Introduction
Chronic rhinosinusitis (CRS), a highly prevalent
health condition, is characterized by mucosal inflammation of the
nose and paranasal sinuses. CRS is an inflammatory disorder
involving the paranasal sinuses and nasal passages which persists
for a minimum of 2–3 months, despite attempts at medical management
(1–3). Based on the presence and absence of
nasal polyps (NPs), CRS is generally divided into two categories,
CRS with and CRS without NPs (CRSwNP and CRSsNP, respectively).
Although CRSsNP is more prevalent, CRSwNP accounts for
approximately 20% of all CRS cases. CRSwNP, which is often
accompanied by asthma, fungal rhinosinusitis, and
aspirin-exacerbated respiratory disease, is considered more
difficult to treat than CRSsNP (4,5).
Despite numerous studies, the detailed pathogenic mechanisms
underlying NP formation in CRS remain unknown (6).
NPs are a common clinical condition, either in
isolation or accompanying CRS. Since 2003, NP formation has been
identified as closely related to epigenetic processes. Zheng et
al (7) identified the top four
genes with altered expression in NP samples (COL18A1,
EP300, GNAS, and SMURF1) from a DNA
methylation microarray; these genes were validated as driven by
promoter methylation using methylation-specific polymerase chain
reaction (PCR). However, unmethylated signals were detected in the
promoter regions of COL18A1, EP300, GNAS, and
SMURF1 in all samples. The methylation frequency of
COL18A1 was significantly higher in NPs than in healthy
control samples. DNA methylation is a key regulator of gene
activity; however, there are many questions regarding this
epigenetic mechanism that remain to be answered.
Epigenetics, which generally refers to heritable
changes in gene expression and the potential for genetic changes
that are not transmitted through alterations in the actual genetic
code (8), has recently been
proposed as an explanation for the key gene/environment
interactions involved in NP formation (3). The mechanisms of epigenetic
regulation underlie many complex diseases and may, therefore,
contribute to the development and heritability of paranasal disease
(9). North and Ellis (10) recently provided an overview of key
studies, indicating an important role for epigenetic modifications
in the developmental origins and pathogenesis of atopy and asthma,
suggesting the potential for future applications of these research
findings to the clinical management of allergy and immunology.
Thus, a better understanding of the role of DNA methylation in NP
formation is required to inform focused epigenetic studies toward
the realization of clinical applications.
As a step toward this goal, the aim of the present
study was to identify DNA methylation changes in specific genes
potentially important in the pathogenesis of NP. To search for
specific genes regulating polyp formation, we performed DNA
methylation analysis using a systematic epigenetic regulatory
approach. The choice of target genes was based on known epigenetic
modulation factors involved in allergies, since changes in the
methylation patterns of these is associated with alterations in
gene expression, phenotype, and, ultimately, polyp formation. The
results of DNA methylation analysis were validated by reverse
transcription-quantitative PCR (RT-qPCR), to compare the DNA
methylation profiles of genes differentially expressed in patients
with CRSwNP and CRSsNP. Our results provide insights into the roles
of specific mRNAs as regulators of polyp formation. Moreover, these
data could inform the development of therapeutic strategies to
modulate the formation of polyps.
Materials and methods
Patients and sample collection
This study was based on data acquired from the
Konyang University Hospital (September 2014 to August 2015) through
a survey conducted by the Centers for Otorhinolaryngology. The
protocol was approved by the Institutional Review Board of Konyang
University Hospital (IRB approval no. 2013-01-036), and all
individuals provided informed consent prior to inclusion. The
diagnosis of CRS was based on the definition of the European
Position Paper on Rhinosinusitis and Nasal Polyps 2012 (3). A total of 18 patients were included
in the study, divided into three groups: CRSwNP (n=7), CRSsNP
(n=7), and healthy controls (n=4). Control tissues were obtained
from patients without any sinus disease, with an average age of
47.5 years. Patients with CRSwNP ranged in age from 15 to 66
(average 44.6) years. Seven patients in the CRSwNP group underwent
revision sinus surgery. All patients were nonsmokers. Uncinate
process (UP) mucosa tissue samples were obtained from control
subjects and those with CRSsNP or CRSwNP. NP tissues from patients
with CRSwNP were also evaluated. Patients selected for the control
group underwent endoscopic transsphenoidal surgery for benign
pituitary tumors and did not have symptoms, imaging, or endoscopic
findings consistent with CRS.
Exclusion criteria were as follows: i) abnormal
atopic status (eosinophil count outside of the normal range and
increased immunocap IgE levels); ii) history of smoking; iii) prior
treatment with oral or spray steroids for 3 months before surgery;
iv) cystic fibrosis, congenital mucociliary problems, systemic
vasculitis, gastroesophageal reflux diseases, antrochoanal polyp,
and fungal sinusitis. Controls with allergic rhinitis were also
excluded. Each tissue sample was divided into two parts, one of
which was fixed in 10% formaldehyde and embedded in paraffin for
histological analysis, and the other was immediately snap-frozen in
liquid nitrogen and stored at −80°C for future RNA, DNA, and
protein extraction. Fresh tissue specimens were quickly cleaned
with 0.9% normal saline and sliced into appropriate sections.
HiSeq MBD-seq library preparation
Genomic DNA was extracted from tissue samples using
a Maxwell 16 MDx Instrument (Promega Inc., Madison, WI, USA). Each
cartridge was placed in a holder with the ridged side of the
cartridge facing toward the numbered side of the rack. The plunger
was placed in the last well of each cartridge so that the bottom of
the plunger reached the bottom of the cartridge. Samples were
transferred to the first well, and the cartridge was placed onto
the Maxwell 16 platform. The system was run according to the
manufacturer protocols and settings for tissue DNA. One microgram
of genomic DNA was sheared to 200–400 bp fragments using a Covaris
LE220 sonicator (Covaris Inc., Woburn, MA, USA). The resulting
fragments were immunoprecipitated using a MethylMiner Methylated
DNA Enrichment Kit (Invitrogen), according to the manufacturer's
recommended protocol. In brief, methylated DNA was isolated from
fragmented whole genomic DNA by binding to the methyl-CpG-binding
domain (MBD) of human MBD2 protein, which was coupled to
paramagnetic streptavidin beads (Dynabeads M-280) via a biotin
linker. Methylated fragments were eluted as a single enriched
population using 2 M NaCl elution buffer. Methylated
double-stranded DNA was end-repaired (i.e., an ‘A’ was ligated to
the 3′ end), and Illumina adapters then ligated to the fragments
with a target size of 300–500 bp products. Size-selected products
were then PCR-amplified, and validated using an Agilent
Bioanalyzer.
Clustering and sequencing
Target gene raw DNA methylation pattern data were
extracted as paired files from Illumina software. To further
explore molecular variation contributing to the differences between
CRSwNP and CRSsNP, we analyzed a flow cell containing millions of
unique clusters loaded onto the HiSeq 2000 system for automated
cycles of extension and imaging. The Illumina system uses a unique
‘bridged’ amplification reaction that occurs on the surface of the
flow cell. A flow cell containing millions of unique clusters is
loaded onto the HiSeq 2000 sequencer for automated cycles of
extension and imaging. The Illumina Sequencing-by-Synthesis system
utilizes four proprietary nucleotides possessing reversible
fluorophore and termination properties. Each sequencing cycle
occurs in the presence of all four nucleotides, leading to higher
accuracy than methods involving only one nucleotide in the reaction
mix at a time. This cycle is repeated, one base at a time,
generating a series of images, each representing a single base
extension at a specific cluster.
Data processing and methylation
profile calling
Paired-end sequencing reads (100 bp) generated by
MBD-sequencing were verified for sequence quality using FastQC
(version 0.10.0) and Trimmomatic (version 0.32) was used to remove
adapter sequences and bases with quality scores <3 from the
reads. In addition, bases that did not meet the criteria, window
size ≥4, mean quality score ≥15, were removed using a
sliding-window trimming method. Next, reads <36 bp were removed
to produce clean data. The cleaned reads were aligned to the human
genome (UCSC hg19) using Bowtie (version 1.1.1 parameter set-n 2-m
1-X500) allowing for up to two nucleotide mismatches to the
reference genome per seed and returning only uniquely mapped reads.
Mapped data (SAM file format) were subjected to sorting and
indexing using SAMtools (version 0.1.19). PCR duplicates were
removed with Picard Mark Duplicates (version 1.118). Analysis of
the MBD data was performed using the MEDIPS package. For each
sample, aligned reads were extended in the sequencing direction to
300 nt. The genome-wide sequencing read coverage of extended reads
was calculated using a 250 bp window size. The resulting coverage
profiles (read count, reads per kilobase of transcript per million
mapped reads, root mean square) at each genomic bin were
calculated. Each differentially methylated region (DMR) was
annotated using the table browser function of the UCSC genome
browser. Annotation included gene structures, transcripts, promoter
regions (defined as −2 kb upstream of the transcription start
site), exons, introns, and CpG islands.
Identification of DMRs
Read counts at each genomic bin were normalized to
the trimmed mean of M-value normalization in the edgeR package. We
applied an exact test to assess the significance of methylation
differences between comparison groups using edgeR. DMRs were
determined by filtering each associated region with a |log2FC|value
≥1 and exact test P<0.05. Hierarchical clustering analysis was
also performed using complete linkage and Euclidean distance as a
measure of similarity to display the methylation patterns of DMRs
that satisfied the significance criteria above for at least one
more comparison pair. Gene enrichment and functional annotation
analyses were performed using the DAVID tool (http://david.abcc.ncifcrf.gov/home.jsp).
All data analysis and visualization of DMRs were conducted using R
3.0.2 (www.r-project.org).
RT-qPCR analysis of mRNAs
Total RNA was isolated from nasal tissues using
TRlzol reagent (Ambion) according to the manufacturer's
instructions. To estimate mRNA expression levels, cDNAs were
synthesized using MMLV reverse transcriptase (Promega). RT-qPCR was
performed (in triplicate) using iQTM SYBR Green Supermix and a
CFX96 qPCR machine (Bio-Rad, Hercules, CA, USA). The primers used
for mRNA detection were as follows: nuclear receptor subfamily 2,
group F (NR2F2), forward 5′-GCCATAGTCCTGTTCACCTCA-3′ and
reverse 5′-AATCTCGTCGGCTGGTTGG-3′; HOXA6, forward
5′-CGGTTTACCCTTGGATGCA-3′ and reverse 5′-GCCCATGGCTCCCATACAC-3′;
ZNF609, forward 5′-TCCTACCTGCCTTCCAGCTA-3′ and reverse
5′-GTGCCTTGTCAGCATCTTCA-3′; ROBO2, forward
5′-TGGAGACCTCACAATCACCA-3′ and reverse 5′-GGCTGGGCCTTGTAGAATTA-3′;
PDE3A, forward 5′-GAACAGATGACACTGCTCAAGTT-3′ and reverse
5′-GAGCAAGAATTGGTTTGTCCAG-3′; a disintegrin and metalloproteinase
with thrombospondin type 1 motif (ADAMTS1), forward
5′-TGTGGTGTTTGCGGGGGAAATG-3′ and reverse
5′-TCGATGTTGGTGGCTCCAGTT-3′; KRT19, forward
5′-CTTCCGAACCAAGTTTGAGAC-3′ and reverse 5′-AGCGTACTGATTTCCTCCTC-3′;
ZNF222, forward 5′-TCAACGAGTCCACACTGGAG-3′ and reverse
5′-AGCTCTTCCCGCAGTTATCA-3′; and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH), forward 5′-ACAGTCAGCCGCATCTTCTT-3′
and reverse 5′-ACGACCAAATCCGTTGACTC-3′. The amplification
conditions were as follows: a pre-denaturation step at 95°C for 3
min, followed by 40 cycles of denaturation at 95°C for 10 sec,
annealing at 58°C for 10 sec, and extension at 72°C for 10 sec. The
Cquantification cycle (Cq) comparison method
2−ΔΔCq was used to calculate relative expression levels,
with GAPDH as the reference gene.
Statistical analysis
Statistical analysis was performed using the MEDIPS
(1.16.0) software package. Methylation data analysis and
visualization of DMRs was conducted using R 3.0.2 (www.r-project.org). qPCR data are presented as means ±
standard deviation. All results were analyzed using Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Clustering of DNA methylation profiles
by Methyl-CpG-binding domain sequencing
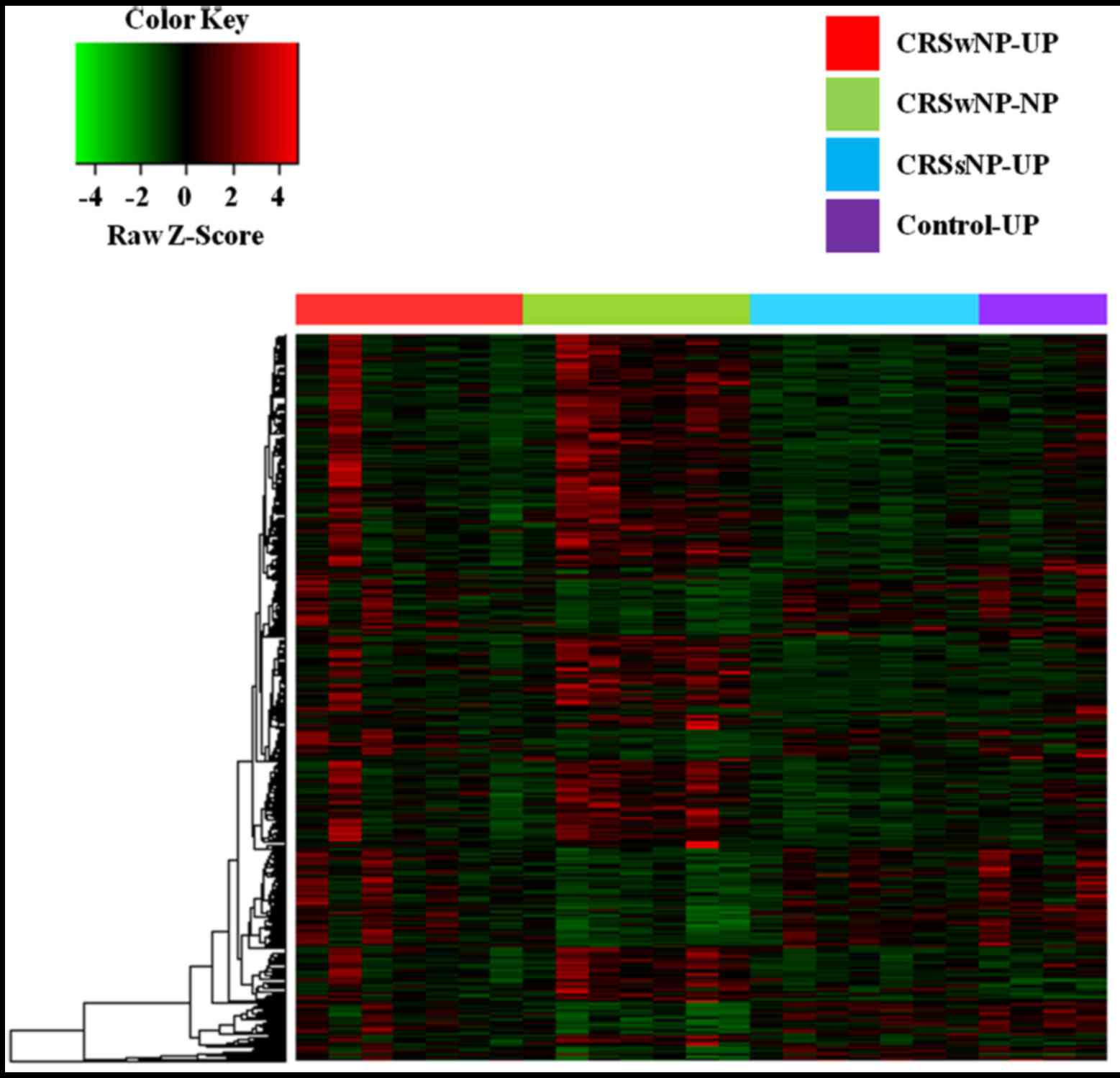
DNA methylation profiling, based on unsupervised
hierarchical clustering, identified four unique clusters with
distinct methylation signatures (Fig.
1). Remarkably, we found that these clusters were correlated
with CRS histological subtypes, genetic abnormalities, and clinical
outcomes. In our analysis, 10 and 30 genes were significantly
hypermethylated and hypomethylated, respectively, in the CRSwNP-NP
clusters compared with controls (Table
I).
 | Table I.Genes with altered methylation
statuses in nasal polyps. |
Table I.
Genes with altered methylation
statuses in nasal polyps.
|
|
|
|
| Relative levels of
methylation |
|---|
|
|
|
|
|
|
|---|
| Description | Gene ID | Chromosome | Ensemble
accession | Control-UP | CRSsNP-UP | CRSwNP-UP | CRSwNP-NP |
|---|
|
|---|
| A,
Hyper-methylation |
|---|
|
|---|
| Retinitis pigmentosa
1 | RP1-202O8.2 | 1 | ENSG00000231868 |
1.089182c |
1.440233c |
0.981016c | 8.466027 |
| Espin | ESPN | 1 | ENSG00000187017 |
0.992249b |
1.135411b |
0.907498b | 6.897929 |
|
Microtubule-associated protein 6 | MAP6 | 11 |
ENSG00000171533 |
2.217911a |
2.030746c |
2.725116b | 6.570451 |
| MPN domain
containing | MPND | 19 |
ENSG00000008382 |
1.187162b |
1.589554b |
2.545118 | 5.575780 |
| Spindle and
kinetochore associated complex subunit 1 | SKA1 | 18 |
ENSG00000154839 |
1.851846a |
1.102924b |
1.712328b | 5.197940 |
| STAM binding
protein like 1 | STAMBPL1 | 10 |
ENSG00000138134 |
1.938985a |
1.662501c |
2.256457b | 5.639432 |
| Rho GTPase
activating protein 31 | ARHGAP31 | 3 |
ENSG00000031081 |
1.784895a |
1.671465c |
2.326500b | 5.367708 |
| Rho GTPase
activating protein 31-antisense RNA 1 | ARHGAP31-AS1 | 3 |
ENSG00000241155 |
1.784895a |
1.671465c |
2.326500b | 5.367708 |
| Prostaglandin E
receptor 4 | PTGER4 | 5 |
ENSG00000171522 |
1.315410b |
3.022724b |
3.166246b | 7.677371 |
| Zinc finger protein
222 | ZNF222 | 19 |
ENSG00000159885 |
1.505544a |
1.383831a |
1.897399a | 4.326746 |
|
| B,
Hypo-methylation |
|
|
|
|
|
| Relative levels
of methylation |
|
|
|
|
|
|
|
Description | Gene ID |
Chromosome | Ensemble
accession |
Control-UP |
CRSsNP-UP |
CRSwNP-UP |
CRSwNP-NP |
|
| Pre-mRNA processing
factor 31 | RP11-120J1.1 | 9 |
ENSG00000225472 |
19.915220c |
16.500749c |
15.645956c | 4.986879 |
| RUN and FYVE domain
containing 4 | RUFY4 | 2 |
ENSG00000188282 |
16.341624c |
16.337111c |
13.857570c | 5.544202 |
| RUN and FYVE domain
containing 5 | RUFY5 | 8 | n/a |
15.768303c |
13.305096c |
11.284723c | 4.726873 |
| RUN and FYVE domain
containing 6 | RUFY6 | 2 | n/a |
11.826268c |
9.432234c |
11.383418c | 3.242687 |
| RUN and FYVE domain
containing 7 | RUFY7 | 8 | n/a |
13.603286c |
10.678649c |
9.672681c | 4.192238 |
| RUN and FYVE domain
containing 8 | RUFY8 | 22 | n/a |
17.220517c |
18.166590c |
16.007680c | 6.757857 |
| RUN and FYVE domain
containing 9 | RUFY9 | 17 | n/a |
9.348106c |
11.361693c |
8.975746c | 3.930507 |
| RUN and FYVE domain
containing 10 | RUFY10 | 8 | n/a |
10.674316c |
7.446879c |
7.634908c | 2.864527 |
| RUN and FYVE domain
containing 11 | RUFY11 | 2 | n/a |
18.865311c |
16.251488c |
13.691127c | 5.308298 |
| RUN and FYVE domain
containing 12 | RUFY12 | 2 | n/a |
14.840328c |
11.996467c |
11.480092c | 5.715029 |
| RUN and FYVE domain
containing 13 | RUFY13 | 11 | n/a |
14.189715c |
6.2297629c |
5.6346066c | 1.839248 |
| RUN and FYVE domain
containing 14 | RUFY14 | 6 | n/a |
9.701559c |
7.746522c |
7.717593c | 3.025173 |
| RUN and FYVE domain
containing 15 | RUFY15 | 6 | n/a |
9.701559c |
7.746522c |
7.717593c | 3.025173 |
| RUN and FYVE domain
containing 16 | RUFY16 | 10 | n/a |
8.791565c |
7.548554c |
6.950384c | 2.123994 |
| Doublesex and mab-3
related transcription factor 3 | DMRT3 | 9 |
ENSG00000064218 |
8.056680c |
6.317158c |
6.723381c | 2.154214 |
| Nuclear receptor
subfamily 2 group E member 1 | NR2E1 | 6 |
ENSG00000112333 |
22.525057c |
16.116569c |
17.970408c | 7.319397 |
| Protein tyrosine
phosphatase, non-receptor type 3 | PTPN3 | 9 |
ENSG00000070159 |
8.833959c |
6.120884b |
6.393193c | 2.302631 |
| Osteopetrosis
associated transmembrane protein 1 | OSTM1 | 6 |
ENSG00000081087 |
19.509941c |
14.291747b |
15.431751c | 6.735904 |
| Amnion associated
transmembrane protein | AMN | 14 |
ENSG00000166126 |
9.728993c |
6.652308b |
8.176856c | 3.172278 |
| Septin 10 | SEPT10 | 2 |
ENSG00000186522 |
18.941045b |
18.182648c |
16.404560b | 8.002855 |
| Zinc finger protein
609 | ZNF609 | 15 |
ENSG00000180357 |
9.909624b |
9.217143b |
8.947813b | 4.347291 |
| Family with
sequence similarity 196 member A | FAM196A | 10 |
ENSG00000188916 |
5.235186a |
5.244315b |
5.655077b | 1.967275 |
| Keratin 19 | KRT19 | 17 |
ENSG00000171345 |
11.305383b |
9.410732b |
4.446763b | 3.627874 |
| Phosphodiesterase
3A | PDE3A | 12 |
ENSG00000172572 |
5.004913b |
3.286156b |
3.498454b | 0.721005 |
| Transmembrane
protein 132E | TMEM132E | 17 |
ENSG00000181291 |
3.908044b |
2.355244a |
2.723931b | 0.490595 |
| Ankyrin repeat
domain 18A | ANKRD18A | 9 |
ENSG00000180071 |
3.932660a |
3.543744b |
3.043928a | 1.086252 |
| Nuclear receptor
subfamily 2 group F member 2 | NR2F2 | 15 |
ENSG00000185551 |
7.370952a |
5.814443a |
6.732305b | 2.443982 |
| Roundabout guidance
receptor 2 | ROBO2 | 3 |
ENSG00000185008 |
5.092977a |
4.044153a |
4.306780a | 1.735872 |
| ADAM with
thrombospondin type 1 motif 1 | ADAMTS1 | 21 |
ENSG00000154734 |
3.037976a |
2.602663a |
3.056663b | 0.691831 |
| Solute carrier
family 18 member A3 | SLC18A3 | 10 |
ENSG00000187714 |
3.300212a |
2.584836a |
3.335012a | 0.991600 |
Functional enrichment analysis
The methylation levels of various genomic features
were identified as associated with biological processes occurring
at the membrane-enclosed lumen level, particularly in polyp tissue.
Gene Ontology analysis indicated that the molecular functions of
the majority of the genes identified as differentially methylated
were associated with DNA binding and cancer pathways in the Kyoto
Encyclopedia of Genes and Genomes pathway database. The results of
DAVID analysis are presented in Fig.
2. Functional annotations of proteins encoded by genes in DMRs
showing significantly (P<0.05) increased or decreased enrichment
compared with controls (Fig. 2)
were classified according to their associated biological processes,
cellular components, and molecular functions. At the level of
biological processes, there were significant differences between
proteins encoded by genes in DMRs associated with the cell cycle,
cytokinesis, and cell division (Fig.
2A), while analysis of cellular components revealed significant
differences between cytoskeletal, vesicular, and synaptic proteins
(Fig. 2B). Analysis according to
molecular function revealed significant differences between
enrichment of encoded proteins associated with translation factor
activity, or nucleic acid, spectrin, or enzyme binding (Fig. 2C). At the level of biological
processes there were significant differences between enrichment of
encoded proteins associated with cell death, intracellular
transport, and cellular morphogenesis (Fig. 2D).
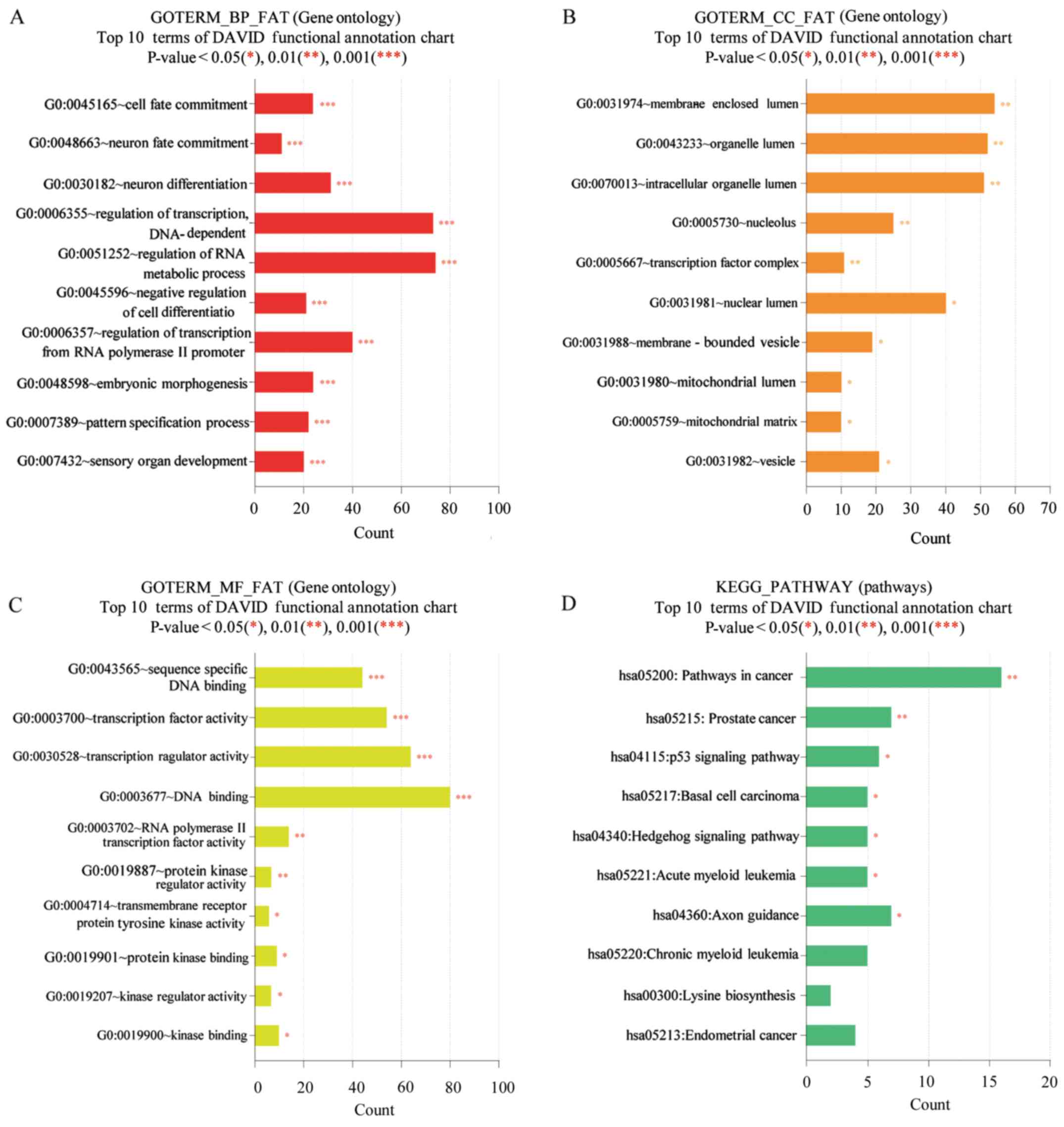 | Figure 2.DAVID functional GO analysis of (A)
BP, (B) CC, (C) MF and (D) KEGG protein enrichment. *P<0.05,
**P<0.01 and ***P<0.001 vs. control. DAVID, Database for
Annotation, Visualization and Integrated Discovery; KEGG, Kyoto
Encyclopedia of Genes and Genomes; GO, gene ontology; BP,
biological processes; CC, cellular components; MF, molecular
function. |
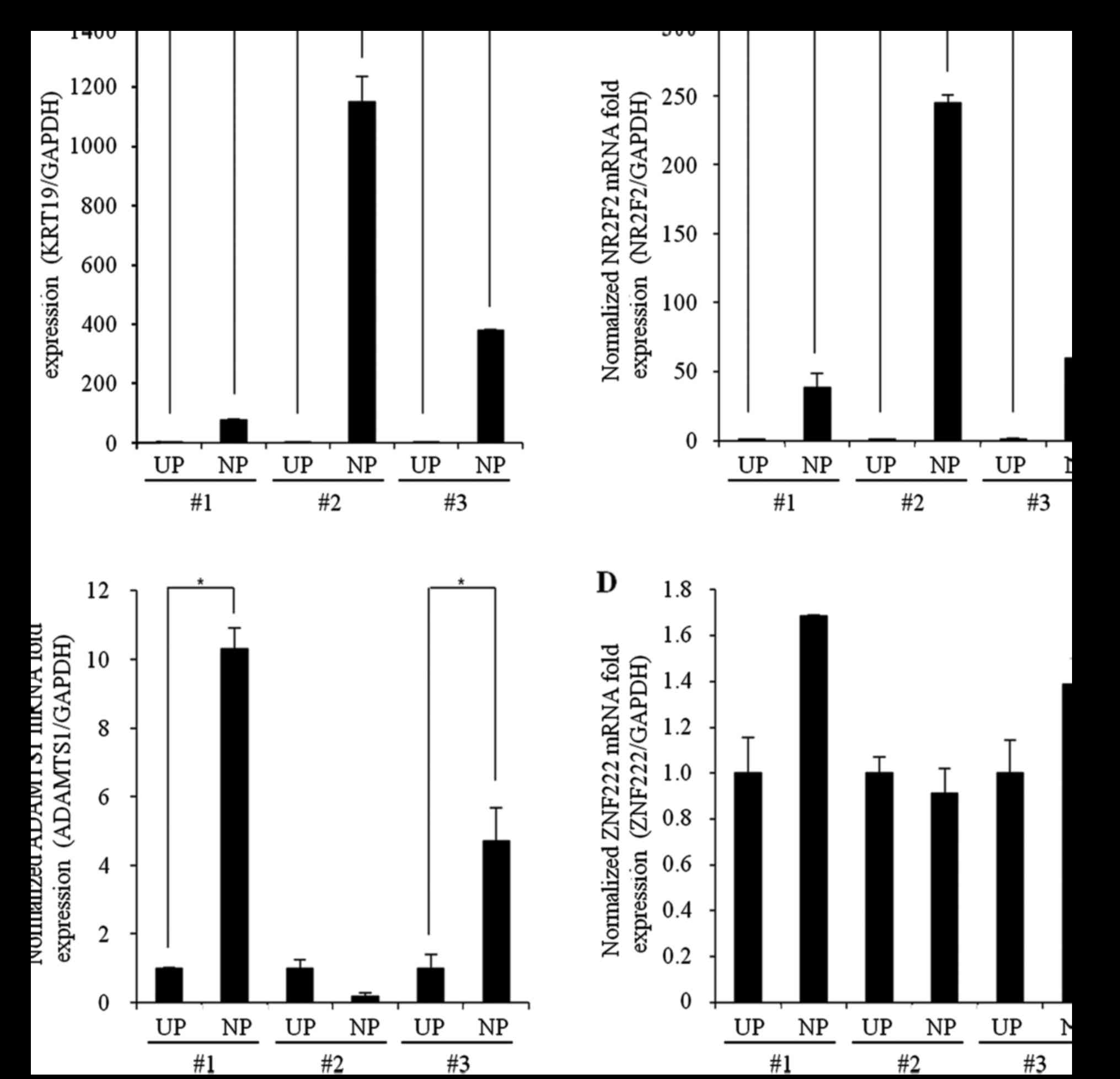
Confirmation of gene expression
changes by qPCR
For validation of the genetic changes associated
with NP formation, we evaluated the mRNA expression levels of genes
that had previously been reported to be closely related to NP
(KRT19, NR2F2, ADAMTS1, and ZNF222) in
samples from three patients with CRSwNP using qPCR. The mRNA
expression levels of KRT19 and NR2F2 were
significantly increased in samples from all three patients with
CRSwNP (CRSwNP-NP) compared with UP control samples (Control,
CRSsNP-UP, and CRSwNP-UP) (Fig. 3A and
B); however, although the mRNA expression level of
ADAMTS1 was significantly increased in two of the patients,
the other patient showed no significant difference in expression
from controls. ZNF222 mRNA levels did not differ
significantly between the NP and UP tissues from any patients.
Discussion
Recent studies have identified genes of unknown
function that are associated with disease as also related to NP
growth. Fritz et al (11)
examined 12,000 human genes transcribed in the nasal mucosa of
patients with allergic rhinitis with and without NP, and identified
34 differentially expressed genes, including those encoding
inflammatory molecules and putative growth factors. Specifically,
the expression levels of 16 genes were increased, and those of 18
genes were decreased in the polyp group. Although genetic studies
based on comparisons with a control group to evaluate epigenetic
changes, gene rearrangement, and changes in chromatin structure.
have been performed for patients with NPs, the results have been
inconsistent. This may be due to heterogeneity among experimental
groups or genetic differences among the ethnic groups included in
the studies; however, the major cause of such variation is expected
to be environmental influences on the regulation of polyp-related
genes (7). Indeed, there is a
growing body of literature suggesting a role for epigenetic factors
in the complex interplay between genes and the environment
(12,13). DNA methylation is a key to
regulation of the genes responsible for polyps; however, the
details of the mechanisms involved remain poorly understood.
To investigate genes differentially transcribed in
the nasal mucosa of patients with allergic rhinitis with and
without nasal polyps, we conducted systematic gene expression
profiling experiments using clinical samples to identify genes
involved in polyp formation. Genomic methylation and expression
analysis of target genes were performed on tissues from patients
with CRSsNP and CRSwNP. To the best of our knowledge, this is the
first study to use epigenetic technology to quantitatively and
simultaneously monitor the expression and DNA methylation status of
genes involved in allergic rhinitis with and without NP.
In this study, the methylation levels of 19,256
genes were analyzed in CRSwNP-UP, CRSsNP-UP, and control-UP samples
compared with those in CRSwNP-NP samples. Among these genes, 518
exhibited differential methylation, primarily those functionally
characterized as related to inflammation. Several DNA methylation
patterns were similarly regulated in the CRSwNP-UP, CRSsNP-UP, and
control-UP samples compared with the CRSwNP-NP samples. These
results confirm that the UP mucosa can be a suitable control tissue
for epigenetic studies. Indeed, the UP mucosa is one of the common
sites of NP development. Moreover, our results support the findings
of previous studies of differences in innate immune cells during
disease progression in non-asthmatic CRS patients by comparison of
the UP mucosa and NPs (14,15).
Patients without any nasal or sinus disease were
recruited as a control group to detect differences between NP
tissue and healthy nasal mucosa. Validation of the expression
levels of genes exhibiting differences in methylation patterns
between the groups revealed that the expression levels of
KRT19, NR2F2, and ADAMTS1 were significantly
different between the CRSwNP-NP and UP mucosa (Control-UP,
CRSwNP-UP, and CRSsNP-UP) tissues. This represents the first
demonstration of the direct involvement of a translational
regulation mechanism in polyp formation. The detected P-value could
be used for quality control of the data. In this study, 10 and 30
genes were found to be hypermethylated and hypomethylated,
respectively, in CRSwNP-NP compared with UP mucosa tissue
samples.
Previous studies have also indicated that the
genetics of NP formation may be affected by environmental factors
(7). Treatment with local
glucocorticoids is generally prescribed to alleviate the symptoms
of NPs. Benson et al (16)
identified altered expression of 203 genes between patients treated
with glucocorticoids compared with those who did not receive
glucocorticoid treatment; 54 genes were downregulated, and 85
upregulated in the glucocorticoid-treated group. Of the 139 genes
with known functions, 22 pro-inflammatory genes were downregulated,
and a number of anti-inflammatory genes were found to be
upregulated in the glucocorticoid-treated group. In addition,
hypomethylation of the phosphodiesterase (PDE) gene was
detected in the polyp group in this study. Similar to the present
study, the expression levels of NR2F2 and ADAM28 were
among those that increased after glucocorticoid treatment, and
these were also identified as hypomethylated using
methyl-CpG-binding domain sequencing.
Esselens et al (17) studied the potential of
Staphylococcus aureus enterotoxin B to induce changes in the
gene DNA methylation pattern in inflamed nasal tissue. They
generated a list of 43 genes exhibiting altered methylation states
after 24 h of culture with S. aureus enterotoxin B; 33 genes
were hypermethylated (including ROBO1, FAM59A,
SLC25A24, TMEM138, ADAMTS16, and
ZNF541) and 10 were hypomethylated (including
ANKRD18A, ROBO2, FAM196A, SLC18A3,
TMEM132E, ADAMTS1, and ZNF609).
ADAMTS1, encoding a matrix metalloprotease, was also found
to be differentially methylated in the present study. The ADAMTS1
protein is involved in modification of the extracellular matrix,
and also acts as an inhibitory protein present in neural scars that
cleaves extracellular matrix proteins.
Moreover, differential methylation patterns were
detected in a genome-wide analysis of polyp tissue from patients
with aspirin-intolerant asthma compared with aspirin-tolerant
asthma; hypermethylation was detected at 332 loci in 296 genes,
while hypomethylation was detected at 158 loci in 141 genes
(18). Gene ontology analysis
revealed that the hypomethylated genes were involved in cell
communication, proliferation, cytokine biosynthesis, cytokine
secretion, immune responses, inflammation, and immunoglobulin
binding, whereas the hypermethylated genes were involved in
ectoderm development, hemostasis, wound healing, calcium ion
binding, and oxidoreductase activity. The genes KRT4,
KRT5, KRT8, KRT15, KRT19, and
KRT24 were hypomethylated and enriched in biological
pathways (19). Our results
confirmed that KRT19 was hypomethylated in polyp tissue.
In the present study, the zinc-finger protein genes
ZNF609 and ZNF22 were identified as hypomethylated
and hypermethylated in NP tissue, respectively. Similarly, patients
with allergic fungal sinusitis exhibited expression differences in
10 genes, including ZNF146 (20). Among the other genes identified as
hypomethylated in polyps, those of the RUFY gene family were
particularly highly expressed. RUFY4 is involved in metal
ion and protein binding. This is the first report of an association
of a member of the RUFY gene family with NPs; further
studies are required to confirm whether other members of this
family have roles in polyp formation.
The aim of this study was to detect differences in
DNA methylation patterns between patients with CRS with and without
NPs. Although we detected clear differences, they were subtle, and
involved only a few characters. In addition, only a few differences
in gene expression were identified between the groups, and we were
unable to distinguish between NP subtypes, such as eosinophilic and
neutrophilic. Nevertheless, this study confirmed that epigenetic
variation has a major role in the formation of polyps. While our
study provides baseline reference data indicating a role for
methylation in polyp formation and the candidate genes involved,
more studies with larger sample sizes are clearly needed to better
elucidate the mechanisms of formation and epigenetics of polyps. In
particular, further studies should focus on the specific roles of
the KRT19, NR2F2, and ADAMTS1 genes on NP
development, and on other genes identified as demonstrating
differential methylation patterns in this study.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education, Science and Technology
(grant nos. 2016R1C1B2012384 and 2015R1D1A3A01019948).
References
|
1
|
Casale M, Pappacena M, Potena M, Vesperini
E, Ciglia G, Mladina R, Dianzani C, Degener AM and Salvinelli F:
Nasal polypsosis: From pathogenesis to treatment, an update.
Inflamm Allergy Drug Targets. 10:158–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hamilos DL: Chronic rhinosinusitis:
Epidemiology and medical management. J Allergy Clin Immunol.
128:693–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fokkens WJ, Lund VJ, Mullol J, Bachert C,
Alobid I, Baroody F, Cohen N, Cervin A, Douglas R, Gevaert P, et
al: European position paper on rhinosinusitis and nasal polyps
2012. Rhinol Suppl. 3:1–298. 2012.
|
|
4
|
Dykewicz MS and Hamilos DL: Rhinitis and
sinusitis. J Allergy Clin Immunol. 125:S103–S115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chaaban MR, Walsh EM and Woodworth BA:
Epidemiology and differential diagnosis of nasal polyps. Am J
Rhinol Allergy. 27:473–478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alexiou A, Sourtzi P, Dimakopoulou K,
Manolis E and Velonakis E: Nasal polyps: Heredity, allergies, and
environmental and occupational exposure. J Otolaryngol Head Neck
Surg. 40:58–63. 2011.PubMed/NCBI
|
|
7
|
Zheng YB, Zhao Y, Yue LY, Lin P, Liu YF,
Xian JM, Zhou GY and Wang DY: Pilot study of DNA methylation in the
pathogenesis of chronic rhinosinusitis with nasal polyps.
Rhinology. 53:345–352. 2015.PubMed/NCBI
|
|
8
|
Hamilos DL: Chronic rhinosinusitis
patterns of illness. Clin Allergy Immunol. 20:1–13. 2007.PubMed/NCBI
|
|
9
|
Slavin RG, Spector SL, Bernstein IL,
Kaliner MA, Kennedy DW, Virant FS, Wald ER, Khan DA, Blessing-Moore
J, Lang DM, et al: The diagnosis and management of sinusitis: A
practice parameter update. J Allergy Clin Immunol. 116 6
Suppl:S13–S47. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
North ML and Ellis AK: The role of
epigenetics in the developmental origins of allergic disease. Ann
Allergy Asthma Immunol. 106:355–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fritz SB, Terrell JE, Conner ER,
Kukowska-Latallo JF and Baker JR: Nasal mucosal gene expression in
patients with allergic rhinitis with and without nasal polyps. J
Allergy Clin Immunol. 112:1057–1063. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White GP, Hollams EM, Yerkovich ST, Bosco
A, Holt BJ, Bassami MR, Kusel M, Sly PD and Holt PG: CpG
methylation patterns in the IFNgamma promoter in naive T cells:
Variations during Th1 and Th2 differentiation and between atopics
and non-atopics. Pediatr Allergy Immunol. 17:557–564. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baron U, Floess S, Wieczorek G, Baumann K,
Grützkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, et
al: DNA methylation in the human FOXP3 locus discriminates
regulatory T cells from activated FOXP3(+) conventional T cells.
Eur J Immunol. 37:2378–2389. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fokkens W, Lund V, Bachert C, Clement P,
Helllings P, Holmstrom M, Jones N, Kalogjera L, Kennedy D, Kowalski
M, et al: EAACI position paper on rhinosinusitis and nasal polyps
executive summary. Allergy. 60:583–601. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meltzer EO, Hamilos DL, Hadley JA, Lanza
DC, Marple BF, Nicklas RA, Bachert C, Baraniuk J, Baroody FM,
Benninger MS, et al: Rhinosinusitis: Establishing definitions for
clinical research and patient care. J Allergy Clin Immunol. 114 6
Suppl:S155–S212. 2004. View Article : Google Scholar
|
|
16
|
Benson M, Carlsson L, Adner M, Jernås M,
Rudemo M, Sjögren A, Svensson PA, Uddman R and Cardell LO: Gene
profiling reveals increased expression of uteroglobin and other
anti-inflammatory genes in glucocorticoid-treated nasal polyps. J
Allergy Clin Immunol. 113:1137–1143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Esselens C, Malapeira J, Colomé N, Casal
C, Rodríguez-Manzaneque JC, Canals F and Arribas J: The cleavage of
semaphorin 3C induced by ADAMTS1 promotes cell migration. J Biol
Chem. 285:2463–2473. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheong HS, Park SM, Kim MO, Park JS, Lee
JY, Byun JY, Park BL, Shin HD and Park CS: Genome-wide methylation
profile of nasal polyps: Relation to aspirin hypersensitive in
asthmatics. Allergy. 66:637–644. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sridhar S, Schembri F, Zeskind J, Shah V,
Gustafson AM, Steiling K, Liu G, Dumas YM, Zhang X, Brody JS, et
al: Smoking-induced gene expression changes in the bronchial airway
are reflected in nasal and buccal epithelium. BMC Genomics.
9:2592008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orlandi RR, Thibeault SL and Ferguson BJ:
Microarray analysis of allergic fungal sinusitis and eosinophilic
mucin rhinosinusitis. Otolaryngol Head Neck Surg. 136:707–713.
2007. View Article : Google Scholar : PubMed/NCBI
|