Introduction
It is widely acknowledged that atherosclerosis is
the principal cause of cardiovascular disease, which ultimately
contributes to mortality rates (1,2).
Although hypercholesterolemia, smoking, male gender, hypertension,
diabetes and age are traditionally considered to be risk factors,
accumulating evidence indicates that hyperlipidemia is the major
risk factor underlying the formation and development of
atherosclerosis (3,4). During the initiation of
atherosclerosis, plasma low-density lipoprotein (LDL) accumulates
in the arterial wall, oxidized, and recruits circulating monocytes
(5). When monocytes transform into
macrophages, they uptake oxidized (ox) LDL, resulting in
substantial cholesterol accumulation and the formation of foam
cells, which ultimately lead to formation of the atherosclerotic
lesion. In this multifactorial process, endothelial cells, smooth
muscle cells, macrophages and lymphocytes are involved and
interact. Increasing experimental evidence suggests that during
this process, an array of cytokines and adhesion molecules are
involved and promote atherogenesis (6,7). The
innate and adaptive immune responses are also pivotal in the
pathogenesis of atherosclerosis (8). Therefore, as a result of the multiple
interactions, atherosclerotic plaques are formed, which ultimately
results in cerebrovascular events and acute coronary syndromes.
Amygdalin (vitamin B17, also known as Laetrile), is
extracted from Semen Persicae, the seed of Prunus Persica
(L.) Batsch. Amygdalin is also found in abundance in the seeds
of apricots, almonds, peaches and other rosaceous plants. To date,
amygdalin has been widely used for the treatment of asthma,
bronchitis, emphysema, leprosy, diabetes and cancer. The authors'
previous study on amygdalin suggested that it may stimulate the
immune system and exhibits immune modulation functions by the
regulation of regulatory T cells (Tregs) in apolipoprotein
E-deficient (ApoE)−/− mice. As ApoE−/− mice
are spontaneous atherosclerotic mice, in order to determine whether
amygdalin can alleviate high-fat/high-cholesterol induced
atherosclerosis, LDLR−/− mice were used in the present
study. It has been demonstrated that the cytokines produced by T
cells, including interleukin (IL)-6 and tumor necrosis factor
(TNF)-α, and macrophages, including IL-1β, affect the extent and
nature of the atherosclerotic plaque (9,10).
The authors' previous study on the anti-atherosclerotic effect of
amygdalin demonstrated that the expression levels of IL-10 and
transforming growth factor (TGF)-β were markedly increased in the
serum of the amygdalin-treated mice. Therefore, it was hypothesized
that amygdalin possesses a broader immune-regulative function on
atherogenesis and is critical in the regulation of Tregs in
high-fat/high-cholesterol diet-induced atherosclerotic
LDLR−/− mice. To confirm this hypothesis, the present
study aimed to determine whether the expression levels of TNF-α,
IL-1β and IL-6 can be regulated by amygdalin. It is well known that
Tregs have a protective role in the progression of atherosclerosis
(11) and they are considered to
be a therapeutic target for the treatment of atherosclerosis. The
authors' previous studies suggested that Tregs can be induced and
expanded by amgydalin (12).
Therefore, to further substantiate the hypothesis, the expression
of forkhead box P3 (Foxp3) was silenced through Foxp3-specific
short hairpin (sh)RNAs and the expression levels of the cytokines
were observed. Previous clinical investigations of matrix
metalloproteinase (MMP)-2 and MMP-9 demonstrated that MMP-9
activity was found in macrophage-rich lesions, whereas the
expression of MMP-2 was higher in lesions rich in smooth muscle
cells, indicating that MMP-2 and MMP-9 are tightly associated with
stable and vulnerable lesions (13). In order to elucidate the mechanism
underlying the anti-atherosclerotic function of amygdalin, the
present study also examined the expression levels of MMP-2 and
MMP-9. Therefore, in order to determine whether amygdalin treatment
attenuates plaque progression and alleviates the symptom of
atherosclerosis, the present study focused on the immune regulatory
function of amygdalin.
Materials and methods
Animals
The LDLR−/− mice were purchased from
Jackson Laboratories (Bar Harbor, ME, USA) and maintained in a 12-h
light/dark cycle in an atmosphere of 0.03% CO2 with free
access to water and food. In accordance with the individual
ventilated cage requirements of Sichuan Academy of Medical Science
& Sichuan Provincial People's Hospital (Sichuan, China), all
mice were raised under specific pathogen-free conditions with
controlled temperature (23±2°C). The use and handling of animals
were in accordance with the Ethics Committee of Sichuan Academy of
Medical Science & Sichuan Provincial People's Hospital.
Study design
Amygdalin was purchased from Changsha Staherb
Natural Ingredients Co., Ltd. (Changsha, Hunan, China). The
8-week-old male mice were divided into five groups (n=30 in each
group, and body weight range of ~22–24 g): i) LDLR−/−
control group (control): 8-week-old male LDLR−/−mice
were fed a standard laboratory diet (10% fat, 15% protein and 75%
carbohydrate); ii) LDLR−/− high-fat diet (HFD) control
group (LDLR−/−): 8-week-old male LDLR−/− mice
were fed an HFD containing 40% fat, 14% protein and 46%
carbohydrate for 12 weeks; iii) Amygdalin (Low) group: 8-week-old
male LDLR−/− mice were fed the HFD for 12 weeks and
amygdalin (1 mg/kg/day for 4 weeks consecutively) was injected
intraperitoneally; iv) Amygdalin (medium) group: 8-week-old male
LDLR−/− mice were fed the HFD for 12 weeks and amygdalin
(3 mg/kg/day for 4 weeks consecutively) was injected
intraperitoneally; v) Amygdalin (high) group: 8-week-old male
LDLR−/− mice were fed the HFD for 12 weeks and amygdalin
(10 mg/kg/day for 4 weeks consecutively) was injected
intraperitoneally. All groups of mice were sacrificed following 4
weeks of drug delivery.
Lipid profile and cytokine
measurements
For measuring total cholesterol content, blood
samples were collected from each mouse at the beginning and end of
amygdalin treatment. Total plasma cholesterol (TC) and triglyceride
(TG) levels were determined using commercial kits (Applygen
Technologies, Inc., Beijing, China). The levels of murine IL-1β,
TGF-α and IL-6 were assayed using ELISA kits with paired antibodies
according to the manufacturer's protocol (R&D Systems, Inc.
Minneapolis, MN, USA).
Assessment of aortic sinus
atherosclerosis
The hearts and upper sections of the aorta were
removed from the mice, fixed, embedded in paraffin and sectioned
(5-µm). The sections were then stained with hematoxylin and eosin
(cat. no. C0105; Beyotime Institute of Biotechnology, Suzhou,
China) and oil red O (cat. no. O0625; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). Atherosclerotic lesions were quantified by
calculating the lesion size in the aortic sinus, as previous
described (14,15). The vessel areas were measured using
ImageJ software (version 1.47; National Institutes of Health;
Bethesda, MD, USA) from images obtained with a Nikon 80i microscope
(Nikon Corporation, Tokyo, Japan). For comparisons of plaque areas
between the amygdalin groups and control groups, 100 and 200 µm
distant sites were used.
Western blot analysis
The proteins were lysed by radioimmunoprecipitation
lysis buffer (Beyotime Institute of Biotechnology) and quantified
with bicinchoninic method (Beyotime Institute of Biotechnology) as
previously described (12). A
total of 20 µg protein was loaded onto a NuPageNovex 10% Bis-Tris
gel (Thermo Fisher Scientific, Inc.) for electrophoresis. Following
electrophoresis, the proteins were transferred onto polyvinylidene
fluoride membranes (Pall Life Sciences, Port Washington, NY, USA).
The membranes were blocked bovine serum albumin (Sigma-Aldrich;
Merck KGaA), and then incubated with primary antibodies overnight
at 4°C followed by incubation with horseradish peroxidase
(HRP)-conjugated secondary antibody at room temperature for 2 h.
Chemiluminescence detection was performed using Immobilon Western
Chemiluminescent HRP substrate (EMD Millipore, Billerica, MA, USA),
and measured directly using a BioSpectrum imaging system (UVP,
Inc., Upland, CA, USA). The mouse monoclonal CD68 (cat. no.
sc-20060; 1:1,000), mouse monoclonal MMP-2 (cat. no. sc-13594;
1:500), mouse monoclonal MMP-9 (cat. no. sc-393859; 1:500) and
mouse monoclonal β-actin (cat. no. sc-47778; 1:5,000) were
purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit
polyclonal monocyte chemoattractant protein-1 (MCP-1; cat. no.
ab9899; 1:500) was purchased from Abcam (Cambridge, MA, USA). T
cells from the spleen were isolated from the mice in each group.
Western blot analysis was performed with mouse monoclonal
anti-Foxp3 antibody (cat. no. ab20034; 1:500; Abcam). For the
secondary antibody, HRP-conjugated goat anti-mouse IgG polyclonal
antibody (cat. no. 115-035-003) and HRP-conjugated goat anti-rabbit
polyclonal IgG (cat. no. 111-035-003) were used, which were
purchased from Jackson ImmunoResearch Laboratories, Inc. (West
Grove, PA, USA). The levels of β-actin were determined as the
loading control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA from the splenocytes and lymphocyte cells
was isolated using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The primer sequences: FOXP3 forward,
5′-CCCAGGAAAGACAGCAACCTT-3′ and reverse,
5′-TTCTCACAACCAGGCCACTTG-3′; MCP-1 forward,
5′-TTAAAAACCTGGATCGGAACCAA-3′ and reverse,
5′-GCATTAGCTTCAGATTTACGGG-3′; MMP-2 forward,
5′-ACCCAGATGTGGCCAACTAC-3′ and reverse, 5′-TACTTTTAAGGCCCGAGCAA-3′;
and MMP-9 forward, 5′-ATGATGGAGGAGAAGCAGTC-3′ and reverse,
5′-AGGTGAAGGGAAAGTGACAT-3′ were used as previously described
(12,16,17).
The RT-qPCR analysis was performed on an ABI 7900 system using
Applied Biosystems Power SYBR® Green PCR Master mix
(Thermo Fisher Scientific, Inc.) in triplicate (18). In brief, 0.5 µM forward and reverse
primes, 2 µl buffer and 100 ng cDNA templates were added to each
tube and the total volume was adjusted to 20 µl by RNase free water
(Thermo Fisher Scientific Inc.). Following 3 min denaturation at
95°C; amplification was performed for 35 cycles, including 95°C for
10 sec for denaturation and 55°C for 30 sec for annealing and
extension. The copy number was calculated using the
2−ΔΔCq method. The Cq value for GAPDH was used to
normalize the samples (19).
Lentivirus preparation and
injection
The Foxp3 target sequences were as follows: forward,
5′-GGACACUCAAUGAAAUCUATT-3′; reverse, 5′-UAGAUUUCAUUGAGUGUCCTC-3′
(20). The shRNAs were synthesized
and cloned into pTY linkers. Third-generation vectors were used in
this experiment. The shRNA lentiviral vector was transiently
transfected into 293T cells (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China). Briefly, the 293T cells were
co-transfected with appropriate quantities of vector plasmids,
including the helper construct, envelope plasmid, tat plasmid and
pTYlinker containing shRNAs. The viral supernatant was harvested at
48 h, filtered through a 0.45-µm filter, subjected to
ultracentrifugation (113,000 × g at 4°C for 2 h) for 100-fold
concentration and stored at −80°C. Then lentiviral supernatant was
thawed at 37°C and diluted in 0.9% saline (Sichuan Kelun
Pharmaceuticals, Co., Ltd., Chengdu, Sichuan, China) and polybrene
(8 µg/ml final concentration; Sigma-Aldrich; Merck KGaA) to give a
dose of 1.6×107 or 1.6×108 transducing units
in a 50 µl injection volume. The virus was injected intravenously
into the mice with a 30-gauge needle on a 1-cc syringe. After the
LDLR−/− mice were confirmed as atherosclerotic, 3
consecutive injections were administered at 3-day intervals
(n=6).
Statistical analysis
The effects of treatment on the lesions, plaque
compositions, levels of IL-1β, IL-6 and TNF-α, and mRNA levels were
calculated using one-way analysis of variance and Bonferroni/Dunn's
test or Student's t-test by GraphPad Prism™ software
(version 5.0; GraphPad Software Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Amygdalin regulates lipid composition
and the development of atherosclerosis
The authors' previous data indicated that amygdalin
attenuated atherosclerosis in ApoE−/− mice. To further
examine the therapeutic effect of amygdalin in atherosclerosis, an
atherosclerotic mouse model was established in the present study by
feeding LDLR−/− mice with an HFD. It was evident from
analyzing the lipid profile (Table
I), that the TG levels and TC levels were increased in the
LDLR−/− mice. However, these changes were alleviated
when the mice were treated with amygdalin. A high dose of amygdalin
resulted in the 2-fold decrease in TG levels, compared with those
in the LDLR−/− mice (P<0.05). By comparing the TC and
LDL levels between the drug delivery groups and corresponding
control groups, it was found that the mice treated with amygdalin
had decreased levels of TC and LDL (P<0.05). In addition,
increased levels of HDL were observed in the amygdalin-treated mice
(P>0.05). Therefore, amygdalin regulated lipid contents in the
HFD-treated LDLR−/− mice.
 | Table I.Lipid profile of different groups of
mice (n=10 in each group). |
Table I.
Lipid profile of different groups of
mice (n=10 in each group).
| Group | TG (mmol/l) | TC (mmol/l) | HDL (mmol/l) | LDL (mmol/l) |
|---|
| Control |
1.05±0.14a |
1.62±0.43a | 0.75±0.04 |
0.82±0.29a |
|
LDLR−/− | 4.96±1.21 | 4.13±2.73 | 0.72±0.13 | 5.42±0.36 |
| Amygdalin
(low) |
3.71±0.51a |
3.62±0.38a |
0.95±0.08a |
4.56±0.26a |
| Amygdalin
(medium) |
2.18±0.48a |
3.36±0.26a |
1.27±0.11a |
3.65±0.30a |
| Amygdalin
(high) |
1.75±0.33a |
2.61±0.58a |
1.55±0.25a |
2.74±0.27a |
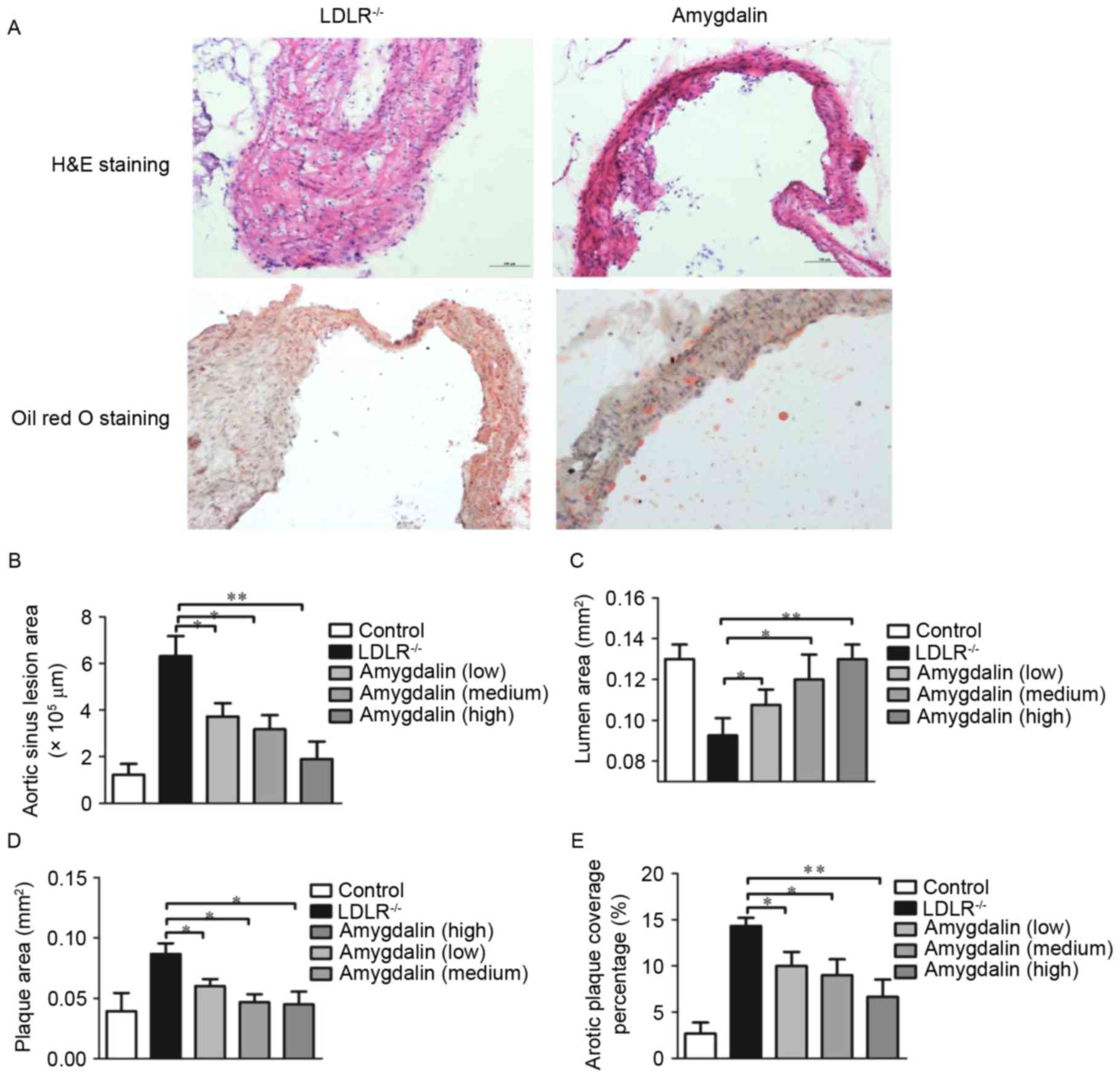
As the atherosclerotic lesions in the
LDLR−/− mice fed with the HFD demonstrated the same
morphological characteristics as those of humans, and the mouse
model imitated the progression of atherosclerosis, the aortic sinus
was stained with H&E and oil red O in the present study
(Fig. 1A). The atherosclerotic
plaques were well developed in the LDLR−/− mice. By
contrast, no atherosclerotic plaques were found in the
amygdalin-treated mice. The, vessel areas, lumen areas and plaque
areas were also analyzed. As exhibited in Fig. 1B, with the progression of
atherosclerosis, the diameters of the aortic sinus increased. In
addition, the areas of the lumen were decreased (Fig. 1C) and those of the plaques were
increased (Fig. 1D) in the
LDLR−/− mice. By contrast, the mice treated with
amygdalin exhibited a significantly reduced extent of
atherosclerosis, evident by the smaller aortic sinus plaques,
smaller aortic sinus and expanded lumen are as. This finding was
confirmed by the analysis of atherosclerosis surface coverage areas
(Fig. 1E), showing decreased
lesions in the amygdalin-treated LDLR−/− mice.
Therefore, these data indicated that amygdalin inhibited the
HFD-induced increase in atherosclerotic lesions in the
LDLR−/− mice.
Amygdalin inhibits the inflammatory
response
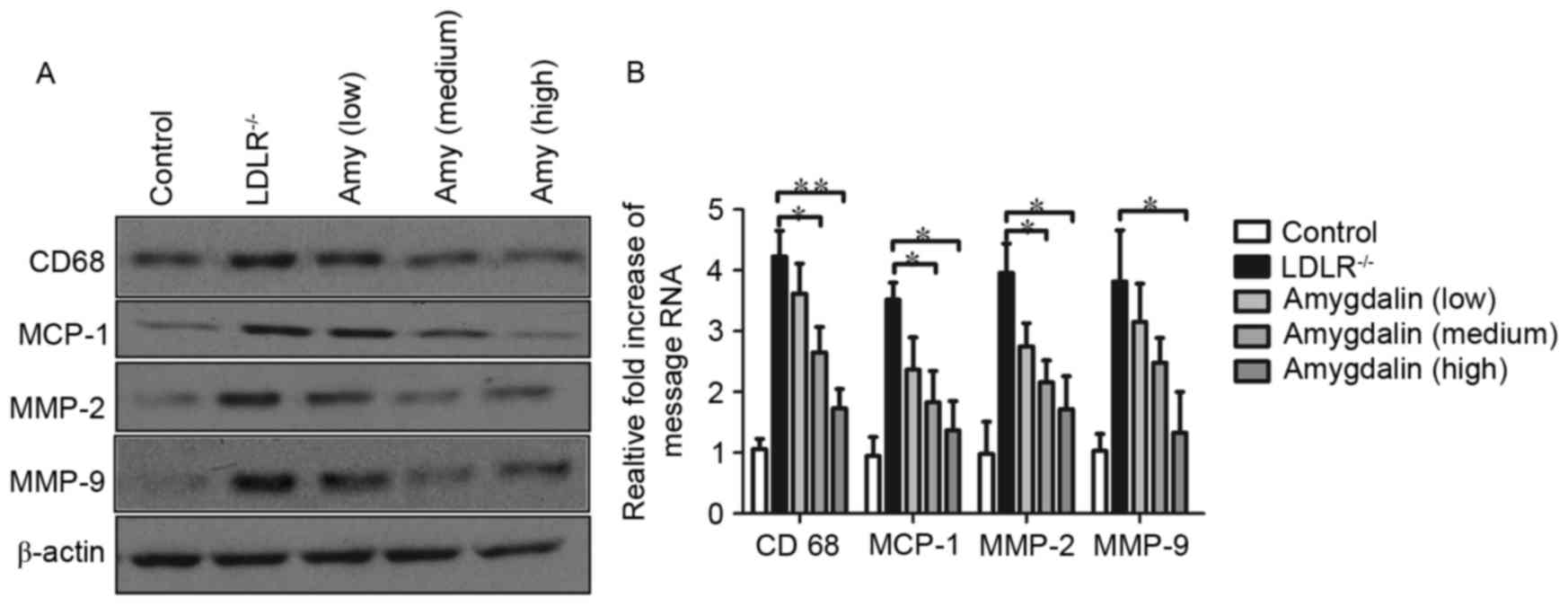
To investigate whether amygdalin inhibits
inflammation in the aorta of LDLR−/− mice, the present
study compared the mRNA and protein levels of MCP-1, CD68, MMP-2
and MMP-9 in the atherosclerotic arteries. As shown in Fig. 2A and B, the LDLR−/− mice
exhibited a marked increase in the expression levels of CD68,
MCP-1, MMP-2 and MMP-9. As expected, decreased activities of CD68,
MCP-1, MMP-2 and MMP-9 were observed following amygdalin treatment.
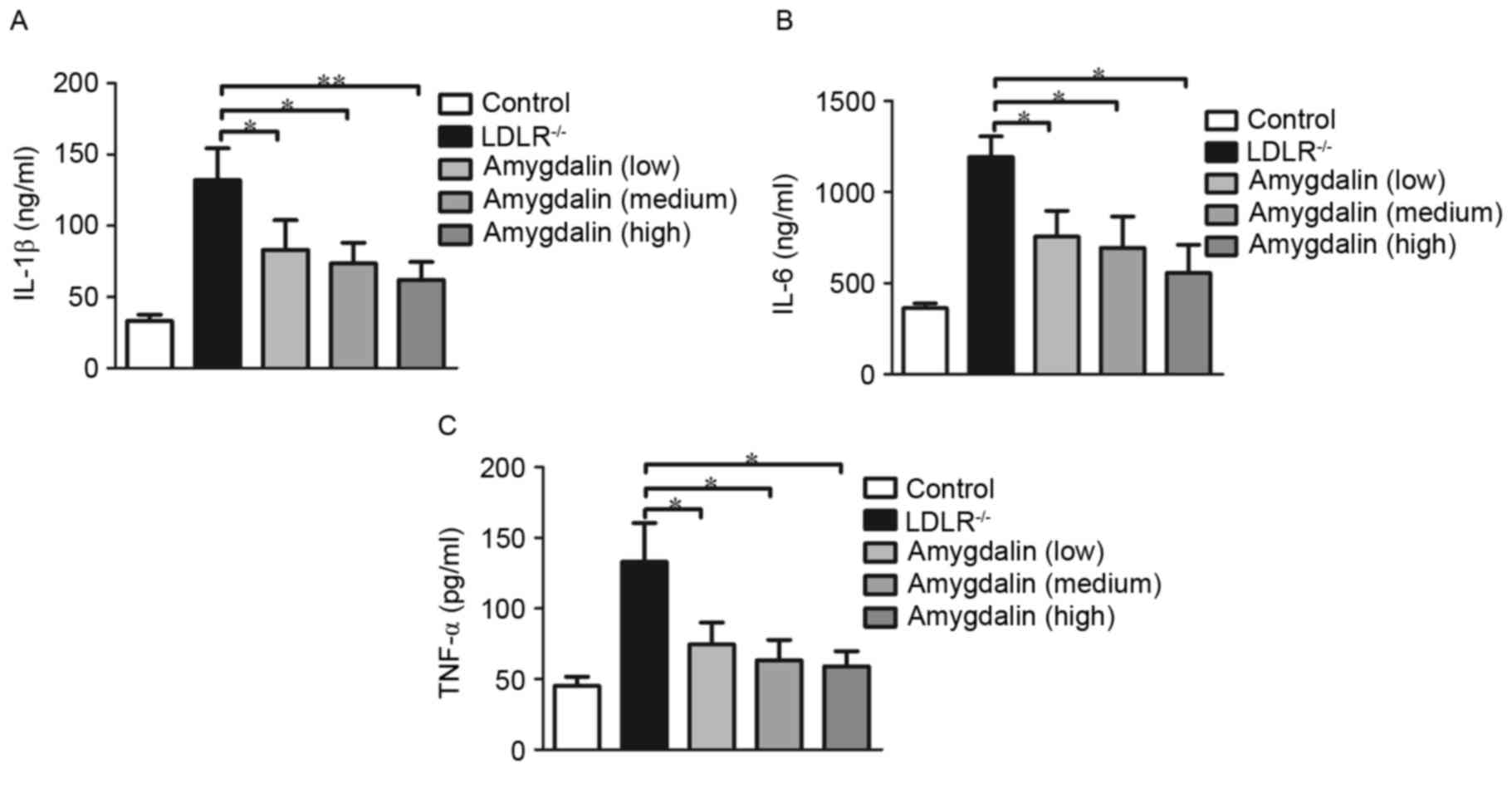
In addition, to further confirm the inhibitory function of
amygdalin in inflammation, plasma inflammatory markers were
assessed. As predicted, the analysis of serum inflammatory
cytokines revealed that amygdalin markedly reduced the secretion of
IL-1β (Fig. 3A), IL-6 (Fig. 3B), and TNF-α (Fig. 3C).
Amygdalin inhibits the progression of
atherosclerosis via the induction of Tregs
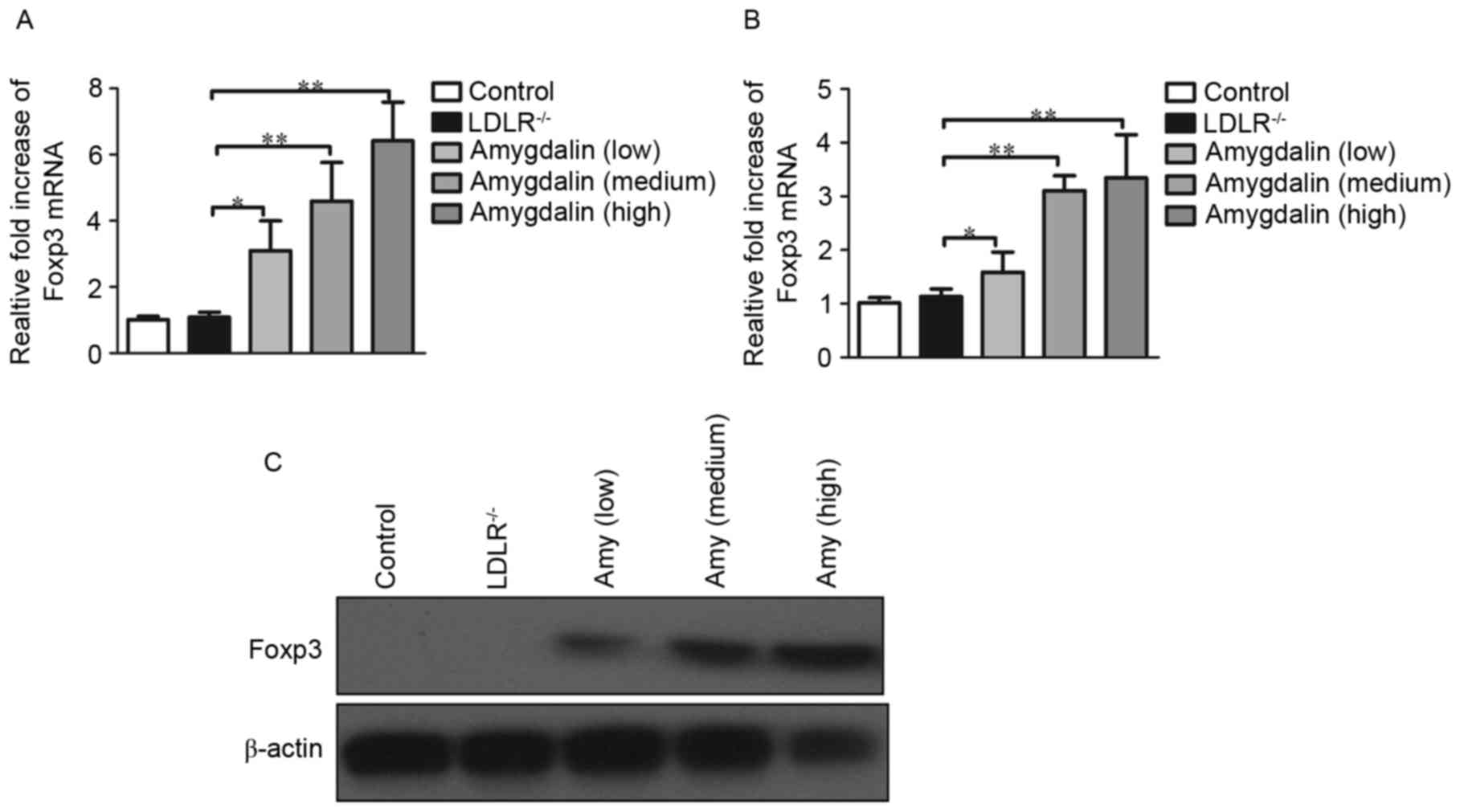
To determine whether the presence of Tregs is
associated with lipid profile changes and inhibited inflammatory
responses following amygdalin delivery, the present study
investigated the mRNA and protein levels of Foxp3 in splenocytes
and peripheral blood cells. Following quantification of them RNA
levels of Foxp3, the mRNA levels in peripheral blood cells were
increased by 6.3-fold (Fig. 4A),
and the mRNA levels in splenocytes were increased by 3.5-fold
(Fig. 4B) in the groups treated
with a high-dose of amygdalin, compared with the untreated groups
(P<0.05). Even in the groups treated with a low dose of
amygdalin, the mRNA levels of Foxp3 in the peripheral blood cells
and splenocytes were markedly elevated. There was also a marked
increase in the protein levels of Foxp3in the amygdalin-treated
groups (Fig. 4C).
A previous report indicated that the deletion of
Foxp3 results in the absence of functional Tregs in the periphery
and a conditionally active form of Foxp3 induces the reprogramming
of human T cells into Tregs (21).
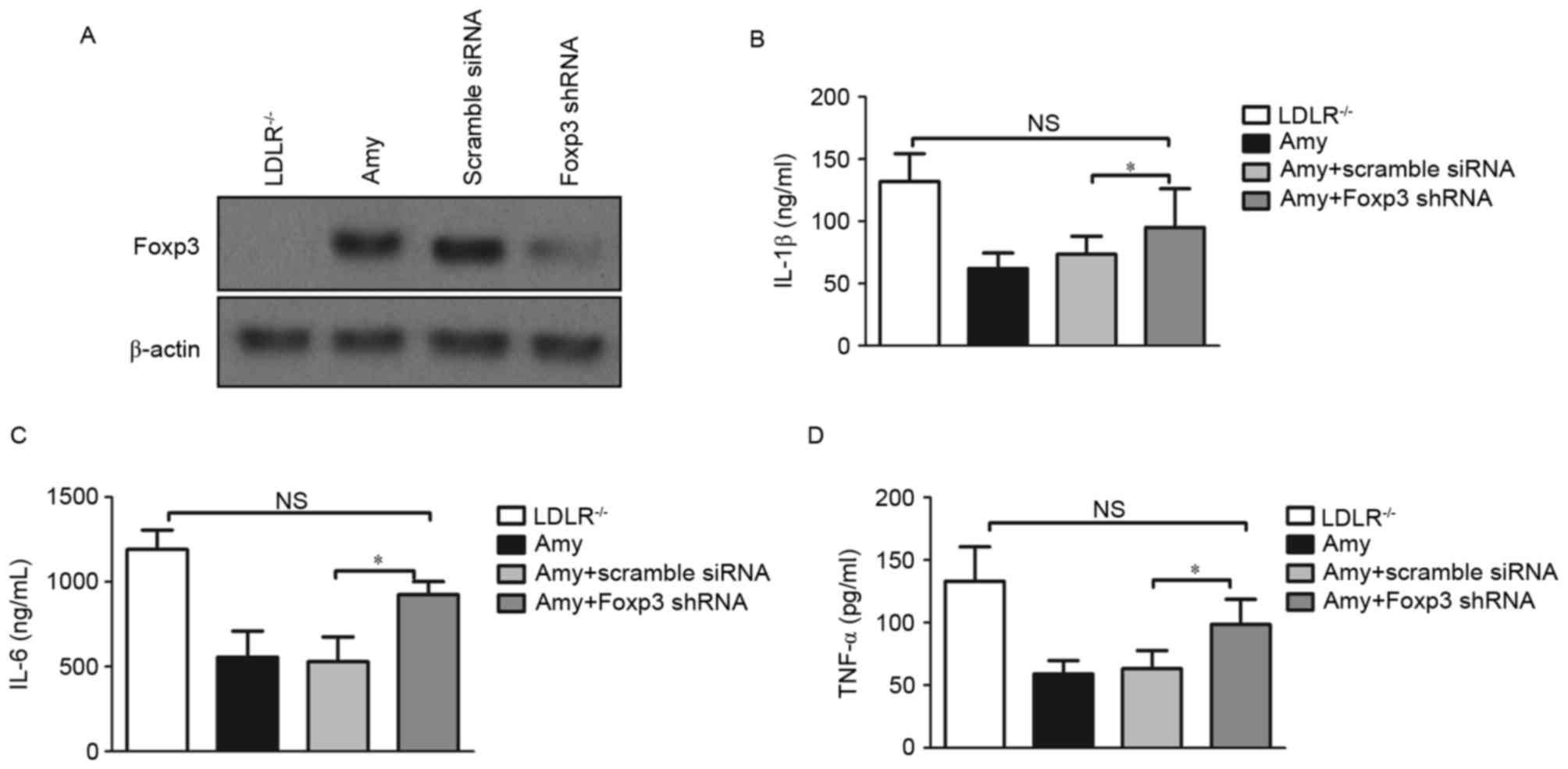
To further establish that Tregs are induced by amygdalin, the
knockdown of Foxp3mRNA was achieved using a lentiviral vector
carrying shRNA (Fig. 5A),
following which serum proinflammatory cytokines were assessed. As
expected, the levels of IL-1β (Fig.
5B), IL-6 (Fig. 5C) and TNF-α
(Fig. 5D) were significantly
increased in the amygdalin-treated mice bearing Foxp3 shRNA. These
data indicated that amygdalin attenuated the development of
atherosclerosis via the regulation of Tregs and proinflammatory
cytokines in LDLR−/− mice.
Discussion
The authors' previous studies on amygdalin in
ApoE−/− mice demonstrated that amygdalin ameliorated the
progression of atherosclerosis by the regulation of Tregs (12). However, to assess the regulatory
role of amygdalin on atherosclerosis induced by a western diet, the
LDLR−/− mouse model was used in the present study.
Unlike ApoE−/− mice, a spontaneous atherosclerotic
model, LDLR−/− mice do not develop atherosclerosis if
they are not fed with an HFD. A previous study demonstrated that
the oral administration of oxLDL induced attenuated atherosclerosis
in LDL−/− mice (22),
indicating that the abnormal lipid composition is involved in the
progression of atherosclerosis. The present study demonstrated that
amygdalin significantly inhibited the development of
atherosclerosis in LDLR−/− male mice fed an HFD as a
western-style diet. These mice, in addition to exhibiting
hypercholesterolemia, exhibited hypertriglyceridemia, therefore,
they exhibited the clinical features of human hypercholesterolemia
and hypertriglyceridemia. On comparing the blood cholesterol
levels, blood lipids were significantly decreased following
amygdalin treatment. This observation was consistent with our
previous study in ApoE−/− mice, indicating that
amygdalin is critical in the regulation of lipid profiles. Of note,
in the present study, the HFD produced more advanced
atherosclerotic lesions in the LDLR−/− mice.
There is increasing evidence that cytokines produced
by CD4+ T cells and macrophages, including TNF-α, IL-1β
and IL-6, affect the extent and nature of the atherosclerotic
plaque. A substantial number of clinical studies have shown that
the plasma concentration of TNF-α is associated with the degree of
atherosclerosis (23,24). To date, epidemiological studies
have found that several inflammatory mediators, including IL-6 and
TNF-α, appear to be elevated in association with increased vascular
risk, indicating cytokine-mediated inflammation is present in the
early stages of atherogenesis (25,26).
Huber et al demonstrated that the injection of recombinant
IL-6 exacerbated development of the early lesion of atherosclerosis
in mice (27). Studies on IL-1β
have also indicated that the overexpression of IL-1β occurs in
various inflammatory diseases, including atherosclerosis, and a
reduction of IL-1β decreases the severity of atherosclerosis in
ApoE−/− mice (28,29).
Studies by Kirii et al demonstrated a significant decrease
in atherosclerotic lesions at the aortic sinus in
ApoE−/−/IL-1β−/− mice, and the mRNA levels of
MCP-1 in the aorta were improved in these mice (28). Their results indicated that the
reduction of IL-1β decreases the severity of atherosclerosis
through increasing the expression of MCP-1. It has also been
reported that macrophage foam cells and smooth muscle cells express
IL-6, indicating that IL-6, in addition to IL-1β and TNF-α, are
critical in the progression of atherosclerosis (9). In the present study, it was
demonstrated that even a low dose of amygdalin markedly reduces the
expression levels of circulating cytokines, including IL-6, IL-1β
and TNF-α. However, compared with the low dose group, no
significant difference in the reduction of proinflammatory
cytokines was observed in when the mice were treated with a high
dose of amygdalin.
It is widely acknowledged that Tregs are important
in the development of atherosclerosis, confirmed by studies
investigating the adoptive transfer of lymphocytes (15,30)
and Th2 cytokines produced by Tregs. In the authors' previous
study, the anti-atherosclerotic function of amygdalin was
demonstrated by its successful induction of Tregs in
ApoE−/− mice. However, in the high-fat/high-cholesterol
diet-induced atherosclerosis of LDLR−/− mice, whether
the anti-atherosclerotic effect of amygdalin is via the regulation
of Tregs remains to be elucidated. Foxp3 has been demonstrated to
govern the development and function of Tregs (31). Mutations in Foxp3 eliminate
CD25+ Tregs and cause autoimmune disorders (32,33).
Therefore, the present study also focused on Tregs, and further
substantiation of our hypothesis was achieved by the silencing of
Foxp3. As expected, elevated mRNA and protein expression levels of
Foxp3 were observed in the amygdalin-treated mice. Following
silencing of Foxp3, a marked increase in cytokines was observed in
the circulation, indicating that the inhibition of Tregs
exacerbated the atherosclerotic situation. These results provide
evidence that the inhibition of Foxp3 acceleratedthe development of
atherosclerosis, even with amygdalin treatment, suggesting that the
anti-atherosclerotic effect of amygdalin occurred through the
regulation of Tregs.
Although it is widely acknowledged that lipids are
central in the pathogenesis of plaques, macrophages, and their
secreted cytokines contribute substantially to atherogenesis. It
has been reported that M1 macrophages in atheroma may have
pro-inflammatory functions, which produce high levels of effectors,
including cytokines IL-1β and TNF-α (13). According to the results of the
present study, demonstrated in Fig.
3, amygdalin significantly reduced the cytokine secretion of M1
macrophages, suggesting the anti-atherosclerotic effect of
amygdalin may be partly due to the inhibition of proinflammatory
cytokines. Specifically, the mice treated with a high dose of
amygdalin showed significantly decreased expression of CD68,
indicating that the high dose of amygdalin exerted a more marked
inhibitory effect on proinflammatory cytokines. As atheroma
formation also involves the recruitment of T cells and chemokines,
the present study examined other factors contributing to
atherosclerosis. It has been documented that MCP-1 has a unique and
crucial role in the initiation and evolution of atherosclerosis by
regulating the migration of monocytes and T-cells into the vessel
wall (34,35). In the present study, an increased
expression level of MCP-1 was observed in the atherosclerotic mice,
however, treatment with amygdalin markedly reduced the chemokine
secretion in the region of the lesion. The migration of SMCs also
contributes to vascular remodeling during the development and
complication of human atherosclerotic lesions. Clinical data show
that MMP-2 facilitates the migration and proliferation of SMCs and
may be important in atherogenesis (34). Cumulative evidence has demonstrated
that MMP-2-deficiency reduces atherosclerotic plaque lesion
formation in ApoE−/− mice (7). In clinical studies, that increased
MMP-2 and MMP-9 staining was observed in the plaques of expansively
remodeled segments, indicating that MMP-2 and MMP-9 may be involved
in plaque vulnerability and in expansive arterial remodeling
(36). By contrast, deficiency of
MMP-9 has also been associated with reduced plaque size, macrophage
content and collagen deposition in aortic lesions of
ApoE−/− mice (37) and
LDLR−/−/Apob100/100 mice (19). In the present study, it was
demonstrated that a high dose of amygdalin markedly reduced MMP-9
at the transcription and translation levels, indicating the
anti-atherosclerotic effect of amygdalin via the inhibition of
MMP-9.
In conclusion, the present study demonstrated that,
in the development of atherosclerosis in LDLR−/− mice
fed with an HFD (western-style diet), amygdalin inhibited the
progression of atherosclerosis. Amygdalin also markedly alleviated
hypercholesterolemia and hypertriglyceridemia in the
LDLR−/− mice and affected the extent of atherosclerotic
lesions. Subsequent investigations on the mechanism of
amygdalin-induced anti-atherosclerotic effects revealed that
amygdalin inhibited the inflammatory response and induced Tregs.
These results demonstrated the potential of amygdalin in the
control and treatment of atherosclerosis, and support the potential
of the clinical application of amygdalin as a novel
anti-inflammatory drug in the prevention of coronary heart
disease.
Acknowledgements
The present study was supported in part by the
Guangxi Natural Science Foundation for Youth (grant no.
2013GXNSFBA019139), Guangxi Colleges and Universities Science and
Technology Research Funding (grant no. ZD2014067), the Guangxi
Health and Family Planning Commission Funding (grant no.
GZPT13-05), the Guangxi Key Laboratory of Chinese Medicine
Foundation Research Funding, Guangxi University of Chinese Medicine
(grant no. 13-051-35) and Guangxi Key Laboratory of Common
Technology of Traditional Chinese Medicine Preparation Funding,
Guangxi University of Chinese Medicine to Dr Jianzhen Lv. The
present study was also supported by the Sichuan Health and Family
Planning Commission Funding (grant no. 16ZD0253), the Sichuan
National Science Research Funding (grant no. 2015JY0183) and the
Sichuan Scientific Research Foundation of the Returned Overseas
Chinese Scholars to Dr Yi Wang.
References
|
1
|
Glass CK and Witztum JL: Atherosclerosis.
The road ahead. Cell. 104:503–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ross R and Harker L: Hyperlipidemia and
atherosclerosis. Science. 193:1094–1100. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tell GS, Crouse JR and Furberg CD:
Relation between blood lipids, lipoproteins, and cerebrovascular
atherosclerosis. A review. Stroke. 19:423–430. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Napoli C, D'Armiento FP, Mancini FP,
Postiglione A, Witztum JL, Palumbo G and Palinski W: Fatty streak
formation occurs in human fetal aortas and is greatly enhanced by
maternal hypercholesterolemia. Intimal accumulation of low density
lipoprotein and its oxidation precede monocyte recruitment into
early atherosclerotic lesions. J Clin Invest. 100:2680–2690. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tedgui A and Mallat Z: Cytokines in
atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev.
86:515–581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Binder CJ, Chang MK, Shaw PX, Miller YI,
Hartvigsen K, Dewan A and Witztum JL: Innate and acquired immunity
in atherogenesis. Nat Med. 8:1218–1226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Whitman SC, Ravisankar P, Elam H and
Daugherty A: Exogenous interferon-gamma enhances atherosclerosis in
apolipoprotein E-/- mice. Am J Pathol. 157:1819–1824. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frostegård J, Ulfgren AK, Nyberg P, Hedin
U, Swedenborg J, Andersson U and Hansson GK: Cytokine expression in
advanced human atherosclerotic plaques: Dominance of
pro-inflammatory (Th1) and macrophage-stimulating cytokines.
Atherosclerosis. 145:33–43. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mallat Z, Gojova A, Brun V, Esposito B,
Fournier N, Cottrez F, Tedgui A and Groux H: Induction of a
regulatory T cell type 1 response reduces the development of
atherosclerosis in apolipoprotein E-knockout mice. Circulation.
108:1232–1237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiagang D, Li C, Wang H, Hao E, Du Z, Bao
C, Lv J and Wang Y: Amygdalin mediates relieved atherosclerosis in
apolipoprotein E deficient mice through the induction of regulatory
T cells. Biochem Biophys Res Commun. 411:523–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sluijter JP, Pulskens WP, Schoneveld AH,
Velema E, Strijder CF, Moll F, de Vries JP, Verheijen J,
Hanemaaijer R, de Kleijn DP and Pasterkamp G: Matrix
metalloproteinase 2 is associated with stable and matrix
metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic
lesions: A study in human endarterectomy specimen pointing to a
role for different extracellular matrix metalloproteinase inducer
glycosylation forms. Stroke. 37:235–239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
George J, Harats D, Gilburd B, Afek A,
Shaish A, Kopolovic J and Shoenfeld Y: Adoptive transfer of
beta(2)-glycoprotein I-reactive lymphocytes enhances early
atherosclerosis in LDL receptor-deficient mice. Circulation.
102:1822–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
George J, Afek A, Gilburd B, Shoenfeld Y
and Harats D: Cellular and humoral immune responses to heat shock
protein 65 are both involved in promoting fatty-streak formation in
LDL-receptor deficient mice. J Am Coll Cardiol. 38:900–905. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cobbold SP, Castejon R, Adams E, Zelenika
D, Graca L, Humm S and Waldmann H: Induction of Foxp3+
regulatory T cells in the periphery of T cell receptor transgenic
mice tolerized to transplants. J Immunol. 172:6003–6010. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang L, Chu Y, Wang Y, Zhao X, Xu W, Zhang
P, Liu X, Dong S, He W and Gao C: siRNA-mediated silencing of Wnt5a
regulates inflammatory responses in atherosclerosis through the
MAPK/NF-kB pathways. Int J Mol Med. 34:1147–1152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang G, Lv J, Li T, Huai G, Li X, Xiang
S, Wang L, Qin Z, Pang J, Zou B and Wang Y: Notoginsenoside R1
ameliorates podocyte injury in rats with diabetic nephropathy by
activating the PI3K/Akt signaling pathway. Int J Mol Med.
38:1179–1189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brattelid T, Winer LH, Levy FO, Liestøl K,
Sejersted OM and Andersson KB: Reference gene alternatives to Gapdh
in rodent and human heart failure gene expression studies. BMC Mol
Biol. 11:222010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsai BY, Suen JL and Chiang BL:
Lentiviral-mediated Foxp3 RNAi suppresses tumor growth of
regulatory T cell-like leukemia in a murine tumor model. Gene Ther.
17:972–979. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ziegler SF: FOXP3: Of mice and men. Annu
Rev Immunol. 24:209–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Puijvelde GH, Hauer AD, de Vos P, van
den Heuvel R, van Herwijnen MJ, Van der Zee R, van Eden W, van
Berkel TJ and Kuiper J: Induction of oral tolerance to oxidized
low-density lipoprotein ameliorates atherosclerosis. Circulation.
114:1968–1976. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Skoog T, Dichtl W, Boquist S,
Skoglund-Andersson C, Karpe F, Tang R, Bond MG, de Faire U, Nilsson
J, Eriksson P and Hamsten A: Plasma tumour necrosis factor-alpha
and early carotid atherosclerosis in healthy middle-aged men. Eur
Heart J. 23:376–383. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bruunsgaard H, Skinhøj P, Pedersen AN,
Schroll M and Pedersen BK: Ageing, tumour necrosis factor-alpha
(TNF-alpha) and atherosclerosis. Clin Exp Immunol. 121:255–260.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ridker PM, Rifai N, Stampfer MJ and
Hennekens CH: Plasma concentration of interleukin-6 and the risk of
future myocardial infarction among apparently healthy men.
Circulation. 101:1767–1772. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ridker PM, Rifai N, Pfeffer M, Sacks F,
Lepage S and Braunwald E: Elevation of tumor necrosis factor-alpha
and increased risk of recurrent coronary events after myocardial
infarction. Circulation. 101:2149–2153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huber SA, Sakkinen P, Conze D, Hardin N
and Tracy R: Interleukin-6 exacerbates early atherosclerosis in
mice. Arterioscler Thromb Vasc Biol. 19:2364–2367. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kirii H, Niwa T, Yamada Y, Wada H, Saito
K, Iwakura Y, Asano M, Moriwaki H and Seishima M: Lack of
interleukin-1beta decreases the severity of atherosclerosis in
ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 23:656–660.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou X, Nicoletti A, Elhage R and Hansson
GK: Transfer of CD4(+) T cells aggravates atherosclerosis in
immunodeficient apolipoprotein E knockout mice. Circulation.
102:2919–2922. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ochs HD, Ziegler SF and Torgerson TR:
FOXP3 acts as a rheostat of the immune response. Immunol Rev.
203:156–164. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mor A, Luboshits G, Planer D, Keren G and
George J: Altered status of CD4(+)CD25(+) regulatory T cells in
patients with acute coronary syndromes. Eur Heart J. 27:2530–2537.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brunkow ME, Jeffery EW, Hjerrild KA,
Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF
and Ramsdell F: Disruption of a new forkhead/winged-helix protein,
scurfin, results in the fatal lymphoproliferative disorder of the
scurfy mouse. Nat Genet. 27:68–73. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gu L, Okada Y, Clinton SK, Gerard C,
Sukhova GK, Libby P and Rollins BJ: Absence of monocyte
chemoattractant protein-1 reduces atherosclerosis in low density
lipoprotein receptor-deficient mice. Mol Cell. 2:275–281. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harrington JR: The role of MCP-1 in
atherosclerosis. Stem Cells. 18:65–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pasterkamp G, Schoneveld AH, Hijnen DJ, de
Kleijn DP, Teepen H, van der Wal AC and Borst C: Atherosclerotic
arterial remodeling and the localization of macrophages and matrix
metalloproteases 1, 2 and 9 in the human coronary artery.
Atherosclerosis. 150:245–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gough PJ, Gomez IG, Wille PT and Raines
EW: Macrophage expression of active MMP-9 induces acute plaque
disruption in apoE-deficient mice. J Clin Invest. 116:59–69. 2006.
View Article : Google Scholar : PubMed/NCBI
|