protein 1 | 68.52 |
|
| Erwinia induced
protein 2 | 362.79 | Erwinia induced
protein 2 | 46.68 |
|
| Fungal
elicitor-induced protein | 243.06 | Disease
resistance-responsive | 23.21 |
|
| Disease
resistance-responsive | 161.67 | Avr9/Cf-9 rapidly
elicited protein | 20.19 |
|
| Erwinia induced
protein 1 | 156.82 | Fungal
elicitor-induced protein | 12.04 |
| Redox-related
proteins | Peroxidase | 7,838.47 | Catalase-1
precursor | 3,442.13 |
|
| Catalase-1
precursor | 3,804.35 | Peroxidase | 389.15 |
|
| glutaredoxin | 283.11 | glutaredoxin | 125.08 |
| Ginsenoside
skeleton | HMGR | 80.05 | HMGR | 539.98 |
| biosynthesis
key | GPS | 21.76 | GPS | 136.23 |
| enzymes | SS | 34.06 | SS | 77.70 |
|
| SE | 31.32 | SE | 96.68 |
|
| β-AS | 5.92 | β-AS | 8.9034 |
|
| Cytochrome
P450 | 371.32 | Cytochrome
P450 | 209.75 |
|
| GT | 76.36 | GT | 48.96 |
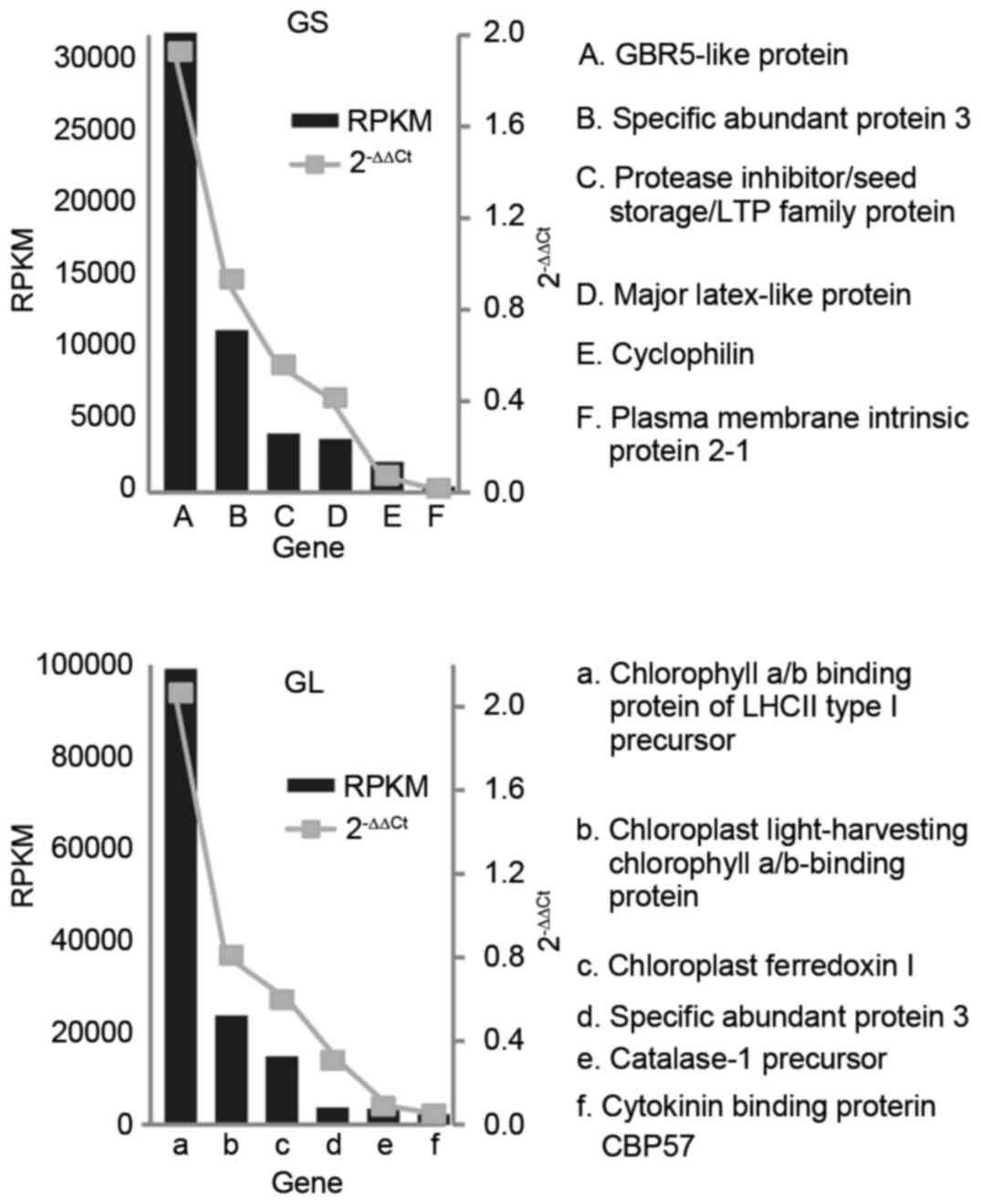
RT-qPCR analysis
RT-qPCR analysis was used to determine the
expression levels of target genes using the 2−ΔΔCq
method, which is a convenient way of analyzing the relative changes
in gene expression levels. A total of six genes were randomly
selected in each library. Statistical analysis of the RT-qPCR
results showed that the RPKM value was consistent with the
2−ΔΔCq value. The expression profiles of the six genes
were consistent with the gene expression results (Fig. 5), supporting the reliability of the
RNA-sequencing data.
Discussion
Early developmental processes in plants often occur
as a consequence of the initiation and changes in protein
expression, and active protein folding during this critical period
(31). Investigating plant
proteins may assist in identifying important signals in
developmental pathways and to understand the physiological
functions of specific genes during plant growth. The expression of
genes in the aerial regions of ginseng, particular during the
leaf-expansion period, remain to be fully elucidated.
In the present study, GS and GL tissues were
selected for transcriptome analysis. Illumina-generated sequencing
was applied to the characterization and assembly of the
transcriptome, which successfully generated and assembled a draft
sequence. Prior to the present study, ginseng was represented by
only 561 sequences in the NCBI protein database and 12,071
sequences in the NCBI EST database (http://www.ncbi.nlm.nih.gov/protein?term=panax%20ginseng%20)
In the present study, >57,372 and 50,523 sequences were produced
in GS and GL, respectively. These data provide a substantial
contribution to existing sequence resources on GS and GL, and are
beneficial to ginseng genetic investigations.
To evaluate the completeness of the transcriptome
libraries produced in the present study, and the effectiveness of
the annotation process, a search was performed of the annotated
sequences for genes classified into GO and COG classifications, and
in KEGG pathways. Sequences were assigned to 44 GO and 25 COG
classifications, and 121 KEGG pathways. These annotations provide a
valuable resource for investigating specific processes, functions,
and pathways in ginseng investigations.
The top 10 most frequent transcripts in GS were
predominantly genes involved in resisting environmental pressure,
and in promoting and organizing rapid growth. The GBR-like protein
is the most frequent transcript promoting plant growth. Its high
expression has been closely linked to rapid elongation of the GS
(32). Chlorophyll a/b binding
protein is associated with photosynthetic functions, and is
beneficial in storing energy and growth elongation in the GS
(33). Specific abundant protein 3
is a stress-related gene, which is involved in altering cell wall
characteristics to tolerate water deficit stress under abiotic
stress conditions (34,35). Peroxidase assists in increasing
plant defenses against pathogens and acts as a catalyst to
facilitate a variety of biological processes. As a naturally
occurring by-product of oxygen metabolism in the body, peroxidase
breaks down hydrogen peroxide into water and oxygen to reduce
cytotoxicity (36,37). Phloem protein 2 is a phloem lectin
conserved in plants, which is considered to assist in the
establishment of phloem-based defenses induced by insect attacks
and other stresses, including wounding and oxidative conditions
(38). The findings of the present
study suggested that in GS, several genes are expressed to promote
rapid growth of tissue and reduce the effect of environmental
pressure. In GL, the most abundant gene was chlorophyll a/b binding
protein, which is involved in photosynthesis. Photosynthesis is a
vital source of energy for almost all living organisms and, during
the leaf-expansion period, this provides a direct energy source for
the growth of plants through the photosynthetic pathway (Table V) (39).
The present study also found several biological
process-related proteins in the GS and GL libraries, including
growth-associated, stress-related, pathogenesis-related, and
redox-related proteins (Table
VI). The same growth-associated genes were expressed in GS and
GL, however stress-related, pathogenesis-related and redox-related
genes were expressed at significantly higher levels in GS, compared
with GL. The RPKM value of pathogenesis-related protein 10 was
5,465.76 in GS, but 823.96 in GL, and peroxidase was 7,838.47 in
GS, but only 389.15 in GL. These results suggested that GS has a
higher defense tolerance and anti-stress ability, compared with
GL.
The leaf-expansion period is the initial growth
stage in which there is a high incidence of disease, which is a
limiting factor in ginseng growth and reproduction (11). It is reported that
pathogenesis-related proteins, defined as host-plant proteins,
which are induced specifically in disease or associated
pathological situations, are associated with the development of
systemic acquired resistance against further infection by fungi,
bacteria and viruses (40). In the
transcription data of the present study, these types of protein,
including pathogenesis-related protein 10, Avr9/Cf-9 rapidly
elicited protein, defensin-like protein 1, fungal elicitor-induced
protein, and Erwinia-induced protein, were found in GS and
GL (Table VI).
Pathogenesis-related protein 10 is structurally related to
ribonucleases and may be active against viruses (39). However, whether they are all
required for resistance or are involved in defense gene activation
remains to be elucidated (41).
Avr9/Cf-9 is induced in response to microbial organisms and is
involved in signaling and/or other aspects of the defense response
(42). Fungal elicitor-induced
protein is induced by fungal infections. Defensin-like protein 1,
also known as cysteine-rich antifungal protein 1, possesses
antifungal activity (43).
Erwinia-induced protein induces hypersensitive responses,
including necrosis (44). These
data suggested that ginseng, during the leaf-expansion period, has
already been infected by viral, fungal and bacterial pathogens, and
that appropriate preventative measures be taken prior to this stage
to improve the yield and quality of ginseng.
The present study also identified ginsenoside
biosynthesis enzymes, including 3-hydroxy-3-methylglutaryl coenzyme
A reductase, geranylgeranyl pyrophosphate synthase, squalene
synthase, squalene epoxidase, oxidosqualene cyclase, β-amyrin
synthase, cytochrome P450 and glucosyltransferase, which are
involved in the mevalonate pathway (45). However, their RPKM values were low.
Additionally, the transcript encoding dammarendiol synthase, a
rate-limiting enzyme in the ginsenoside biosynthesis pathway, was
not present in our transcriptome dataset, indicating that
ginsenosides were not actively biosynthesized in either the GS or
the GL during the leaf-expansion period.
In conclusion, the present study performed de
novo transcriptome sequencing of GS and GL during the
leaf-expansion period using the Illumina platform. The data showed
that >38,000,000 and 118,000,000 sequencing reads were produced
in GS and GL, respectively, and these reads were assembled into
99,809 and 50,523 unique sequences, respectively. By performing
BLAST analysis of the unigenes against public databases (Nr,
Swiss-Prot, KEGG, and COG), functional annotations and
classifications were obtained. The substantial number of
transcriptomic sequences and their functional annotations provide
useful resources for molecular investigations of GS and GL. In
addition, several candidate genes were identified, which may be
involved in growth and environmental stress responses.
Acknowledgements
The present study was supported by grants from
National Natural Foundation of China (grant nos. 81373937, 81503212
and 20140520042JH), the Scientific and Technological Development
Program of Jilin, China (grant no. 20140622003JC) and the Strategic
Adjustment of the Economic Structure of Jilin Province to Guide the
Capital Projects (grant no. 2014N155).
References
|
1
|
Yamada N, Araki H and Yoshimura H:
Identification of antidepressant-like ingredients in ginseng root
(Panax ginseng CA Meyer) using a menopausal depressive-like state
in female mice: Participation of 5-HT2A receptors. Psychopharmacol
(Berl). 216:589–599. 2011. View Article : Google Scholar
|
|
2
|
Chen F, Chen Y, Kang X, Zhou Z, Zhang Z
and Liu D: Anti-apoptotic function and mechanism of ginseng
saponins in Rattus pancreatic β-cells. Biol Pharm Bull.
35:1568–1573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park JD, Rhee DK and Lee YH: Biological
activities and chemistry of saponins from Panax ginseng CA Meyer.
Phytochemistry Rev. 4:159–175. 2005. View Article : Google Scholar
|
|
4
|
Chang YS, Seo EK and Gyllenhaal C: Panax
ginseng: A role in cancer therapy? Integr Cancer Ther. 2:13–33.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiang YZ, Shang HC, Gao XM and Zhang BL: A
comparison of the ancient use of ginseng in traditional Chinese
medicine with modern pharmacological experiments and clinical
trials. Phytother Res. 22:851–858. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li S, Li J, Yang XL, Cheng Z and Zhang WJ:
Genetic diversity and differentiation of cultivated ginseng (Panax
ginseng CA Meyer) populations in North-east China revealed by
inter-simple sequence repeat (ISSR) markers. Gen Resour Crop
Evolution. 58:815–824. 2011. View Article : Google Scholar
|
|
7
|
Wu D, Austin RS and Zhou S: The root
transcriptome for North American ginseng assembled and profiled
across seasonal development. BMC Genomics. 14:5642013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xing YZ and Ma FR: The research of trends
of each growing periods in ginseng. J Northeast Normal Univ.
1:57–62. 1981.
|
|
9
|
Dale JE: The control of leaf expansion.
Ann Rev Plant Physiol Plant Mol Biol. 39:267–295. 1988. View Article : Google Scholar
|
|
10
|
Punja ZK, Wan A and Rahman M: Growth,
population dynamics and diversity of Fusarium equiseti in ginseng
fields. Eur J Plant Pathol. 121:173–184. 2008. View Article : Google Scholar
|
|
11
|
Jeger MJ: Analysis of disease progress as
a basis for evaluating disease management practices. Annu Rev
Phytopathol. 42:61–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi HI, Waminal NE, Park HM, Kim NH, Choi
BS, Park M, Choi D, Lim YP, Kwon SJ, Park BS, et al: Major repeat
components covering one-third of the ginseng (Panax ginseng C.A.
Meyer) genome and evidence for allotetraploidy. Plant J.
77:906–916. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jung CH, Seog HM, Choi IW, Choi HD and
Choi HY: Effects of wild ginseng (Panax ginseng C.A. Meyer) leaves
on lipid peroxidation levels and antioxidant enzyme activities in
streptozotocin diabetic rats. J Ethnopharmacol. 98:245–250. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van Dijk EL, Auger H, Jaszczyszyn Y and
Thermes C: Ten years of next-generation sequencing technology.
Trends Genet. 30:418–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gupta P, Goel R, Pathak S, Srivastava A,
Singh SP, Sangwan RS, Asif MH and Trivedi PK: De novo assembly,
functional annotation and comparative analysis of Withania
somnifera leaf and root transcriptomes to identify putative genes
involved in the with anolides biosynthesis. PLoS One. 8:e627142013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chan AI, McGregor LM and Liu DR: Novel
selection methods for DNA-encoded chemical libraries. Curr Opin
Chem Biol. 26:55–61. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang N, Zhang L, Tao Y, Guo L, Sun J, Li
X, Zhao N, Peng J, Li X, Zeng L, et al: Construction of a high
density SNP linkage map of kelp (Saccharina japonica) by sequencing
Taq I site associated DNA and mapping of a sex determining locus.
BMC Genomics. 16:1892015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schroeder A, Mueller O, Stocker S,
Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M
and Ragg T: The RIN: An RNA integrity number for assigning
integrity values to RNA measurements. BMC Mol Biol. 7:32006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Li S, Li J, Li C and Zhang Y: De
novo transcriptome sequencing in Pueraria lobata to identify
putative genes involved in isoflavones biosynthesis. Plant Cell
Rep. 34:733–743. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsanakas GF, Manioudaki ME, Economou AS
and Kalaitzis P: De novo transcriptome analysis of petal senescence
in Gardenia jasminoides Ellis. BMC Genomics. 4:5542014. View Article : Google Scholar
|
|
21
|
Camacho C, Coulouris G, Avagyan V, Ma N,
Papadopoulos J, Bealer K and Madden TL: BLAST+: Architecture and
applications. BMC Bioinformatics. 10:4212009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gene Ontology Consortium: The gene
ontology (GO) project in 2006. Nucleic Acids Res. 34(Database
issue): D322–D326. 2006.PubMed/NCBI
|
|
23
|
Conesa A, Götz S, García-Gómez JM, Terol
J, Talón M and Robles M: Blast2GO: A universal tool for annotation,
visualization and analysis in functional genomics research.
Bioinformatics. 21:3674–3676. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tatusov RL, Fedorova ND, Jackson JD,
Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov
SL, Nikolskaya AN, et al: The COG database: An updated version
includes eukaryotes. BMC Bioinformatics. 4:412003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanehisa M, Goto S, Hattori M,
Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M and
Hirakawa M: From genomics to chemical genomics: New developments in
KEGG. Nucleic Acids Res. 34(Database issue): D354–D357. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mortazavi A, Williams BA, McCue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-Seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bustin SA, Beaulieu JF, Huggett J, Jaggi
R, Kibenge FS, Olsvik PA, Penning LC and Toegel S: MIQE précis:
Practical implementation of minimum standard guidelines for
fluorescence-based quantitative real-time PCR experiments. BMC Mol
Biol. 11:742010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Raso A, Mascelli S, Nozza P, Ugolotti E,
Vanni I, Capra V and Biassoni R: Troubleshooting fine-tuning
procedures for qPCR system design. J Clin Lab Anal. 25:389–394.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ye J, Fang L, Zheng H, Zhang Y, Chen J,
Zhang Z and Wang J, Li S, Li R, Bolund L and Wang J: WEGO: A web
tool for plotting GO annotations. Nucleic Acids Res. 34(Web Server
Issue): W293–W297. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takáč T, Pechan T and Šamaj J:
Differential proteomics of plant development. J Proteomics.
74:577–588. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Khan Z, Kim SG, Jeon YH, Khan HU, Son SH
and Kim YH: A plant growth promoting rhizobacterium, Paenibacillus
polymyxa strain GBR-1, suppresses root-knot nematode. Bioresource
Technol. 99:3016–3023. 2008. View Article : Google Scholar
|
|
33
|
Nagao R, Yokono M, Teshigahara A, Akimoto
S and Tomo T: Light-harvesting ability of the fucoxanthin
chlorophyll a/c-binding protein associated with photosystem II from
the Diatom Chaetoceros gracilis as revealed by picosecond
time-resolved fluorescence spectroscopy. J PhysChem B.
118:5093–5100. 2014.
|
|
34
|
Ha YI, Lim JM, Ko SM, Liu JR and Choi DW:
A ginseng-specific abundant protein (GSAP) located on the cell wall
is involved in abiotic stress tolerance. Gene. 386:115–122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang T, Zhou B, Luo M, Abbas HK, Kemerait
R, Lee RD, Scully BT and Guo B: Expression analysis of
stress-related genes in kernels of different maize (Zea mays L.)
inbred lines with different resistance to aflatoxin contamination.
Toxins (Basel). 3:538–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Karthikeyan M, Jayakumar V, Radhika K,
Bhaskaran R, Velazhahan R and Alice D: Induction of resistance in
host against the infection of leaf blight pathogen (Alternaria
palandui) in onion (Allium cepa var. aggregatum). Indian J Biochem
Biophys. 42:3712005.PubMed/NCBI
|
|
37
|
Salleh FM, Evans K, Goodall B, Machin H,
Mowla SB, Mur LA, Runions J, Theodoulou FL, Foyer CH and Rogers HJ:
A novel function for a redox-related LEA protein (SAG21/AtLEA5) in
root development and biotic stress responses. Plant Cell Environ.
35:418–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang C, Shi H, Chen L, Wang X, Lü B,
Zhang S, Liang Y, Liu R, Qian J, Sun W, et al: Harpin-induced
expression and transgenic overexpression of the phloem protein gene
AtPP2-A1 in Arabidopsis repress phloem feeding of the green peach
aphid Myzuspersicae. BMC Plant Biol. 11:112011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Barber J: Photosynthetic energy
conversion: Natural and artificial. Chem Soc Rev. 38:185–196. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Van Loon LC and Van Strien EA: The
families of pathogenesis-related proteins, their activities and
comparative analysis of PR-1 type proteins. Physiol Mol Plant
Pathol. 55:85–97. 1999. View Article : Google Scholar
|
|
41
|
Varet A, Parker J, Tornero P, Nass N,
Nürnberger T, Dangl JL, Scheel D and Lee J: NHL25 and NHL3, two
NDR1/HIN1-like genes in Arabidopsis thaliana with potential role(s)
in plant defense. Mol Plant Microbe Interact. 15:608–616. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Durrant WE, Rowland O, Piedras P,
Hammond-Kosack KE and Jones JD: cDNA-AFLP reveals a striking
overlap in race-specific resistance and wound response gene
expression profiles. Plant Cell. 12:963–977. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cole AM, Ganz T, Liese AM, Burdick MD, Liu
L and Strieter RM: Cutting edge: IFN-inducible ELR−CXC chemokines
display defensin-like antimicrobial activity. J Immunol.
167:623–627. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kelman A and Sequeira L: Resistance in
plants to bacteria. R Soc Lond Proc. 181:247–266. 1972; View Article : Google Scholar
|
|
45
|
Chen S, Luo H, Li Y, Sun Y, Wu Q, Niu Y,
Song J, Lv A, Zhu Y, Sun C, et al: 454 EST analysis detects genes
putatively involved in ginsenoside biosynthesis in Panax ginseng.
Plant Cell Rep. 30:1593–1601. 2011. View Article : Google Scholar : PubMed/NCBI
|