Introduction
Hepatic fibrosis is a common chronic disease caused
by long-term stimulation of one or more physical, chemical or
microbial factors in the liver. Hepatic fibrosis is characterized
by an aberrant accumulation of fibroblasts and excessive
extracellular matrix (ECM) deposition, with evident inflammatory
lesions and structural alterations (1). Despite the unclear pathogenesis of
hepatic fibrosis, early diagnosis and treatment may reduce the
mortality rate of patients (2).
Therefore, attenuating or reversing hepatic fibrosis has become an
important factor to be considered in the prevention and treatment
of chronic hepatic injury and cirrhosis.
Hepatic stellate cells (HSCs) are the cells which
primarily contribute to fibrogenesis during liver injury. These
oval-shaped cells are located in the hepatic sinusoidal space and
the space of Disse (3). Numerous
retinoid lipid droplets may be observed in the cytoplasm of HSCs,
indicating that the primary functions of HSCs are to store and
metabolize vitamin A, secrete ECM and produce collagenase.
Therefore, these cells are primarily involved in collagen synthesis
in the liver. The activation and proliferation of HSCs are
important events in hepatic fibrosis (4). A previous study suggested that the
activation of HSCs may include three important stages: The initial
stage, permanent stage and inflammation resolution stage (5). In the initial stage, ECM and other
products, including peroxides, are released from the damaged
hepatocytes. In the permanent stage, cellular behaviors are
categorized into at least six types: Proliferation, chemotaxis,
fibrogenesis, contraction, ECM degradation and vitamin A depletion.
In the inflammation resolution stage, HSC apoptosis is promoted, or
HSCs may be transformed into a quiescent state. Hepatocyte
apoptosis and necrosis caused by various harmful factors, including
the inflammatory response in liver tissues, activate Kupffer cells
to secrete pro-inflammatory cytokines. These cytokines, in addition
to various chemical transmitters, activate and transform HSCs into
myofibroblasts, resulting in an alteration to the functional
phenotype of HSCs (6). In
addition, activated HSCs may promote the proliferation of
myofibroblasts through paracrine or autocrine mechanisms, thereby
synthesizing large amounts of collagen fibers and other ECM
components. During this process, regulators, including tumor
necrosis factor-α (TNF-α) (7,8),
transforming growth factor β1 (TGF-β1) (9) and platelet-derived growth factor
(PDGF) (10), and the ECM,
interact with each other (11,12)
to form a sophisticated network and serve roles in hepatic
fibrogenesis.
Among these cytokines, PDGF is the most potent
factor involved in stimulating HSC proliferation, differentiation,
and migration (10). PDGF
additionally promotes collagen production and deposition, and
transforms HSCs into myofibroblasts. Blocking PDGF signaling
inhibits HSC proliferation and ameliorates liver fibrogenesis
(12). Clinical studies have
additionally confirmed that excessive activation of PDGF and its
downstream molecules appears to be associated with the extent of
necroinflammation and fibrosis in patients with hepatic damage
(13–15). Therefore, the PDGF signaling
pathway serves important roles in the development and prognosis of
hepatic fibrosis.
PDGF and the PDGF receptor (PDGFR)
PDGF family
PDGF is a polypeptide growth factor with a relative
molecular weight of 28–35 kDa; PDGF effectively promotes cell
division and proliferation. PDGF is primarily produced by
platelets, vascular endothelial cells, pericytes and Kupffer cells.
PDGF, as the product of oncogenesis, was first identified in
platelet α-granules at 1,000 PDGF molecules/platelet. Under
physiological conditions, PDGF is primarily secreted by Kupffer
cells. Once a tissue has been damaged, various diploid cells may
synthesize and secrete PDGF, which is associated with the
proliferation of connective tissues, including fibroblasts, and
vascular endothelial cells, via autocrine and paracrine mechanisms
(16).
A total of four PDGF subunits, termed PDGF-A, -B, -C
and -D, have been identified. These subunits contain highly
conservative homologous structural domains of PDGF/vascular
endothelial growth factor (VEGF), and they produce five homologous
or heterogeneous biopolymers, termed PDGF-AA, -BB, -AB, -CC, and
-DD, via a disulfide-bond linkage (17,18).
Among the dipolymers, PDGF-A, which exhibits a molecular weight of
16 kDa, is composed of 211 amino acids (aa), and located at the
chromosomal site 7p22. PDGF-A is highly-expressed in the muscle,
aorta and heart (19). PDGF-B,
with a molecular weight of 14 kDa, is located at the chromosomal
site 22q13 and is highly-expressed in the placenta and heart. These
two dipolymers are able to form three homologous or heterologous
dipolymers, termed PDGF-AA, -BB and -AB. PDGF-AA primarily binds
with PDGFR-αα to control the proliferation and chemotaxis of cells,
while PDGF-AB binds with PDGFR-αα and PDGFR-αβ, and PDGF-BB binds
with all subunits (PDGFR-αα, -αβ and -ββ); PDGFR-AB and -BB are
able to promote collagen synthesis and cellular adhesion (17).
The remaining PDGF subunits, PDGF-C and PDGF-D, were
discovered by comparing sequence data in the Expressed Sequence
Tags database (20,21). The structures and mechanisms of
these subunits remain to be elucidated. PDGF-C is composed of 345
aa, exhibits a molecular weight of 36.7 kDa, and consists of three
N-linked glycosylation sites at the 22nd, 55th and 254th aa
residues. PDGF-D consists of 370 aa, exhibits a molecular weight of
40.2 kDa, and consists of an N-linked glycosylation site at its
276th aa residue.
Unlike the first two isoforms, PDGF-C and PDGF-D
only produce homologous biopolymers, termed PDGF-CC and PDGF-DD,
the activity of which may be promoted via specific protease
cleavage of their CUB domains. PDGF-C and -D are highly-expressed
in the kidneys and heart.
PDGFR
PDGFR, which belongs to the receptor tyrosine kinase
(RTK) family and serves the function of a protein tyrosine kinase,
is primarily located in vascular endothelial cells, fibroblasts and
Kupffer cells (22). PDGFs have
two types of receptor, termed PDGFR-α and PDGFR-β, thereby forming
three subtypes of dipolymers, termed PDGFR-αα, -αβ, and -ββ.
PDGFR-α and PDGFR-β each contain five structural domains:
Immunoglobulin-like domain, transmembrane domain, ATP binding site,
intracellular hydrophilic kinase insert domain, and cytoplasmic
tail; these receptors are respectively located at the chromosomal
sites 4q12 and 5q33. PDGFR-α and PDGFR-β display different
expression patterns and physiological functions. Additionally, the
PDGFR-α signal and the PDGF-A, PDGF-B and PDGF-C chains, which
exhibit close binding affinities, are associated with the early
hematopoietic system and vascularization (23); however, PDGF-B and PDGF-D exhibit
high binding tendencies with PDGFR-β (24). Therefore, PDGF-B is more sensitive
to PDGFR-α and PDGFR-β compared with other subunits.
Biological functions of PDGF and PDGFR
In physiological conditions, PDGF is primarily
expressed in the α-granules of platelets. However, when liver
damage occurs, PDGF may be highly-expressed in macrophages, injured
endothelial cells and activated HSCs. During the early stages of
various chronic liver diseases, increased PDGFR expression on the
membranes of activated HSCs, in addition to activation of HSCs by
the synthesized PDGF via the autocrine mechanism, enhances cellular
chemotaxis and decreases the amount of intracellular vitamin A,
demonstrating that PDGF is involved in ECM production (22,23).
Further studies have also demonstrated that the four PDGF subunits
serve different roles in the pathogenesis of hepatic fibrosis. In
particular, PDGF-B has been demonstrated to be the most potent
factor associated with HSC activation (23,25).
PDGF-A mRNA expression exhibits a minor fluctuation during the
process of activated HSC transdifferentiation to myofibroblast-like
cells; PDGF-B is elevated during the early stage, although it is
markedly reduced from day 3 (17).
By contrast, PDGF-C and -D mRNA levels continuously rise during the
whole process of transdifferentiation (17). In addition, quiescent HSCs only
express PDGFR-α, whereas activated HSCs display a marked
upregulation of the expression of PDGFR. The expression profile of
PDGFR-β mRNA is consistent with that of PDGF-C and -D during
transdifferentiation (17). This
finding indicated that signal transduction is primarily mediated by
PDGF-B at the early stage, and by PDGF-C and PDGF-D at the late
stage of hepatic fibrogenesis (17).
A previous study demonstrated that the
PDGFR-β-mediated PDGF-B and PDGF-D signaling pathways are the most
important proliferation signaling pathways in the development of
hepatic fibrosis (26). In a rat
model of fibrotic liver disease induced by bile duct ligation
(BDL), the expression of PDGF-B, PDGF-D, and PDGFR-β mRNA were
respectively increased compared with the other isotypes; PDGF-B-
and PDGF-D-stimulated HSC activation and proliferation markedly
altered, exhibiting apparent activation morphologically. By
contrast, the proliferation of cells treated with PDGF-A or PDGF-C
did not alter (27). This previous
result indicated that PDGF-B and -D may induce more apparent
hepatic fibrosis compared with PDGF-A and -C. In addition, hepatic
fibrosis and HSC activation have been demonstrated to be markedly
increased in transgenic mice overexpressing PDGF-BB compared with
normal controls; the expression levels of α-smooth muscle actin
(α-SMA) and PDGFR-β in the liver were also elevated, demonstrating
that PDGF-B and PDGFR-β may promote fibrosis in an animal model and
at the cellular level (28).
Therefore, PDGFR-β expression may be most apparent in the
cytomembranes of HSCs. PDGFR-β may additionally exhibit relatively
strong affinities with PDGF-B and -D, and these three factors serve
important roles in hepatic fibrosis.
PDGF signaling pathway
The majority of the substrates of PDGF exhibit
similar structures to the Src homology 2 (SH2) domain. These
substrates bind to a corresponding phosphorylation site of the
activated PDGFR, enabling receptor dimerization and
autophosphorylation, and leading to phosphorylation of tyrosine
residues at specific intracellular sites (29). The currently identified substrates
primarily include phospholipase Cγ (PLCγ), Ras,
phosphatidylinositol 3-kinase (PI3K), and the signal transducer and
activator of transcription (STAT) pathway (Fig. 1).
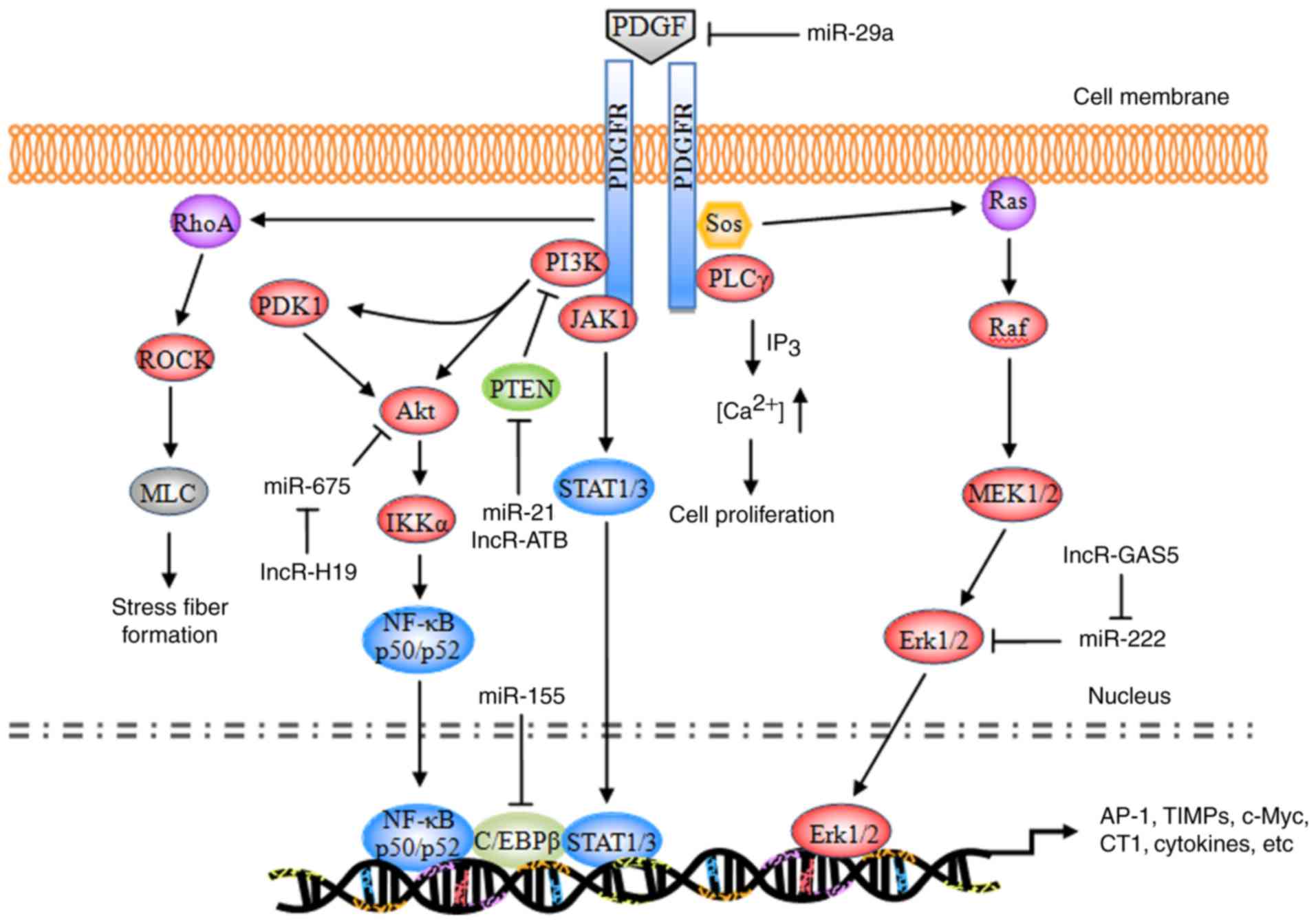 | Figure 1.Schematic representation of the PDGF
signaling pathway. PDGF, platelet-derived growth factor; PDGFR,
PDGF receptor; miR, microRNA; PI3K, phosphatidylinositol 3-kinase;
PLCγ, phospholipase Cγ; Sos, son of sevenless homolog; JAK1,
tyrosine-protein kinase JAK1; PDK1, 3-phosphoinositide-dependent
protein kinase 1; Akt, RAC-α serine/threonine protein kinase; PTEN,
phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and
dual-specificity protein phosphatase PTEN; ROCK, Rho-associated
protein kinase 1; MLC, membrane protein MLC; NF-κB, nuclear
factor-κB; IKKα, inhibitor of NF-κB kinase subunit α; C/EBPβ,
CCAAT/enhancer-binding protein β; STAT, signal transducer and
activator of transcription; MEK, dual specificity mitogen-activated
protein kinase kinase; Erk, extracellular signal-regulated kinase;
AP-1, transcription factor AP-1; TIMP, metalloproteinase inhibitor;
c-Myc, myc proto-oncogene protein; CT1, cardiotrophin 1. |
During the development of hepatic fibrosis,
autophosphorylated PDGFR primarily activates the Ras system, which
in turn activates the RAF proto-oncogene serine/threonine protein
kinase (Raf-1), dual-specificity mitogen-activated protein kinase
kinaseN (MEK) and extracellular signal-regulated protein kinase
(ERK) signaling pathways. The activated signaling pathways enhance
the phosphorylation of cytoplasmic target proteins, regulate the
activity of various proteases, promote the phosphorylation of a
number of transcription factors [including activator protein 1
(AP-1) and nuclear factor-κB (NF-κB)] and enable them to bond with
the target gene promoters of the corresponding response elements.
Consequently, these downstream elements increase the
transcriptional activity, regulate the expression levels of the
products of target genes [including type I collagen (CT I),
metalloproteinase inhibitors (TIMPs), matrix metalloproteinases
(MMPs), apoptosis regulator Bcl-2 (Bcl-2), and E3 ubiquitin-protein
ligase XIAP (XIAP)], resulting in cell division, proliferation and
apoptosis (30–32).
Ras pathway
Ras is a small GTPase with a relative molecular
weight of 21 kDa. Ras has been demonstrated to be a ‘crossing
point’ or molecular switch for a variety of cell signal
transduction processes. The protein possesses endogenous GTP enzyme
activity, facilitating extracellular-to-intracellular signaling,
and is additionally an upstream protein of the Raf-MEK-ERK pathway
(33). Ras, combined with c-Raf-1,
facilitates the transport of Raf from the cytoplasm to the
cytomembrane; Raf is subsequently activated by the corresponding
kinase on the cytomembrane and its C-end catalytic domain bonded
with MEK phosphorylates (via MAPK) two Ser residues in the MEK
catalytic domain, thereby activating MEK.
PDGF, combined with a corresponding receptor on the
cytomembrane, induces the phosphorylation of intracellular tyrosine
residues of the receptor; the phosphorylated tyrosine may recruit
GF receptors containing SH structural domains from the cytoplasm to
bond with the protein 2 (Grb2); by virtue of its SH2 structural
domain, Grb2 binds with the guanine nucleotide exchange factor [son
of sevenless (SOS)] and forms a Grb2-SOS compound, which
dissociates the GDP of Ras to bind with GTP and converts the
quiescent Ras-GDP into an active Ras-GTP to activate Ras. The
activated Ras in turn activates Raf1, MEKl/2 and ERKl/2 and
transfers the corresponding signals into the cell nucleus, thereby
promoting the phosphorylation of various transcription factors,
increasing transcriptional activity, and triggering cell growth,
differentiation, migration, angiogenesis, anti-apoptosis, drug
tolerance and other processes (34,35).
During the induction of hepatic fibrosis by PDGF, ERK1/2 primarily
regulates the mitosis and chemotaxis of HSCs. The ERK1/2 pathway
inhibitor is able to completely inhibit mitosis in HSCs and
decrease the mitogenesis and chemotaxis of the cells, thus reducing
the concentration of inflammatory sites (36–38).
PLCγ pathway
PLCγ is a 145-kDa enzyme consisting of two SH2
domains and one SH3 domain. In hepatic fibrosis, PDGF mediates the
cell proliferation via the regulatory role of PLCγ in the mitosis
of HSCs (39). The primary
mechanism of action is as follows: When PDGF is bonded with PDGFR,
PLCγ is activated by a protein tyrosine kinase; the activated PLCγ
hydrolyzes phosphatidylinositol diphosphate, producing inositol
triphosphate (IP3) and diacylglycerol (DAG). IP3 primarily acts on
the Ca2+ repository of the endoplasmic reticulum,
promoting the release of cytoplasmic Ca2+; the
intracellular Ca2+ thus increases and induces mitosis in
HSCs. DAG, together with Ca2+, acts on the protease
protein kinase C (PKC) and induces the phosphorylation of Ser and
Thr residues of the ribosome, in addition to the proliferation of
cells (40,41). In addition, DAG is able to activate
the intracellular Na+/H+ proton pump, leading
to the frequent intracellular H+ and extracellular
Na+ interchanges; DAG additionally decreases the
quantity of cellular H+, which is conducive to mitosis
(42,43).
PI3K pathway
PI3K is a heterogeneous dipolymer consisting of two
subunits, termed p85 and p110. p85 is the subunit which regulates
the primary functions of PI3K (44). The RAC-α serine/threonine protein
kinase (Akt/PKB) pathway is currently considered to be the primary
downstream signaling pathway of PI3K. PKB is a serine/threonine
protease and it encompasses three domains: The N-terminal
regulatory domain, the catalytic domain, and the C-terminal tail
domain. The specific signaling pathway is as follows: The p85
subunit, which is bonded with the phosphorylated PDGFR,
phosphorylates the third group of the phosphatidylinositol ring,
producing PI (3,4) P2, PI (3–5) P3,
and certain secondary messengers for downstream signaling; the
latter may bind with the group via the N-terminal regulatory domain
of PKB, transporting it to the cytomembrane or partially activating
it. Notably, phosphorylated sites respectively activate
3-phosphoinositide-dependent protein kinase 1 (PDK1) and PDK2;
subsequently, PDK1 and PDK2, or an unknown Ser473 kinase,
phosphorylates the Ser473 and Thr308 ion of the PKB protein,
thereby completely activating the group. The PDGFR-activated
PI3K/Akt pathway may promote actin recombination, increase cell
migration, mediate metabolic regulation, stimulate cell growth and
inhibit cellular apoptosis (45).
ERK signaling is involved in PI3K/Akt-mediated HSC chemotaxis and
proliferation (46).
Tyrosine protein kinase JAK (JAK)/STAT
pathway
STAT signaling is a process wherein an 84–113-kDa
cytoplasmic protein, combined with the regulatory domain DNA of the
target gene, regulates transcription. A total of seven STAT family
members have been identified, termed STATl, STAT2, STAT3, STAT4,
STAT5a, STAT5b, and STAT6. STAT is an intracellular signal
transduction protein, and additionally acts as a transcription
factor (47). Following
phosphorylation by tyrosine protein kinases of the JAK family,
STATs produce homologous or heterogeneous dipolymers; the
dipolymers are subsequently translocated, enter the nucleus, and
trigger gene transcription following DNA-binding (48). Activated PDGFR primarily triggers
the phosphorylation of the C-terminal tyrosine residues (Tyr705)
and serine residues (Ser727) of the STAT protein via JAKs, thereby
activating STAT5, leading to the production of homologous or
heterologous STAT protein dipolymers. The dipolymers are
subsequently translocated, enter the nucleus, activate the target
genes, and promote cell growth and division (49,50).
Rho pathway
Using two intracellular signaling pathways (PKC and
RhoA), PDGF alters the cytoskeleton distribution in HSCs,
stimulates the transformation of HSCs into muscle-like fibroblasts,
and promotes type I and III collagen formation (51).
Non-coding RNA (ncRNA) regulation
Altered profiles of ncRNAs, including microRNAs
(miRNAs/miRs), long non-coding RNAs (lncRNAs) and circular RNAs
(circRNAs), have been demonstrated to be associated with hepatic
fibrosis. Different alterations of miRNA expression have been
observed. Several specific miRNAs (including let-7d, miR-29a,
miR-21, miR-155 and miR-214) (52–56)
and lncRNAs (lncRNA-ATB, GAS5 and H19) (57–59),
have been identified to be diagnostic biomarkers and the
therapeutic targets in the progression of hepatic fibrosis. miRNAs
induced or repressed by PDGF challenge exhibit a feedback mechanism
balancing multiple growth factor receptor signaling in HSC
activation (52–56,60,61),
while lncRNAs are able to competitively bind to anti-fibrogenic
miRNAs leading to increased collagen I production (57–59).
Since a single ncRNA may affect the expression of various mRNAs, it
may simultaneously modulate various cellular events. At present,
the multiple molecular mechanism through which ncRNAs may serve a
role in hepatic fibrogenesis have not been completely
elucidated.
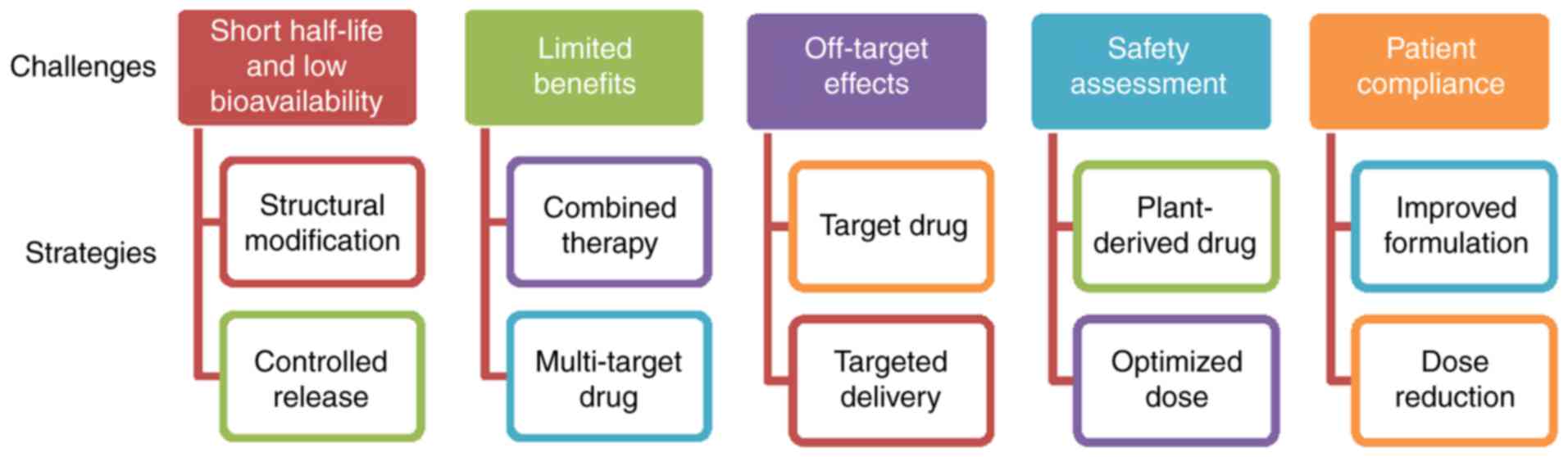
Targeting the PDGF signaling pathway in
hepatic fibrosis treatment
With the increased understanding of the biological
functions of the PDGF signaling pathway in liver fibrogenesis,
antifibrotic drugs have become a focus of research. A number of
strategies to regulate the PDGF signaling pathway have been
employed in preclinical and clinical settings (Table I). These may be categorized as: i)
PDGF isoform antagonists; ii) blocking of PDGFR activation; and
iii) downstream regulation of the PDGF pathway.
 | Table I.PDGF inhibitors and
downregulators. |
Table I.
PDGF inhibitors and
downregulators.
| Author, year | Drug | Targets | Action | Applications | Groups | (Refs.) |
|---|
| Gounder et
al, 2011; Zahavi et al, 2016; Lin et al, 2016;
Hong et al, 2013; Hao et al, 2016; Liu et al,
2015 | Sorafenib | Raf kinase,
PDGFR-β, VEGFR2/3, c-kit | Receptor tyrosine
kinase inhibitor | Advanced renal cell
carcinoma and unresectable hepatocellular carcinoma | Approved | (66–69,71,72) |
| Qu et al,
2016; Karuppagounder et al, 2014; Elsherbiny et al,
2015; Shaker et al, 2011; Lemos et al, 2015 | Imatinib | Bcr-Abl, PDGFR-β,
c-kit | Protein-tyrosine
kinase inhibitor | Chronic myelogenous
leukemia | Approved | (74–78) |
| Moawad, 2015; Kuo
et al, 2012; Kim et al, 2012 | Nilotinib | PDGFR-α/β | Tyrosine kinase
inhibitor | Chronic myelogenous
leukemia | Approved | (82–84) |
| Hutson et
al, 2010 | Pazopanib | PDGFR-α/β,
VEGFR1/2/3, c-kit | Protein-tyrosine
kinase inhibitor | Advanced renal cell
cancer and advanced soft tissue sarcoma | Approved | (86) |
| Eisen et al,
2012 | Regorafenib | PDGFR-α/β,
VEGFR1/2/3, RET, BRAF, FGFR1/2 | Multiple
membrane-bound and intracellular kinases inhibitor | Metastatic
colorectal cancer and advanced gastrointestinal stromal tumors | Approved | (87) |
| Iwamoto et
al, 2000; Rice et al, 1999 | AG1295/1296 | PDGFR-β | Tyrosine kinase
inhibitor | Unknown | Preclinical | (85,88) |
| Venè et al,
2016; Soininen et al, 2007; Raval et al, 2010; Gao
et al, 2013 | Celecoxib | COX-2, Akt | COX-2
inhibitor | Rheumatoid
arthritis, osteoarthritis, acute pain, colon and familial
adenomatous polyposis | Approved | (89–92) |
| Trappoliere et
al, 2009; Serviddio et al, 2014 | Silymarin | Phosphorylation of
IkB and Raf/MEK/ERK | Unknown | Liver related
diseases including hepatic fibrosis and cirrhosis | Approved | (96,97) |
| Zhou et al,
2015; Lian et al, 2015; Taverna et al, 2015; El-Bahr,
2013; Zhang et al, 2015 | Curcumin | PI3K/Akt, ERK | Unknown | Edible pigment;
aging, irradiation, hepatic fibrosis and hepatitis | Approved | (100–104) |
| Wang et al,
2012; Lv et al, 2010; Gao et al, 2012; Xu et
al, 2012 | Salvianolic acid
B | JAK2/STAT3, TGF-β1
and RhoA | Unknown | Myocardial
infarction, cerebral ischemia, hepatic fibrosis and hepatitis | Approved | (110–113) |
PDGF-B kinoid vaccines, prepared using
PDGF-B-derived polypeptides bonded to carrier protein
heterocomplexes, displayed marked antifibrotic effects in
CCl4-induced hepatic fibrosis (62). Notably, the normal function of the
hepatocytes was not altered by the vaccine. Although the side
effects were not observed in this previous study and in other
reports (63–65), the safety of PDGF vaccine requires
investigation in a systematic toxicity assay.
Sorafenib is a first-line oral chemotherapy drug for
patients with advanced hepatocellular carcinoma. As an RTK
inhibitor, sorafenib may target a number of kinases on the
cytomembrane, including Raf, VEGFR2/3, and PDGFR-β (66,67).
Despite the narrow therapeutic window and adverse effects (68,69),
sorafenib has previously been demonstrated to be a potential
antifibrotic agent, due to its multi-targeting of the Ras/MEK/ERK
pathway (70). Sorafenib may
induce HSC autophagy and apoptosis through activation of the
Akt/protein kinase mTOR/p70S6K and MAPK signaling pathways
(71). It may additionally
suppress collagen deposition, neovascularization and oxidative
stress through PDGF downregulation, STAT3 inhibition and
mitochondrial respiration in fibrosis mouse models consuming a high
fat diet, and undergoing BDL and dimethylnitrosamine injection
(72,73).
Nilotinib, a selective breakpoint cluster region
protein (Bcr)-tyrosine-protein kinase ABL (Abl) non-receptor
tyrosine kinase (nRTK) inhibitor, is well-tolerated and has been
approved for the treatment of leukemia. Since RTKs may be activated
by nRTKs, the interaction between RTKs and nRTKs is involved in the
regulation of HSC differentiation and proliferation (74). In addition to its anticancer
activity, nilotinib therefore exerts numerous beneficial
therapeutic outcomes, including neuroprotective, vasodilatory and
antifibrotic effects (75–78). Nilotinib inhibited α-SMA,
procollagen-(I), TIMP-1, phosphorylated (p)-ERK and p-Akt
expression, and reduced collagen deposition in activated HSCs and
in the liver tissues of CCl4- and BDL-induced fibrosis
rats (79–81). Nilotinib inhibited PDGFR
activation, in addition to TGFβ receptor type II via Src (81). These previous results indicated
that nilotinib may represent a putative antifibrotic treatment due
to its combined inhibition of nRTK and RTK.
Imatinib (also termed Gleevec® and
STI-571) is a selective tyrosine kinase inhibitor (TKI), which is
able to specifically target PDGFR, Abl, Abelson tyrosine-protein
kinase 2, mast/stem cell growth factor receptor, and their
oncogenic forms Bcr-Abl (82). At
concentrations required for Bcr-Abl inhibition, imatinib is
additionally able to attenuate hepatic fibrosis by blocking the
expression of PDGFR-β and decreasing the levels of proinflammatory
cytokines. Therefore, imatinib has been demonstrated to induce HSC
apoptosis in vitro and to control liver fibrosis in
CCl4- and thioacetamide (TAA)-treated mice (80,82–84).
Compared with a decreased of 60% mediated by anti-PDGF antibodies,
imatinib has been demonstrated to exert an 85% decrease in HSC
migration triggered by bile duct segments (27). However, unlike nilotinib, animals
treated with imatinib (20 mg/kg) exhibited a degree of
hepatotoxicity evidenced by increased levels of serum
aminotransferases and total bilirubin (79).
Other TKIs, including pazopanib, regorafenib, AG1295
and AG1296, may selectively inhibit the tyrosine phosphorylation of
PDGFR-β (85–87) and the PDGF-BB-induced activation of
its downstream signaling pathway in HSCs (85). additionally, AG1295 inhibits
PDGF-induced thymidine uptake by pulmonary myofibroblasts in
vitro (88).
Celecoxib, etoricoxib and DFU, selective
cyclooxygenase-2 (COX-2) inhibitors (coxibs), are widely-used in
the management of osteoarthritis and rheumatoid arthritis, in
addition to the treatment of colon cancer, atherosclerosis and
Alzheimer's disease, due to their analgesic, anticoagulant and
anti-inflammatory effects (89–91).
During the development of steatohepatitis and hepatic fibrosis,
COX-2 and its products prostaglandin E2
(PGE2) and prostacyclin (PGI) may upregulate the
expression of VEGF, PDGF and fibroblast growth factor receptor 1,
resulting in ERK activation, and COX-2 may be activated by these
factors (92). COX-2 inhibitors
may alter the metabolism of arachidonic acid and, subsequently,
PGE2 and PGI. Therefore, coxibs may inhibit PDGF-induced
HSC proliferation; however, in contrast to NS-398 and DFU, only
celecoxib (≥50 mM) is able to induce HSC apoptosis and inhibit Akt
activation (93). Oral
administration of celecoxib significantly decreased hepatic
collagen deposition and α-SMA expression in CCl4- and
TAA-treated rats due to its dual inhibitory effects on intrahepatic
fibrosis and angiogenesis (94).
A number of active substances from traditional
herbal and ethnobotanical medicine [e.g., silymarin, quercetin,
proanthocyanidins, curcumin and salvianolic acid B (Sal B)] have
come into use as putative treatments for liver disease. Silymarin,
a mixture of flavonolignans obtained from the edible milk thistle
plant [Silybum marinaum (L) Gaenrt], has been used as a
natural medicine for the treatment of liver diseases. The four
principal isomers within silymarin are silybin, isosilybin,
siliehristin and silydianin (95).
Among these isomers, silybin is the most bioactive substance, which
is able to directly inhibit the phosphorylation of the Raf/MEK/ERK
pathway, decrease the activation of sodium/hydrogen exchanger 1 and
inhibitor of NF-κBα phosphorylation, thereby preventing oxidative
anomalies and fibrosis (96,97).
Treatment with silybin or silybin-vitamin E phospholipid complexes
has been demonstrated to ameliorate hepatic fibrosis in patients
with chronic hepatitis C, who have been treated previously with
pegylated-interferon a and ribavirin (98,99).
Curcumin, the principal component of the spice
turmeric and certain herbal medicines (Zedoariae rhizome and
Radix Curcumae), is able to inhibit
epithelial-to-mesenchymal transition and affect numerous
intracellular targets, involving certain miRNAs, and the estrogen
receptor, nuclear factor-like 2, insulin-like growth factor-1 and
PI3K/Akt signaling pathways (100–103). Due to its numerous potential
effects, including anti-inflammatory, hypolipidemic, hypoglycemic
and chemopreventive activity, curcumin may increase antioxidant
enzyme activities, activate cytochrome P4502E1 and concomitantly
suppress the activity of fatty acid synthase, β-catenin
transactivation and DNA-binding (104,105). In addition, curcumin has been
demonstrated to markedly reverse PDGF-BB-induced hepatic
myofibroblast cell proliferation and the expression of collagen I
and collagen IV mRNA. Curcumin may alleviate hepatic fibrosis by
modulating lipid metabolism and inducing HSC apoptosis, in part via
the PDGF- and ERK-dependent pathway (101,106).
Sal B, and its metabolite danshensu, the major
active substances from Salvia miltiorrhiza, are widely-used
as commercial anticoagulant drugs in the treatment of myocardial
infarction and cerebral ischemia (107–109). The two substances exhibit
hepatoprotective effects against CCl4-induced lipid
peroxidation, collagen accumulation and necroinflammation in liver
tissues, the probable mechanism underlying which is associated with
the regulation of the intrahepatic JAK2/STAT3 and TGF-β1/mothers
against decapentaplegic homolog pathways for maintaining collagenic
homoeostasis (110–112). In vitro, Sal B has been
demonstrated to inhibit endothelin-induced HSC activation by
regulating RhoA/cardiac myosin light chain 2 signaling activation
and the phosphorylation of downstream protein phosphatase 1
regulatory subunit 12A at Thr(696) (113).
Future challenges and prospects
PDGF-B-mediated PDGFR-β signaling has been
demonstrated to serve an important role in hepatic fibrosis
(17,22). Although the numerous approaches
mentioned above have been applied to modulate this pathway, no
ideal treatment for liver fibrosis has been employed in clinical
practice at present (Fig. 2).
TKIs, coxibs and natural products exhibit short half-lives and low
bioavailabilities, and therefore require long-time repeated
administration to achieve therapeutic benefits (114,115). In addition, the majority of TKIs
are only approved for cancer therapy, and coxibs for arthritis.
Although the effectiveness of TKIs and coxibs has been demonstrated
in animal models and cultured HSCs, the outcomes of patients
treated in early-phase clinical trials have not been systematically
analyzed. Notably, due to the high similarity of the homologous
domain, TKIs, including sorafenib, sunitinib and pazopanib, may
inhibit PDGFR expression and regulate the expression of VEGF
receptors (VEGFR), which are involved in the maintenance of vessel
diameters and the integrity of endothelial cells (116); therefore, the inhibition of PDGFR
by these approved targeted drugs may impair non-target normal cells
or tissues. The primary concerns about the safety of TKIs and
coxibs are bleeding and myocardial ischemia. Congestive liver
failure, QT prolongation and severe fatigue induced by TKIs have
additionally been observed in certain clinical trials (117–119). With the development of molecular
biotechnology methods, gene therapy may be used to transport
genetic materials to the specific cells for correcting or targeting
the genetic defects, thereby achieving the goal of disease
treatment. However, there are significant potential safety and
target-specific concerns associated with small interfering RNA- and
adenovirus-mediated gene therapies involving genetic modification
(120).
An additional challenge is that the inhibition of a
single profibrotic molecule may exert little or no impact on
fibrogenesis and overall recovery. Hepatic fibrogenesis is a
complex process involving a variety of pathogenic and host-specific
signal transduction processes (4).
Certain stimuli, including TGF-β1, TNF-α and hepatitis viruses,
serve roles in HSC activation. Different stages of fibrosis
development may depend on different growth factors (7–9).
Notably, PDGF mediates the actions of these stimuli in liver cells,
including the regulation of hepatocyte growth and death, in
addition to the differentiation of activated HSCs to
myofibroblasts; conversely, TGF-β1, TNF-α and hepatitis-mediated
oxidative stress may upregulate PDGF expression (121,122). In particular, hepatitis B virus-
or hepatitis C virus-associated fibrosis is considered to be
irreversible prior to the control of the ongoing viral replication
and inflammation by combined treatment (123). In addition, it remains unknown
whether the normal biological functions of the liver may be
affected by the inhibition of PDGF signaling. Considering that that
liver is a non-immunological organ engaged primarily in metabolism,
nutrient storage and detoxification, novel therapies are required
to deliver longer lasting benefits directly to the targeted cells.
Combination strategies (including multi-agent regimens and improved
drug delivery) are the future of antifibrotic therapy, and the
primary focus for medicinal chemistry is the reduction of toxicity
and the maintenance (or enhancement) of potency. Novel combinations
may simultaneously target multiple profibrotic factors to induce
HSC apoptosis, improve the microenvironment, prolong drug release
and reverse drug resistance. Co-delivery systems modified by
nanotechnology may be a promising strategy to maximize the additive
or synergistic effects of drugs, since various agents may
specifically bind to the target together. These multi-target
delivery systems, which may more specifically deliver inhibitors to
the target, may be able to increase the antifibrotic action and
decrease the exposure of normal and non-targeted cells or tissues.
However, epidemiological evidence for the association between plant
remedies (involving the mixed herbal formulations) and the long
latency of hepatic fibrosis progression has led to the active
investigation of natural chemicals from different
naturally-occurring substances in various preclinical and clinical
studies. In general, the potential of natural compounds to exhibit
low toxicity and low drug-resistance, with pleiotropy and
synergistic action, makes them promising candidates for the
treatment of hepatic fibrosis. A number of plant-derived active
compounds have been approved for the treatment of hepatic fibrosis
and may provide valuable resources for the development of novel
formulations and treatment modalities in future.
Conclusion
In conclusion, excessive PDGF expression is an
important factor in HSC activation. Hepatic fibrosis may be
effectively reversed by inhibiting PDGF and PDGFR expression, or by
inhibiting the phosphorylation of its downstream signaling
pathways, although complete elucidation of the mechanisms
underlying signaling transduction and crosstalk between the
different pathways requires additional research. A number of PDGF
antagonists and receptor inhibitors have been previously
investigated for their potential to treat hepatic fibrosis,
although further clinical trials to examine their safety and
effectiveness may be considered.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant no. 81573591), the Zhejiang
Science and Technology Foundation (grant nos. 2014F10033 and
2015F50056), Zhejiang Innovation discipline ‘Laboratory Animal
Genetic Engineering’ (grant no. 201604) and the Zhejiang Foundation
for the Cultivation of High-level Innovative Health Talents (grant
no. 201206).
References
|
1
|
Bonder A, Tapper EB and Afdhal NH:
Contemporary assessment of hepatic fibrosis. Clin Liver Dis.
19:123–134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Trautwein C, Friedman SL, Schuppan D and
Pinzani M: Hepatic fibrosis: Concept to treatment. J Hepatol. 62(1
Suppl): S15–S24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jacobs F, Wisse E and De Geest B: The role
of liver sinusoidal cells in hepatocyte-directed gene transfer. Am
J Pathol. 176:14–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tacke F and Weiskirchen R: Update on
hepatic stellate cells: Pathogenic role in liver fibrosis and novel
isolation techniques. Expert Rev Gastroenterol Hepatol. 6:67–80.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin C, Evason KJ, Asahina K and Stainier
DY: Hepatic stellate cells in liver development, regeneration, and
cancer. J Clin Invest. 123:1902–1910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu W, Baker RD, Bhatia T, Zhu L and Baker
SS: Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life
Sci. 73:1969–1987. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Robert S, Gicquel T, Bodin A, Lagente V
and Boichot E: Characterization of the MMP/TIMP imbalance and
collagen production induced by IL-1β or TNF-α release from human
hepatic stellate cells. PLoS One. 11:e01531182016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ceccarelli S, Panera N, Mina M, Gnani D,
De Stefanis C, Crudele A, Rychlicki C, Petrini S, Bruscalupi G,
Agostinelli L, et al: LPS-induced TNF-α factor mediates
pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty
liver disease. Oncotarget. 6:41434–41452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shah R, Reyes-Gordillo K,
Arellanes-Robledo J, Lechuga CG, Hernández-Nazara Z, Cotty A,
Rojkind M and Lakshman MR: TGF-β1 up-regulates the expression of
PDGF-β receptor mRNA and induces a delayed PI3K-, AKT- and
p70(S6K)-dependent proliferative response in activated hepatic
stellate cells. Alcohol Clin Exp Res. 37:1838–1848. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kocabayoglu P, Lade A, Lee YA, Dragomir
AC, Sun X, Fiel MI, Thung S, Aloman C, Soriano P, Hoshida Y and
Friedman SL: β-PDGF receptor expressed by hepatic stellate cells
regulates fibrosis in murine liver injury, but not carcinogenesis.
J Hepatol. 63:141–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baig MS, Yaqoob U, Cao S, Saqib U and Shah
VH: Non-canonical role of matrix metalloprotease (MMP) in
activation and migration of hepatic stellate cells (HSCs). Life
Sci. 155:155–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang N, Zhang YP, Ying W and Yao XX:
Interleukin-1β upregulates matrix metalloproteinase-13 gene
expression via c-Jun N-terminal kinase and p38 MAPK pathways in rat
hepatic stellate cells. Mol Med Rep. 8:1861–1865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jain MK, Adams-Huet B, Terekhova D,
Kushner LE, Bedimo R, Li X and Holodniy M: Acute and chronic immune
biomarker changes during interferon/ribavirin treatment in HIV/HCV
co-infected patients. J Viral Hepat. 22:25–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bai Q, An J, Wu X, You H, Ma H, Liu T, Gao
N and Jia J: HBV promotes the proliferation of hepatic stellate
cells via the PDGF-B/PDGFR-β signaling pathway in vitro. Int J Mol
Med. 30:1443–1450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okada H, Honda M, Campbell JS, Sakai Y,
Yamashita T, Takebuchi Y, Hada K, Shirasaki T, Takabatake R,
Nakamura M, et al: Acyclic retinoid targets platelet-derived growth
factor signaling in the prevention of hepatic fibrosis and
hepatocellular carcinoma development. Cancer Res. 72:4459–4471.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Friedman SL, Wei S and Blaner WS: Retinol
release by activated rat hepatic lipocytes: Regulation by Kupffer
cell-conditioned medium and PDGF. Am J Physiol. 264:G947–G952.
1993.PubMed/NCBI
|
|
17
|
Breitkopf K, Roeyen Cv, Sawitza I, Wickert
L, Floege J and Gressner AM: Expression patterns of PDGF-A, -B, -C
and -D and the PDGF-receptors alpha and beta in activated rat
hepatic stellate cells (HSC). Cytokine. 31:349–357. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fredriksson L, Li H and Eriksson U: The
PDGF family: Four gene products from five dimeric isoforms.
Cytokine Growth Factor Rev. 15:197–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sarzani R, Arnaldi G and Chobanian AV:
Hypertension-induced changes of platelet-derived growth factor
receptor expression in rat aorta and heart. Hypertension.
17:888–985. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bergsten E, Uutela M, Li X, Pietras K,
Ostman A, Heldin CH, Alitalo K and Eriksson U: PDGF-D is a
specific, protease-activated ligand for the PDGF beta-receptor. Nat
Cell Biol. 3:512–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Uutela M, Laurén J, Bergsten E, Li X,
Horelli-Kuitunen N, Eriksson U and Alitalo K: Chromosomal location,
exon structure, and vascular expression patterns of the human PDGFC
and PDGFD genes. Circulation. 103:2242–2247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liegl B, Leithner A, Bauernhofer T,
Windhager R, Guelly C, Regauer S and Beham A: Immunohistochemical
and mutational analysis of PDGF and PDGFR in desmoid tumours: Is
there a role for tyrosine kinase inhibitors in c-kit-negative
desmoid tumours? Histopathology. 49:576–581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosenfeld M, Keating A, Bowen-Pope DF,
Singer JW and Ross R: Responsiveness of the in vitro hematopoietic
microenvironment to platelet-derived growth factor. Leuk Res.
9:427–434. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ogawa S, Ochi T, Shimada H, Inagaki K,
Fujita I, Nii A, Moffat MA, Katragadda M, Violand BN, Arch RH and
Masferrer JL: Anti-PDGF-B monoclonal antibody reduces liver
fibrosis development. Hepatol Res. 40:1128–1141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park HJ, Kim HG, Wang JH, Choi MK, Han JM,
Lee JS and Son CG: Comparison of TGF-β, PDGF, and CTGF in hepatic
fibrosis models using DMN, CCl4, and TAA. Drug Chem
Toxicol. 39:111–118. 2016.PubMed/NCBI
|
|
26
|
Borkham-Kamphorst E, Meurer SK, Van de
Leur E, Haas U, Tihaa L and Weiskirchen R: PDGF-D signaling in
portal myofibroblasts and hepatic stellate cells proves identical
to PDGF-B via both PDGF receptor type α and β. Cell Signal.
27:1305–1314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kinnman N, Hultcrantz R, Barbu V, Rey C,
Wendum D, Poupon R and Housset C: PDGF-mediated chemoattraction of
hepatic stellate cells by bile duct segments in cholestatic liver
injury. Lab Invest. 80:697–707. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Czochra P, Klopcic B, Meyer E, Herkel J,
Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle
PR, Lohse AW and Kanzler S: Liver fibrosis induced by hepatic
overexpression of PDGF-B in transgenic mice. J Hepatol. 45:419–428.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Twamley-Stein GM, Pepperkok R, Ansorge W
and Courtneidge SA: The Src family tyrosine kinases are required
for platelet-derived growth factor-mediated signal transduction in
NIH 3T3 cells. Proc Natl Acad Sci USA. 90:pp. 7696–7700. 1993;
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zvibel I, Bar-Zohar D, Kloog Y, Oren R and
Reif S: The effect of Ras inhibition on the proliferation,
apoptosis and matrix metalloproteases activity in rat hepatic
stellate cells. Dig Dis Sci. 53:1048–1053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fischer AN, Fuchs E, Mikula M, Huber H,
Beug H and Mikulits W: PDGF essentially links TGF-beta signaling to
nuclear beta-catenin accumulation in hepatocellular carcinoma
progression. Oncogene. 26:3395–3405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bromann PA, Korkaya H, Webb CP, Miller J,
Calvin TL and Courtneidge SA: Platelet-derived growth factor
stimulates Src-dependent mRNA stabilization of specific early genes
in fibroblasts. J Biol Chem. 280:10253–10263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hennig A, Markwart R, Esparza-Franco MA,
Ladds G and Rubio I: Ras activation revisited: Role of GEF and GAP
systems. Biol Chem. 396:831–848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bera A, Das F, Ghosh-Choudhury N, Li X,
Pal S, Gorin Y, Kasinath BS, Abboud HE and Choudhury G Ghosh: A
positive feedback loop involving Erk5 and Akt turns on mesangial
cell proliferation in response to PDGF. Am J Physiol Cell Physiol.
306:C1089–C1100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiong C, Liu X and Meng A: The kinase
activity-deficient isoform of the protein araf antagonizes
Ras/mitogen-activated protein kinase (Ras/MAPK) signaling in the
zebrafish embryo. J Biol Chem. 290:25512–25521. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan TL, Wang PW, Leu YL, Wu TH and Wu TS:
Inhibitory effects of Scutellaria baicalensis extract on hepatic
stellate cells through inducing G2/M cell cycle arrest and
activating ERK-dependent apoptosis via Bax and caspase pathway. J
Ethnopharmacol. 139:829–837. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park JH, Yoon J, Lee KY and Park B:
Effects of geniposide on hepatocytes undergoing
epithelial-mesenchymal transition in hepatic fibrosis by targeting
TGFβ/Smad and ERK-MAPK signaling pathways. Biochimie. 113:26–34.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Osman I and Segar L: Pioglitazone, a PPARγ
agonist, attenuates PDGF-induced vascular smooth muscle cell
proliferation through AMPK-dependent and AMPK-independent
inhibition of mTOR/p70S6K and ERK signaling. Biochem Pharmacol.
101:54–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Margolis B, Zilberstein A, Franks C,
Felder S, Kremer S, Ullrich A, Rhee SG, Skorecki K and Schlessinger
J: Effect of phospholipase C-gamma overexpression on PDGF-induced
second messengers and mitogenesis. Science. 248:607–610. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mukherjee S, Duan F, Kolb MR and Janssen
LJ: Platelet derived growth factor-evoked Ca2+ wave and
matrix gene expression through phospholipase C in human pulmonary
fibroblast. Int J Biochem Cell Biol. 45:1516–1524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kojima N, Hori M, Murata T, Morizane Y and
Ozaki H: Different profiles of Ca2+ responses to
endothelin-1 and PDGF in liver myofibroblasts during the process of
cell differentiation. Br J Pharmacol. 151:816–827. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benedetti A, Di Sario A, Casini A, Ridolfi
F, Bendia E, Pigini P, Tonnini C, D'Ambrosio L, Feliciangeli G,
Macarri G and Svegliati-Baroni G: Inhibition of the NA(+)/H(+)
exchanger reduces rat hepatic stellate cell activity and liver
fibrosis: An in vitro and in vivo study. Gastroenterology.
120:545–556. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Di Sario A, Bendia E, Taffetani S,
Marzioni M, Candelaresi C, Pigini P, Schindler U, Kleemann HW,
Trozzi L, Macarri G and Benedetti A: Selective
Na+/H+ exchange inhibition by cariporide
reduces liver fibrosis in the rat. Hepatology. 37:256–266. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Perkinton MS, Ip JK, Wood GL, Crossthwaite
AJ and Williams RJ: Phosphatidylinositol 3-kinase is a central
mediator of NMDA receptor signalling to MAP kinase (Erk1/2),
Akt/PKB and CREB in striatal neurons. J Neurochem. 80:239–254.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan H, Ma L, Fan B, Wu J, Yang Z and Wang
L: Role of PDGFR-β/PI3K/AKT signaling pathway in PDGF-BB induced
myocardial fibrosis in rats. Am J Transl Res. 6:714–723.
2014.PubMed/NCBI
|
|
46
|
Niba ET, Nagaya H, Kanno T, Tsuchiya A,
Gotoh A, Tabata C, Kuribayashi K, Nakano T and Nishizaki T:
Crosstalk between PI3 kinase/PDK1/Akt/Rac1 and Ras/Raf/MEK/ERK
pathways downstream PDGF receptor. Cell Physiol Biochem.
31:905–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Villarino AV, Kanno Y, Ferdinand JR and
O'Shea JJ: Mechanisms of Jak/STAT signaling in immunity and
disease. J Immunol. 194:21–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matsui F and Meldrum KK: The role of the
Janus kinase family/signal transducer and activator of
transcription signaling pathway in fibrotic renal disease. J Surg
Res. 178:339–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Neeli I, Liu Z, Dronadula N, Ma ZA and Rao
GN: An essential role of the Jak-2/STAT-3/cytosolic phospholipase
A(2) axis in platelet-derived growth factor BB-induced vascular
smooth muscle cell motility. J Biol Chem. 279:46122–46128. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jiang JX, Mikami K, Venugopal S, Li Y and
Török NJ: Apoptotic body engulfment by hepatic stellate cells
promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent
pathways. J Hepatol. 51:139–186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Di Sario A, Bendia E, Svegliati-Baroni G,
Marzioni M, Ridolfi F, Trozzi L, Ugili L, Saccomanno S, Jezequel AM
and Benedetti A: Rearrangement of the cytoskeletal network induced
by platelet-derived growth factor in rat hepatic stellate cells:
Role of different intracellular signalling pathways. J Hepatol.
36:179–190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Csak T, Bala S, Lippai D, Kodys K,
Catalano D, Iracheta-Vellve A and Szabo G: MicroRNA-155 deficiency
attenuates liver steatosis and fibrosis without reducing
inflammation in a mouse model of steatohepatitis. PLoS One.
10:e01292512015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wei J, Feng L, Li Z, Xu G and Fan X:
MicroRNA-21 activates hepatic stellate cells via PTEN/Akt
signaling. Biomed Pharmacother. 67:387–392. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kwiecinski M, Elfimova N, Noetel A, Töx U,
Steffen HM, Hacker U, Nischt R, Dienes HP and Odenthal M:
Expression of platelet-derived growth factor-C and insulin-like
growth factor I in hepatic stellate cells is inhibited by miR-29.
Lab Invest. 92:978–987. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Noetel A, Kwiecinski M, Elfimova N, Huang
J and Odenthal M: microRNA are central players in anti- and
profibrotic gene regulation during liver fibrosis. Front Physiol.
3:492012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Okada H, Honda M, Campbell JS, Takegoshi
K, Sakai Y, Yamashita T, Shirasaki T, Takabatake R, Nakamura M,
Tanaka T and Kaneko S: Inhibition of microRNA-214 ameliorates
hepatic fibrosis and tumor incidence in platelet-derived growth
factor C transgenic mice. Cancer Sci. 106:1143–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fu N, Niu X, Wang Y, Du H, Wang B, Du J,
Li Y, Wang R, Zhang Y, Zhao S, et al: Role of LncRNA-activated by
transforming growth factor beta in the progression of hepatitis C
virus-related liver fibrosis. Discov Med. 22:29–42. 2016.PubMed/NCBI
|
|
58
|
Yang JJ, Liu LP, Tao H, Hu W, Shi P, Deng
ZY and Li J: MeCP2 silencing of LncRNA H19 controls hepatic
stellate cell proliferation by targeting IGF1R. Toxicology 359–360.
1–46. 2016.
|
|
59
|
Yu F, Zheng J, Mao Y, Dong P, Lu Z, Li G,
Guo C, Liu Z and Fan X: Long non-coding RNA growth arrest-specific
transcript 5 (GAS5) inhibits liver fibrogenesis through a mechanism
of competing endogenous RNA. J Biol Chem. 290:28286–28298. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Niu X, Fu N, Du J, Wang R, Wang Y, Zhao S,
Du H, Wang B, Zhang Y, Sun D and Nan Y: miR-1273g-3p modulates
activation and apoptosis of hepatic stellate cells by directly
targeting PTEN in HCV-related liver fibrosis. FEBS Lett.
590:2709–2724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hyun J, Wang S, Kim J, Rao KM, Park SY,
Chung I, Ha CS, Kim SW, Yun YH and Jung Y: MicroRNA-378 limits
activation of hepatic stellate cells and liver fibrosis by
suppressing Gli3 expression. Nat Commun. 7:109932016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hao ZM, Fan XB, Li S, Lv YF, Su HQ, Jiang
HP and Li HH: Vaccination with platelet-derived growth factor B
kinoids inhibits CCl4-induced hepatic fibrosis in mice.
J Pharmacol Exp Ther. 342:835–842. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Durez P, Vandepapeliere P, Miranda P,
Toncheva A, Berman A, Kehler T, Mociran E, Fautrel B, Mariette X,
Dhellin O, et al: Therapeutic vaccination with TNF-Kinoid in TNF
antagonist-resistant rheumatoid arthritis: A phase II randomized,
controlled clinical trial. PLoS One. 9:e1134652014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jing Q, Yin T, Wan Y, Shi H, Luo S, Li M,
Zhang H, He H, Liu S, Li H, et al: Interleukin-13 peptide kinoid
vaccination attenuates allergic inflammation in a mouse model of
asthma. Int J Mol Med. 30:553–560. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Delavallée L, Le Buanec H, Bessis N,
Assier E, Denys A, Bizzini B, Zagury D and Boissier MC: Early and
long-lasting protection from arthritis in tumour necrosis factor
alpha (TNFalpha) transgenic mice vaccinated against TNFalpha. Ann
Rheum Dis. 67:1332–1338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gounder MM, Lefkowitz RA, Keohan ML,
D'Adamo DR, Hameed M, Antonescu CR, Singer S, Stout K, Ahn L and
Maki RG: Activity of Sorafenib against desmoid tumor/deep
fibromatosis. Clin Cancer Res. 17:4082–4090. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zahavi T, Lanton T, Divon MS, Salmon A,
Peretz T, Galun E, Axelrod JH and Sonnenblick A: Sorafenib
treatment during partial hepatectomy reduces tumorgenesis in an
inflammation-associated liver cancer model. Oncotarget.
7:4860–4870. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lin TsT, Gao DY, Liu YC, Sung YC, Wan D,
Liu JY, Chiang T, Wang L and Chen Y: Development and
characterization of sorafenib-loaded PLGA nanoparticles for the
systemic treatment of liver fibrosis. J Control Release. 221:62–70.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hong F, Chou H, Fiel MI and Friedman SL:
Antifibrotic activity of sorafenib in experimental hepatic
fibrosis: Refinement of inhibitory targets, dosing, and window of
efficacy in vivo. Dig Dis Sci. 58:257–264. 2013.PubMed/NCBI
|
|
70
|
Wang Y, Gao J, Zhang D, Zhang J, Ma J and
Jiang H: New insights into the antifibrotic effects of sorafenib on
hepatic stellate cells and liver fibrosis. J Hepatol. 53:132–144.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hao H, Zhang D, Shi J, Wang Y, Chen L, Guo
Y, Ma J, Jiang X and Jiang H: Sorafenib induces autophagic cell
death and apoptosis in hepatic stellate cell through the JNK and
Akt signaling pathways. Anticancer Drugs. 27:192–203. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu C, Yang Z, Wang L, Lu Y, Tang B, Miao
H, Xu Q and Chen X: Combination of sorafenib and gadolinium
chloride (GdCl3) attenuates dimethylnitrosamine(DMN)-induced liver
fibrosis in rats. BMC Gastroenterol. 15:1592015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Deng YR, Ma HD, Tsuneyama K, Yang W, Wang
YH, Lu FT, Liu CH, Liu P, He XS, Diehl AM, et al: STAT3-mediated
attenuation of CCl4-induced mouse liver fibrosis by the protein
kinase inhibitor sorafenib. J Autoimmun. 46:25–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Qu K, Huang Z, Lin T, Liu S, Chang H, Yan
Z, Zhang H and Liu C: New insight into the anti-liver fibrosis
effect of multitargeted tyrosine kinase inhibitors: From molecular
target to clinical trials. Front Pharmacol. 6:3002016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Karuppagounder SS, Brahmachari S, Lee Y,
Dawson VL, Dawson TM and Ko HS: The c-Abl inhibitor, nilotinib,
protects dopaminergic neurons in a preclinical animal model of
Parkinson's disease. Sci Rep. 4:48742014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Elsherbiny NM, El-Sherbiny M and Said E:
Amelioration of experimentally induced diabetic nephropathy and
renal damage by nilotinib. J Physiol Biochem. 71:635–648. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shaker ME, Zalata KR, Mehal WZ, Shiha GE
and Ibrahim TM: Comparison of imatinib, nilotinib and silymarin in
the treatment of carbon tetrachloride-induced hepatic oxidative
stress, injury and fibrosis. Toxicol Appl Pharmacol. 252:165–175.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lemos DR, Babaeijandaghi F, Low M, Chang
CK, Lee ST, Fiore D, Zhang RH, Natarajan A, Nedospasov SA and Rossi
FM: Nilotinib reduces muscle fibrosis in chronic muscle injury by
promoting TNF-mediated apoptosis of fibro/adipogenic progenitors.
Nat Med. 21:786–794. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shiha GE, Abu-Elsaad NM, Zalata KR and
Ibrahim TM: Tracking anti-fibrotic pathways of nilotinib and
imatinib in experimentally induced liver fibrosis: An insight. Clin
Exp Pharmacol Physiol. 41:788–797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shaker ME, Shiha GE and Ibrahim TM:
Comparison of early treatment with low doses of nilotinib, imatinib
and a clinically relevant dose of silymarin in
thioacetamide-induced liver fibrosis. Eur J Pharmacol. 670:593–600.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu Y, Wang Z, Kwong SQ, Lui EL, Friedman
SL, Li FR, Lam RW, Zhang GC, Zhang H and Ye T: Inhibition of PDGF,
TGF-β, and Abl signaling and reduction of liver fibrosis by the
small molecule Bcr-Abl tyrosine kinase antagonist Nilotinib. J
Hepatol. 55:612–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Moawad EY: Predicting effectiveness of
imatinib mesylate in tumors expressing platelet-derived
growthfactors (PDGF-AA, PDGF-BB), stem cell factor ligands and
their respective receptors (PDGFR-α, PDGFR-β, and c-kit). J
Gastrointest Cancer. 46:272–283. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kuo WL, Yu MC, Lee JF, Tsai CN, Chen TC
and Chen MF: Imatinib mesylate improves liver regeneration and
attenuates liver fibrogenesis in CCL4-treated mice. J Gastrointest
Surg. 16:361–369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kim Y, Fiel MI, Albanis E, Chou HI, Zhang
W, Khitrov G and Friedman SL: Anti-fibrotic activity and enhanced
interleukin-6 production by hepatic stellate cells in response to
imatinib mesylate. Liver Int. 32:1008–1017. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Iwamoto H, Nakamuta M, Tada S, Sugimoto R,
Enjoji M and Nawata H: Platelet-derived growth factor receptor
tyrosine kinase inhibitor AG1295 attenuates rat hepatic stellate
cell growth. J Lab Clin Med. 135:406–412. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hutson TE, Davis ID, Machiels JP, De Souza
PL, Rottey S, Hong BF, Epstein RJ, Baker KL, McCann L, Crofts T, et
al: Efficacy and safety of pazopanib in patients with metastatic
renal cell carcinoma. J Clin Oncol. 28:475–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Eisen T, Joensuu H, Nathan PD, Harper PG,
Wojtukiewicz MZ, Nicholson S, Bahl A, Tomczak P, Pyrhonen S, Fife
K, et al: Regorafenib for patients with previously untreated
metastatic or unresectable renal-cell carcinoma: A single-group
phase 2 trial. Lancet Oncol. 13:1055–1062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rice AB, Moomaw CR, Morgan DL and Bonner
JC: Specific inhibitors of platelet-derived growth factor or
epidermal growth factor receptor tyrosine kinase reduce pulmonary
fibrosis in rats. Am J Pathol. 155:213–221. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Venè R, Tosetti F, Minghelli S, Poggi A,
Ferrari N and Benelli R: Celecoxib increases EGF signaling in colon
tumor associated fibroblasts, modulating EGFR expression and
degradation. Oncotarget. 6:12310–12325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Soininen H, West C, Robbins J and
Niculescu L: Long-term efficacy and safety of celecoxib in
Alzheimer's disease. Dement Geriatr Cogn Disord. 23:8–21. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Raval M, Frank PG, Laury-Kleintop L, Yan G
and Lanza-Jacoby S: combined with atorvastatin prevents progression
of atherosclerosis. J Surg Res. 163:e113–e122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gao JH, Wen SL, Yang WJ, Lu YY, Tong H,
Huang ZY, Liu ZX and Tang CW: Celecoxib ameliorates portal
hypertension of the cirrhotic rats through the dual inhibitory
effects on the intrahepatic fibrosis and angiogenesis. PLoS One.
8:e693092013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Paik YH, Kim JK, Lee JI, Kang SH, Kim DY,
An SH, Lee SJ, Lee DK, Han KH, Chon CY, et al: Celecoxib induces
hepatic stellate cell apoptosis through inhibition of Akt
activation and suppresses hepatic fibrosis in rats. Gut.
58:1517–1527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ekor M, Odewabi AO, Kale OE, Adesanoye OA
and Bamidele TO: Celecoxib, a selective cyclooxygenase-2 inhibitor,
lowers plasma cholesterol and attenuates hepatic lipid peroxidation
during carbon-tetrachloride-associated hepatotoxicity in rats. Drug
Chem Toxicol. 36:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Reina M and Martínez A: Is Silybin the
best free radical scavenger compound in Silymarin? J Phys Chem B.
120:4568–4578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Trappoliere M, Caligiuri A, Schmid M,
Bertolani C, Failli P, Vizzutti F, Novo E, di Manzano C, Marra F,
Loguercio C and Pinzani M: A component of sylimarin, exerts
anti-inflammatory and anti-fibrogenic effects on human hepatic
stellate cells. J Hepatol. 50:1102–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Serviddio G, Bellanti F, Stanca E, Lunetti
P, Blonda M, Tamborra R, Siculella L, Vendemiale G, Capobianco L
and Giudetti AM: Silybin exerts antioxidant effects and induces
mitochondrial biogenesis in liver of rat with secondary biliary
cirrhosis. Free Radic Biol Med. 73:117–126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Malaguarnera M, Motta M, Vacante M,
Malaguarnera G, Caraci F, Nunnari G, Gagliano C, Greco C, Chisari
G, Drago F and Bertino G: Silybin-vitamin E-phospholipids complex
reduces liver fibrosis in patients with chronic hepatitis C treated
with pegylated interferon α and ribavirin. Am J Transl Res.
7:2510–2518. 2015.PubMed/NCBI
|
|
99
|
Bares JM, Berger J, Nelson JE, Messner DJ,
Schildt S, Standish LJ and Kowdley KV: Silybin treatment is
associated with reduction in serum ferritin in patients with
chronic hepatitis C. J Clin Gastroenterol. 42:937–944. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhou M, Fan C and Tian N: Effects of
curcumin on the gene expression profile of L-02 cells. Biomed Rep.
3:519–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lian N, Jiang Y, Zhang F, Jin H, Lu C, Wu
X, Lu Y and Zheng S: Curcumin regulates cell fate and metabolism by
inhibiting hedgehog signaling in hepatic stellate cells. Lab
Invest. 95:790–803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Taverna S, Giallombardo M, Pucci M, Flugy
A, Manno M, Raccosta S, Rolfo C, De Leo G and Alessandro R:
Curcumin inhibits in vitro and in vivo chronic myelogenous leukemia
cells growth: A possible role for exosomal disposal of miR-21.
Oncotarget. 6:21918–21933. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
El-Bahr SM: Curcumin regulates gene
expression of insulin like growth factor, B-cell CLL/lymphoma 2 and
antioxidant enzymes in streptozotocin induced diabetic rats. BMC
Complement Altern Med. 13:3682013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang X, Chen M, Zou P, Kanchana K, Weng
Q, Chen W, Zhong P, Ji J, Zhou H, He L and Liang G: Curcumin analog
WZ35 induced cell death via ROS-dependent ER stress and G2/M cell
cycle arrest in human prostate cancer cells. BMC Cancer.
15:8662015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lee HI, McGregor RA, Choi MS, Seo KI, Jung
UJ, Yeo J, Kim MJ and Lee MK: Low doses of curcumin protect
alcohol-induced liver damage by modulation of the alcohol metabolic
pathway, CYP2E1 and AMPK. Life Sci. 93:693–699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhao Y, Ma X, Wang J, He X, Hu Y, Zhang P,
Wang R, Li R, Gong M, Luo S and Xiao X: Curcumin protects against
CCl4-induced liver fibrosis in rats by inhibiting HIF-1α through an
ERK-dependent pathway. Molecules. 19:18767–18780. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Xue J, Zhang X, Zhang C, Kang N, Liu X, Yu
J, Zhang N, Wang H, Zhang L, Chen R, et al: Protective effect of
Naoxintong against cerebral ischemia reperfusion injury in mice. J
Ethnopharmacol. 182:181–189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lv H, Wang L, Shen J, Hao S, Ming A, Wang
X, Su F and Zhang Z: Salvianolic acid B attenuates apoptosis and
inflammation via SIRT1 activation in experimental stroke rats.
Brain Res Bull. 115:30–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Deng Y, Yang M, Xu F, Zhang Q, Zhao Q, Yu
H, Li D, Zhang G, Lu A, Cho K, et al: Combined salvianolic acid B
and ginsenoside Rg1 exerts cardioprotection against
ischemia/reperfusion injury in rats. PLoS One. 10:e01354352015.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang R, Yu XY, Guo ZY, Wang YJ, Wu Y and
Yuan YF: Inhibitory effects of salvianolic acid B on CCl(4)-induced
hepatic fibrosis through regulating NF-κB/IκBα signaling. J
Ethnopharmacol. 144:592–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lv Z, Song Y, Xue D, Zhang W, Cheng Y and
Xu L: Effect of salvianolic-acid B on inhibiting MAPK signaling
induced by transforming growth factor-β1 in activated rat hepatic
stellate cells. J Ethnopharmacol. 132:384–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Gao HY, Li GY, Lou MM, Li XY, Wei XY and
Wang JH: Hepatoprotective effect of matrine salvianolic acid B salt
on carbon tetrachloride-induced hepatic fibrosis. J Inflamm (Lond).
9:162012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xu H, Zhou Y, Lu C, Ping J and Xu LM:
Salvianolic acid B lowers portal pressure in cirrhotic rats and
attenuates contraction of rat hepaticstellate cells by inhibiting
RhoA signaling pathway. Lab Invest. 92:1738–1748. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Marslin G, Revina AM, Khandelwal VK,
Balakumar K, Prakash J, Franklin G and Sheeba CJ: Delivery as
nanoparticles reduces imatinib mesylate-induced cardiotoxicity and
improves anticancer activity. Int J Nanomedicine. 10:3163–3170.
2015.PubMed/NCBI
|
|
115
|
Younis N, Shaheen MA and Abdallah MH:
Silymarin-loaded Eudragit(®) RS100 nanoparticles
improved the ability of silymarin to resolve hepatic fibrosis in
bile duct ligated rats. Biomed Pharmacother. 81:93–103. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhong H, Wang D, Wang N, Rios Y, Huang H,
Li S, Wu X and Lin S: Combinatory action of VEGFR2 and MAP kinase
pathways maintains endothelial-cell integrity. Cell Res.
21:1080–1087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Srikanthan A, Ethier JL, Ocana A, Seruga
B, Krzyzanowska MK and Amir E: Cardiovascular toxicity of
multi-tyrosine kinase inhibitors in advanced solid tumors: A
population-based observational study. PLoS One. 10:e01227352015.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ghatalia P, Je Y, Mouallem NE, Nguyen PL,
Trinh QD, Sonpavde G and Choueiri TK: Hepatotoxicity with vascular
endothelial growth factor receptor tyrosine kinase inhibitors: A
meta-analysis of randomized clinical trials. Crit Rev Oncol
Hematol. 93:257–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chrisoulidou A, Mandanas S, Margaritidou
E, Mathiopoulou L, Boudina M, Georgopoulos K and
Pazaitou-Panayiotou K: Treatment compliance and severe adverse
events limit the use of tyrosine kinase inhibitors in refractory
thyroid cancer. Onco Targets Ther. 8:2435–2442. 2015.PubMed/NCBI
|
|
120
|
Reichenbach V, Fernández-Varo G, Casals G,
Oró D, Ros J, Melgar-Lesmes P, Weiskirchen R, Morales-Ruiz M and
Jiménez W: Adenoviral dominant-negative soluble PDGFRβ improves
hepatic collagen, systemic hemodynamics, and portal pressure in
fibrotic rats. J Hepatol. 57:967–973. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Shah R, Reyes-Gordillo K,
Arellanes-Robledo J, Lechuga CG, Hernández-Nazara Z, Cotty A,
Rojkind M and Lakshman MR: TGF-β1 up-regulates the expression of
PDGF-β receptor mRNA and induces a delayed PI3K-, AKT- and
p70(S6K)-dependent proliferative response in activated hepatic
stellate cells. Alcohol Clin Exp Res. 37:1838–1848. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Bai Q, An J, Wu X, You H, Ma H, Liu T, Gao
N and Jia J: HBV promotes the proliferation of hepatic stellate
cells via the PDGF-B/PDGFR-β signaling pathway in vitro. Int J Mol
Med. 30:1443–1450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Liang CC, Liu CH, Chung CS, Lin CK, Su TH,
Yang HC, Liu CJ, Chen PJ, Chen DS and Kao JH: Advanced hepatic
fibrosis and steatosis are associated with persistent alanine
aminotransferase elevation in chronic hepatitis C patients negative
for hepatitis C virus RNA during pegylated interferon plus
ribavirin therapy. J Infect Dis. 211:1429–1436. 2015. View Article : Google Scholar : PubMed/NCBI
|