Introduction
Free fatty acid receptor 4 (FFAR4) is activated by
long chain free fatty acids (FFAs), and its activation serves
diverse roles in regulating hormone secretion, inflammatory
responses and cell survival (1–3).
Previous studies that examined the functions of FFAR4 identified a
series of non-FFA agonists, and among them was grifolic acid
(2,4,5).
Grifolic acid is a phenolic compound that was initially isolated
from the fruiting bodies of the mushroom Albatrellus
confluens. The pharmacological actions of grifolic acid have
not been fully characterized, except for its ability to activate
FFAR4. Grifolic acid was reported to activate extracellular
signal-regulated kinase (ERK) responses and to stimulate an
increase in intracellular calcium concentrations
[(Ca2+i)] in FFAR4-expressing cells, but not
in FFAR1-expressing cells (2). In
addition, grifolic acid exhibited stimulatory effects on
glucagon-like peptide-1 secretion in mouse enteroendocrine STC-1
cells that express FFAR4 endogenously (2,6).
Conversely, grifolic acid inhibited the α-linolenic acid-induced
ERK and [Ca2+]i responses in FFAR4-expressing
cells (2). Grifolic acid was also
revealed to attenuate lard oil-induced secretion of
glucose-dependent insulinotropic polypeptide (GIP) from
FFAR4-expressing K cells of the upper small intestine (7).
FFAR4 is expressed in pro-inflammatory
CD11c+ macrophages, in mouse RAW264.7 macrophages and in
primary intraperitoneal macrophages (8). FFAR4 activation was previously
reported to inhibit the lipopolysaccharide (LPS)-stimulated
phosphorylation of inhibitor of nuclear factor-κB kinase subunit β
and c-Jun N-terminal kinase and subsequently inhibited the
secretion of tumor necrosis factor-α and interleukin (IL)-6 in
RAW264.7 cells (8). Another
previous study demonstrated that FFAR4 activation promoted the
actions of cytosolic phospholipase A2 and cyclooxygenase-2 in
RAW264.7 cells and in human primary monocyte-derived macrophages,
which in turn led to prostaglandin E2 release, nuclear
factor (NF)-κB inhibition and the inhibition of LPS-induced IL-6
secretion (9). Therefore, it is
clear that FFAR4 is expressed in macrophages and has
anti-inflammatory effects; however, the effects of grifolic acid on
the function of macrophages remain unknown. In the present study,
the effects of grifolic acid on macrophages were investigated and
an FFAR4-independent function was identified. These finding
demonstrated a novel action of grifolic acid on cells, and it is
recommended that care should be taken when grifolic acid is used as
FFAR4 agonist.
Materials and methods
Chemicals
Grifolic acid was obtained from R&D Systems Inc.
(Minneapolis, MN, USA). Rabbit anti-FFAR4 polyclonal antibody (cat.
no. SAB4501490) and cellular adenosine 5′-triphosphate (ATP) assay
kit were purchased from Sigma-Aldrich (Merck KGaA; Darmstadt,
Germany). Annexin V-fluorescein isothiocyanate (FITC)/propidium
iodide (PI) staining kits were purchased from BD Pharmingen (BD
Biosciences, San Jose, CA, USA). Mouse FFAR4 Silencer Select siRNA
(cat. no. S200889), Silencer Negative Control siRNA (cat. no.
AM4613), Lipofectamine® RNAiMAX reagent, Opti-MEM
medium, Dulbecco's modified Eagle's medium (DMEM), fetal bovine
serum (FBS), JC-1 dye and rabbit anti-b-actin polyclonal antibody
(cat. no. PA1-183) were purchased from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA).
Cell culture
Mouse RAW264.7 cells were purchased from ATCC
(Manassas, VA, USA) and cultured in a humidified incubator with 5%
CO2 at 37°C in DMEM supplemented with 10% FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin. The medium was changed every
3 days, and RAW264.7 cells were subcultured at 80% confluency and
seeded to plates or dishes for each experiment.
Cell viability assay
RAW264.7 cells were cultured in 96-well plates and
treated with grifolic acid (1.25–20 µmol/l for 2–24 h at 37°C) at
90% confluency in serum-free medium. The control cells were treated
with the solvent without grifolic acid. Subsequently, MTT was added
into medium (0.5 mg/ml) and cells were incubated for 4 h at 37°C.
The medium was discarded and 100 µl isopropanol with 0.01 M HCl was
added to each well and plates were agitated to fully dissolve MTT
crystals. The absorbance of each well was measured at 560 nm with
an ELISA microplate reader (Thermo Fisher Scientific, Inc.). The
background absorbance at 690 nm was also measured and subtracted
from the values measured at 560 nm. The experiments were repeated
three times.
Flow cytometry
The Annexin V-FITC/PI Apoptosis Detection kit was
used to measure cell death. Following incubation with 0 µmol/l
(control), 10 or 20 µmol/l grifolic acid, RAW264.7 cells were
detached from dishes by 0.05% trypsin/EDTA. Cells were washed with
cold PBS and resuspended in the binding buffer supplied in the kit
at 1×106 cells/ml. Annexin V-FITC and PI were added to
cell suspension at a dilution of 1:20 and incubated for 15 min at
room temperature in the dark. Subsequently, cells were diluted in
binding buffer and analyzed by flow cytometry with the CFlow Plus
software (BD Biosciences), and the FITC+/PI+
double-positive cells were counted. The experiments were repeated
three times.
Cellular ATP content measurement
RAW264.7 cells (1×107 cells per sample)
were treated with grifolic acid (10 or 20 µmol/l for 2 h).
Subsequently, cells were lysed and intracellular ATP levels were
measured using ATP assay kits (Sigma-Aldrich; Merck KGaA). Briefly,
cell lysates from different groups were obtained by incubating with
detergent and agitation at ~12 Hz for 5 min at room temperature.
The constituted substrate solutions given in the kits were added to
each sample and incubated for 5 min in the dark. The luminescence
of each sample was measured with a luminescence plate reader
(Thermo Fisher Scientific, Inc.). ATP levels of each sample were
calculated according to the constructed standard curve. The total
protein of each sample was extracted using the
radioimmunoprecipitation lysis buffer from the Beyotime Institute
of Biotechnology (Beijing, China) and quantified by bicinchoninic
acid (BCA) assay. The cellular ATP content was corrected to the
total protein levels. The experiments were repeated three
times.
Measurement of mitochondrial membrane
potential (MMP)
RAW264.7 cells (5×105 cells/well) were
plated into 35 mm dishes and stained with JC-1 (5 µg/ml) in
serum-free medium for 15 min at 37°C. Subsequently, cells were
washed with serum-free medium, and the fluorescence was recorded
prior to (0 min) and following grifolic acid treatment every 10 min
using a Leica TCS SP8 confocal microscope (Leica Microsystems,
Inc., Buffalo Grove, IL, USA) in live cell station at 37°C. For
cyclosporine A treatment, the cells were incubated with 20 µmol/l
cyclosporine A for 10 min prior to stimulation with grifolic acid.
The red fluorescence (excitation 560 nm; emission 600 nm) and green
fluorescence (excitation 488 nm; emission 535 nm) were measured
synchronously, and the ratio of fluorescence intensity for each
cell was analyzed by LAS LITE version 3.3 (Leica Microsystems,
Inc., Buffalo Grove, IL, USA). The experiments were repeated three
times.
FFAR4 siRNA transfection
RAW264.7 cells were grown to 80% confluency at the
time of transfection. The mouse FFAR4 siRNA (100 pmol, Thermo
Fisher Scientific, Inc.) or negative control siRNA (100 pmol;
Thermo Fisher Scientific, Inc. was diluted in 50 µl Opti-MEM medium
(Thermo Fisher Scientific, Inc.), and Lipofectamine RNAiMAX (1 µl;
Thermo Fisher Scientific, Inc.) was diluted in 50 µl Opti-MEM, and
they were incubated for 15 min at room temperature. Subsequently,
the siRNA and Lipofectamine RNAiMAX solutions were mixed and
incubated for 15 min at room temperature to allow complexes to
form. The complexes were added to each well at a dilution of 1:4.
Cells were incubated at 37°C in a humidified 5% CO2
incubator for 24 h to induce gene knockdown. Following 24 h
incubation with Opti-MEM medium the cells were used for
experiments. The experiments were repeated three times.
Western blot analysis
Total protein was extracted from RAW264.7 cells
(1×107 cells per sample) using the ReadyPrep Protein
Extraction kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
quantified by bicinchoninic acid assay. Proteins (40 µg per sample)
were separated using a 12% SDS-PAGE and were transferred to
nitrocellulose membranes using a Trans-Blot SD semi-dry
electrophoresis transfer cell (Bio-Rad Laboratories, Inc.).
Membranes were subsequently blocked at room temperature with 5%
skimmed milk for 2 h at room temperature. Then membranes were
incubated with a rabbit-anti FFAR4 antibody (1:1,000) or
rabbit-anti β-actin antibody (1:1,000) at 4°C, overnight. Following
three washes with TBS, the membranes were incubated with a
horseradish peroxidase-conjugated goat-anti rabbit immunoglobulin G
antibody (1:1,000; cat. no. 31460; Thermo Fisher Scientific, Inc.)
for 1 h at room temperature. Following three washes with TBS,
Clarity Max™ Western enhanced chemiluminescence
Substrate (Bio-Rad Laboratories, Inc.) was added and membranes were
incubated at room temperature for 4 min. The luminescence from the
membranes was imaged by a ChemiDoc MP gel imaging and analysis
system (170–8280; Bio-Rad Laboratories, Inc.). The experiments were
repeated three times.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Comparisons of means of multiple groups with each other
or against one control group were analyzed with one-way analysis of
variance followed by Bonferroni post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of grifolic acid on RAW264.7
cell viability
Grifolic acid at 20 µmol/l significantly inhibited
RAW264.7 cell viability at 2 h and led to almost total loss of cell
viability following 6 h incubation (P<0.01). Observations were
obtained at 2, 6, 12 and 24 h for each concentration following
preliminary experiments, which demonstrated that these incubation
lengths were optimal for use in the present study (data not shown).
Grifolic acid at 10 µmol/l exhibited significant inhibition of
viability following 6 h incubation and achieved maximal effects at
24 h (P<0.05). Grifolic acid at 5 and 2.5 µmol/l inhibited
RAW264.7 cell viability from 6 and 12 h, respectively and
demonstrated significantly increased levels with the increased
duration of incubation Treatment with grifolic acid at 1.25 µmol/l
did not exhibit inhibitory effects at any time points investigated
(Fig. 1).
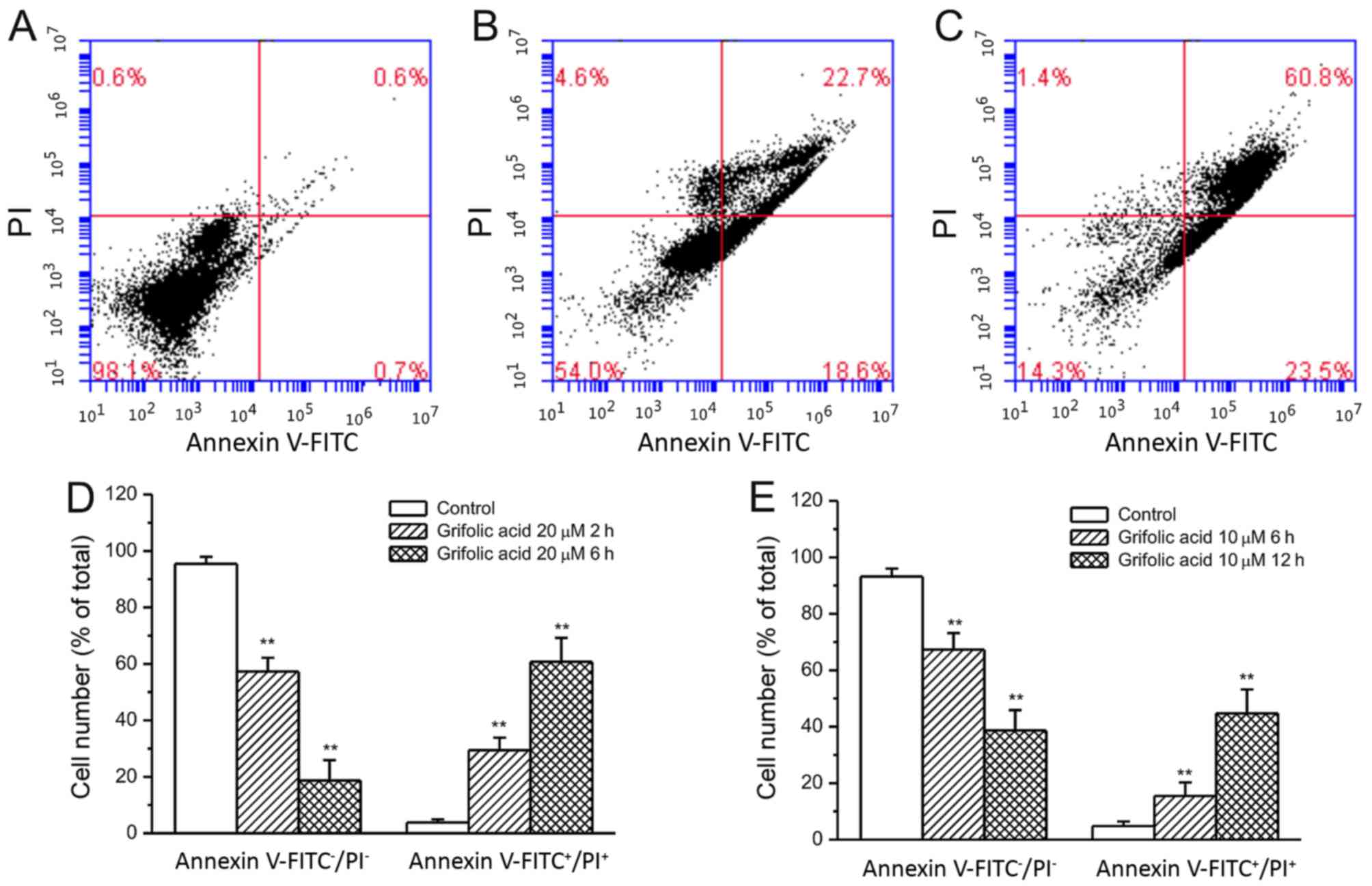
Grifolic acid-treated RAW264.7 cell
death
Based on the MTT assay results, at 10 and 20 µmol/l
grifolic acid demonstrated more significant effects on RAW264.7
cells and were used in the experiments of Annexin V/PI staining.
Observations were obtained at 2 and 6 h for each concentration
following preliminary experiments, which demonstrated that longer
incubations resulted in cell lysis whereas little effect was
observed at shorter incubation times (data not shown). Grifolic
acid (20 µmol/l) treatment for 2 or 6 h lead to a significant
increase in the number of both Annexin V- and PI-staining positive
RAW264.7 cells compared with the control cells that were not
treated with grifolic acid (P<0.01; Fig. 2A-D). Similarly, grifolic acid
treatment at 10 µmol/l for 6 or 12 h also demonstrated a
significant increase in the number of Annexin
V-FITC+/PI+ cells compared with the control
cells (P<0.01; Fig. 2E).
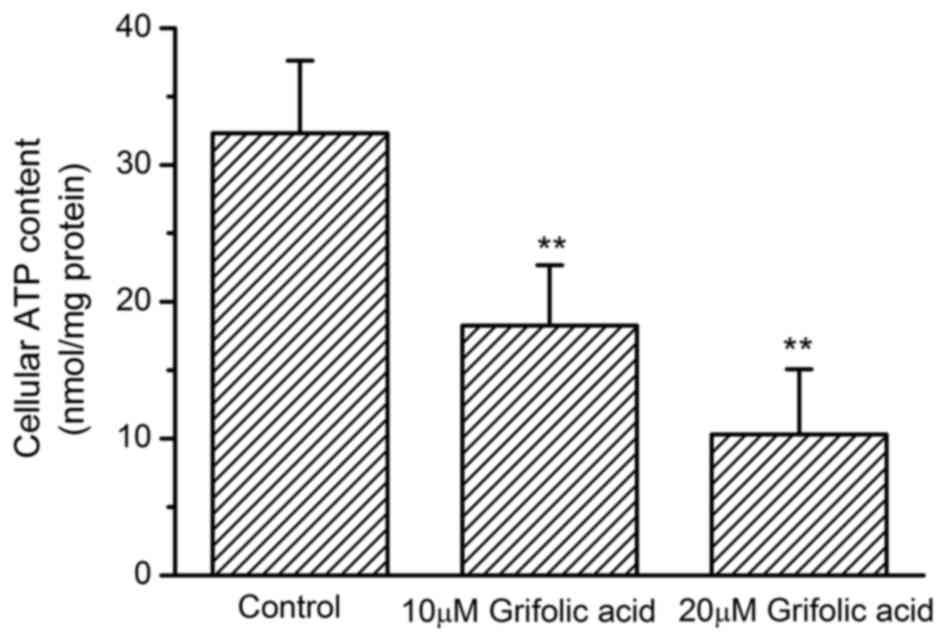
Inhibition of ATP production by
grifolic acid in RAW264.7 cells
The cellular ATP content in untreated control
RAW264.7 cells was 32.31±5.32 nmol/mg protein. ATP content
significantly reduced to 10.29±4.76 nmol/mg protein when cells were
treated with 20 µmol/l grifolic acid for 2 h (P<0.01). Grifolic
acid treatment at 10 µmol/l for 2 h also significantly reduced
cellular ATP content to 18.26±4.41 nmol/mg protein (P<0.01;
Fig. 3).
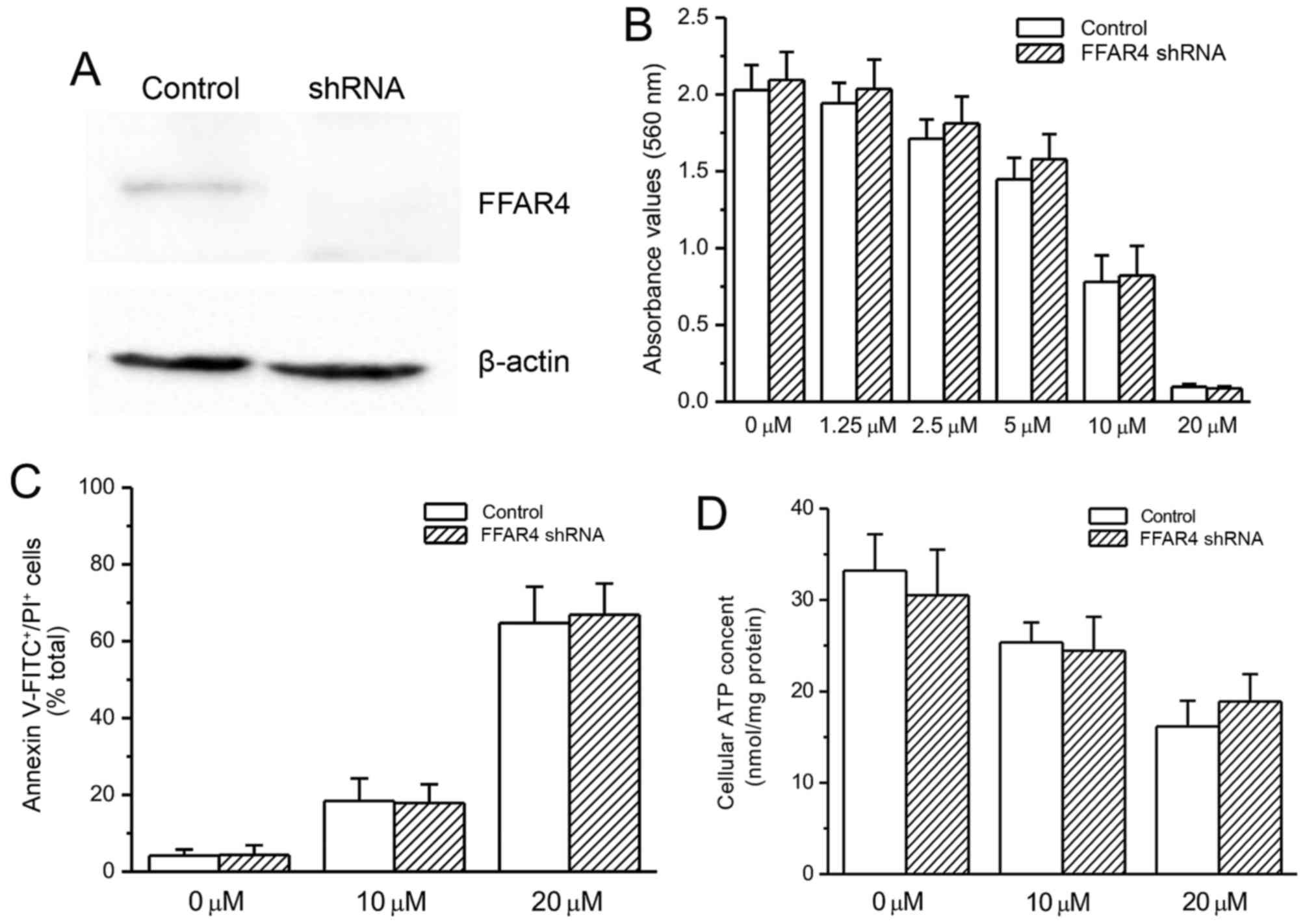
Effects of FFAR4 knockdown on grifolic
acid-induced RAW264.7 cell death
Mouse FFAR4 siRNA transfection resulted in a notable
reduction in FFAR4 protein expression level compared with the
untransfected control cells (Fig.
4A). FFAR4 knockdown did not exhibit a significant influence on
grifolic acid-induced inhibition of cell viability and apoptosis,
as indicated by MTT assay and Annexin V-FITC/PI staining (Fig. 4B and C, respectively). Similarly,
the decrease in cellular ATP content in grifolic acid-treated
RAW264.7 cells was not affected by FFAR4-knockdown (Fig. 4D).
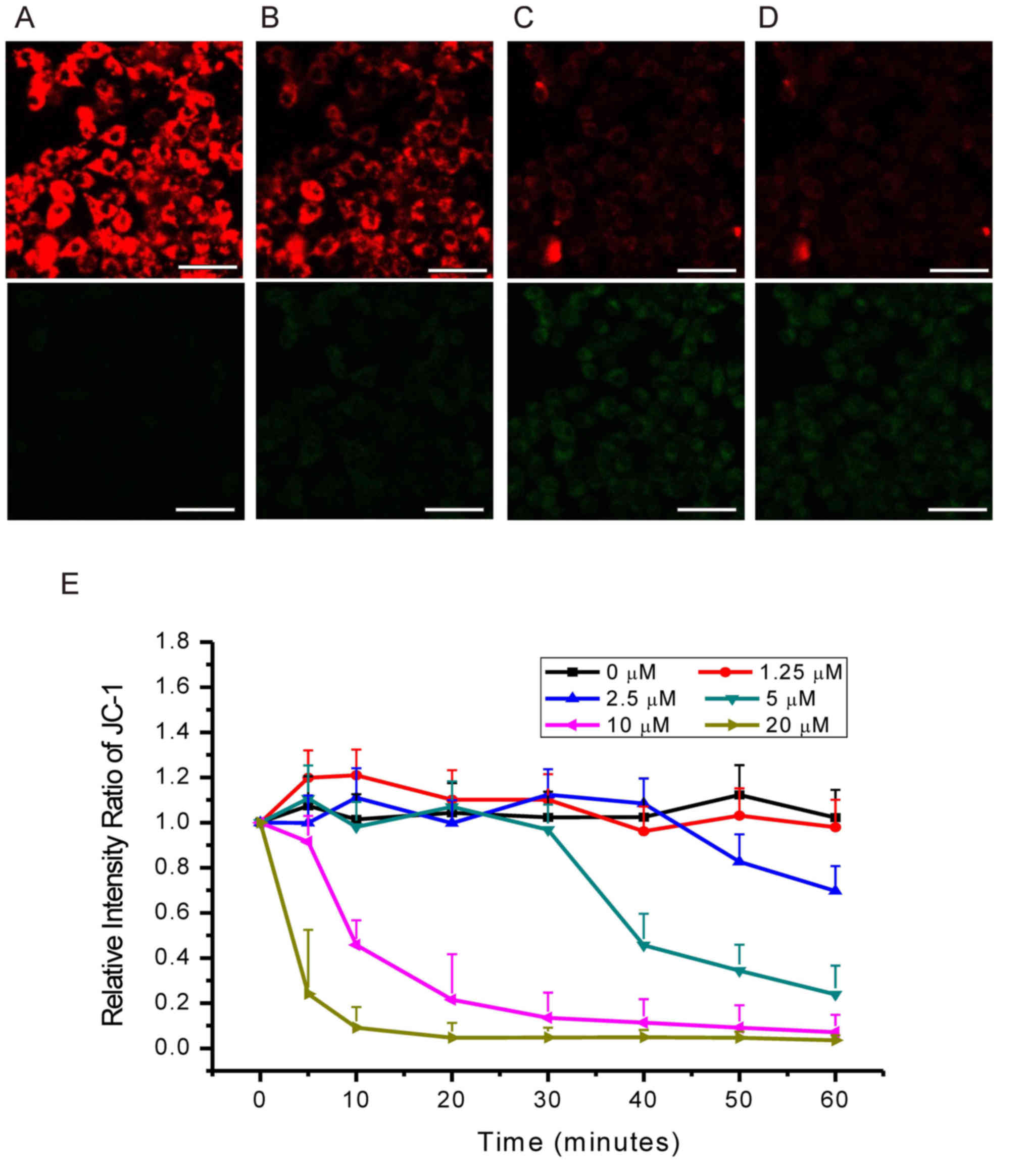
Effects of grifolic acid on MMP of
RAW264.7 cells
JC-1 emits green fluorescence in the cytoplasm and
exhibits membrane potential-dependent accumulation in mitochondria
with a shift of emission wavelength from green to red (10,11);
MMP reduction is indicated by a decrease in the red/green
fluorescence intensity ratio. In the present study, grifolic acid
treatment resulted in a decrease in red/green fluorescence
intensity ratio in a dose- and time-dependent manner (Fig. 5). Cells treated with 20 µmol/l
grifolic acid exhibited a decrease in MMP at 5 min, with the
maximal effects achieved within 20 min (Fig. 5E). Grifolic acid treatment at 10
µmol/l significantly reduced MMP at 10 min and achieved the maximal
effects in 40 min (Fig. 5E). MMP
was also significantly decreased compared with the control 60 min
following treatment with 5 and 2.5 µmol/l grifolic acid at 40 and
50 min, respectively, but not with 1.25 µmol/l treatment (Fig. 5E).
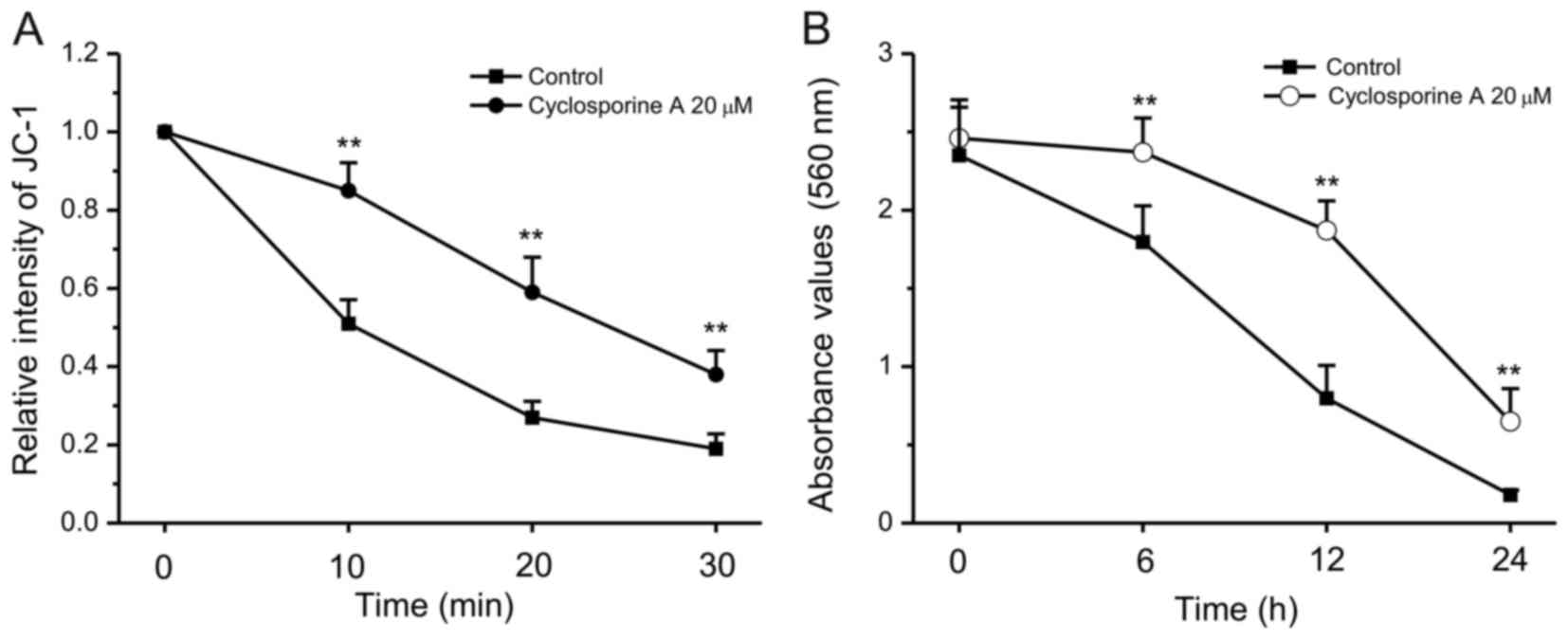
Protective effects of cyclosporine A
against grifolic acid-induced MMP loss and Raw264.7 cell death
Cyclosporine A is able to attenuate mitochondrial
permeability transition and improve mitochondrial respiratory
function (12,13). Therefore, the effects of
cyclosporine A on the grifolic acid-induced decrease in MMP were
investigated. When RAW264.7 cells were pretreated with cyclosporine
A at 20 µmol/l for 10 min, grifolic acid-induced MMP loss was
significantly attenuated compared with control cells that were
treated with grifolic acid but not with cyclosporine A (P<0.01;
Fig. 6A). Untreated control cells
demonstrated no changes during measurement (data not shown). In
addition, cyclosporine A treatment (10 µmol/l) also inhibited
grifolic acid-induced decrease in cell viability of RAW264.7 cells
(Fig. 6B).
Discussion
Grifolic acid was first extracted from the fresh
fruiting bodies of the mushroom A. confluens, but
little is known about its actions. Grifolic acid was previously
demonstrated to activate ERK and to increase intracellular calcium
concentration in FFAR4-expressing cells though FFAR4 signaling
(2). A number of other reports
also revealed grifolic acid's actions on FFAR4 in FFAR4-expressing
mouse intestinal STC-1 cells and K cells that secrete GIP. Grifolic
acid was also used to antagonize the effects of FFAs on FFAR4
(7). Therefore, grifolic acid is
considered as an agonist or antagonist for FFAR4. FFAR4 is
expressed in macrophages including RAW264.7 cells (14). Results from the present study
demonstrated that grifolic acid treatment induced mouse RAW264.7
macrophage apoptosis. However, the effects of grifolic acid on cell
death were unaltered by the inhibition of FFAR4 expression in
RAW264.7 cells. Therefore, the present study suggested that
grifolic acid is not a pure FFAR4 agonist or antagonist, and may
have additional pharmacological targets to inhibit cell
viability.
The effects of grifolic acid to induce RAW264.7 cell
death may be due to the decrease in ATP production following damage
to mitochondrial functions. Grifolic acid-treated RAW264.7 cells
exhibited a significant increase in the percentage of Annexin
V-FITC/PI double-positive cells, which indicated necrosis or late
stage of apoptosis, but this needs to be confirmed. It is known
that cellular ATP levels influence cell viability (15,16).
Cellular ATP reduction leads to cell death, and the rate and speed
of ATP reduction determines the type of cell death (17). A slow reduction of ATP levels leads
to cell apoptosis, whereas rapid reduction of ATP results in cell
swelling and necrosis (15,18,19).
Grifolic acid induced a decrease in ATP levels in 2 h in RAW264.7
cells, which may lead to cell necrosis.
Mitochondria are the cell organelles responsible for
ATP production (20). To function
under physiological conditions, mitochondria need to maintain MMP;
a reduction in MMP may lead to functional damage of mitochondria
and may result in deficiency of ATP production (21). In the present study, it was
demonstrated that grifolic acid significantly reduced MMP of
RAW264.7 cells in a dose- and time-dependent manner, which is in
accordance with the effects of grifolic acid on RAW264.7 cell
death. This is the first study, to the best of the authors'
knowledge, to demonstrate the influence of grifolic acid on
mitochondria, which indicated that mitochondria may be a target of
grifolic acid. The improvement of grifolic acid-induced reduction
of MMP and cell death by cyclosporine A further supported the
observation that grifolic acid affected mitochondrial function.
Mitochondria form permeability transition pores on the membrane
under certain pathological conditions, which reduce MMP and lead to
a release of cell death regulators (22,23).
Cyclosporine A may prevent the formation of permeability transition
pores on the mitochondrial membrane by acting on cyclosporine
A-binding protein in mitochondria (24–27).
Cyclosporine A treatment significantly attenuated the grifolic
acid-induced decrease in MMP and protected RAW264.7 cells against
grifolic acid-induced cell death. The attenuation of grifolic
acid-induced MMP reduction by cyclosporine A, the mitochondria
protector, further indicated that mitochondria may be the target of
grifolic acid. In conclusion, grifolic acid exposure damaged
mitochondrial function, which resulted in MMP reduction and led to
the decrease in ATP production causing RAW264.7 cell death.
The effects of grifolic acid on RAW264.7 cell death
appear to be independent of FFAR4. FFAR4 is expressed in
macrophages such as RAW264.7 cells, and its activation serves an
anti-inflammatory role in macrophages (8,28,29).
In the present study, FFAR4 expression was successfully inhibited
as confirmed by western blot. Knockdown of FFAR4 did not
demonstrate any influence on grifolic acid-induced cell death and
MMP reduction. Therefore, FFAR4 may not take part in the action of
grifolic acid causing RAW264.7 cell death. The molecular mechanism
of grifolic acid-induced damage to mitochondria remains unknown and
needs to be clarified in the future.
In summary, grifolic acid treatment reduced MMP and
ATP production and led to RAW264.7 cell death. Considering that the
concentration of grifolic acid used in the present study was within
the same range as what was used to activate FFAR4 (2,6,7,30),
it was suggested that grifolic acid may not be a pure FFAR4
agonist. Other pharmacological target for grifolic acid that links
grifolic acid to cell mitochondria dysfunction remains to be
clarified.
Acknowledgements
The present study was supported by the grants from
Shaanxi Province (grant no. 2015KTCQ03-03) and Xi'an Medical
University (grant no. 2015RCYJ02).
References
|
1
|
Mobraten K, Haug TM, Kleiveland CR and Lea
T: Omega-3 and omega-6 PUFAs induce the same GPR120-mediated
signalling events, but with different kinetics and intensity in
Caco-2 cells. Lipids Health Dis. 12:1012013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hara T, Hirasawa A, Sun Q, Sadakane K,
Itsubo C, Iga T, Adachi T, Koshimizu TA, Hashimoto T, Asakawa Y and
Tsujimoto G: Novel selective ligands for free fatty acid receptors
GPR120 and GPR40. Naunyn Schmiedebergs Arch Pharmacol. 380:247–255.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu HD, Wang WB, Xu ZG, Liu CH, He DF, Du
LP, Li MY, Yu X and Sun JP: FFA4 receptor (GPR120): A hot target
for the development of anti-diabetic therapies. Eur J Pharmacol.
763:160–168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun Q, Hirasawa A, Hara T, Kimura I,
Adachi T, Awaji T, Ishiguro M, Suzuki T, Miyata N and Tsujimoto G:
Structure-activity relationships of GPR120 agonists based on a
docking simulation. Mol Pharmacol. 78:804–810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimpukade B, Hudson BD, Hovgaard CK,
Milligan G and Ulven T: Discovery of a potent and selective GPR120
agonist. J Med Chem. 55:4511–4515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li AJ, Wang Q, Dinh TT, Simasko SM and
Ritter S: Mercaptoacetate blocks fatty acid-induced GLP-1 secretion
in male rats by directly antagonizing GPR40 fatty acid receptors.
Am J Physiol Regul Integr Comp Physiol. 310:R724–R732. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iwasaki K, Harada N, Sasaki K, Yamane S,
Iida K, Suzuki K, Hamasaki A, Nasteska D, Shibue K, Joo E, et al:
Free fatty acid receptor GPR120 is highly expressed in
enteroendocrine K cells of the upper small intestine and has a
critical role in GIP secretion after fat ingestion. Endocrinology.
156:837–846. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oh DY, Talukdar S, Bae EJ, Imamura T,
Morinaga H, Fan W, Li P, Lu WJ, Watkins SM and Olefsky JM: GPR120
is an omega-3 fatty acid receptor mediating potent
anti-inflammatory and insulin-sensitizing effects. Cell.
142:687–698. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Y, Chen LY, Sokolowska M, Eberlein M,
Alsaaty S, Martinez-Anton A, Logun C, Qi HY and Shelhamer JH: The
fish oil ingredient, docosahexaenoic acid, activates cytosolic
phospholipase A2 via GPR120 receptor to produce prostaglandin E2
and plays an anti-inflammatory role in macrophages. Immunology.
143:81–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reers M, Smith TW and Chen LB: J-aggregate
formation of a carbocyanine as a quantitative fluorescent indicator
of membrane potential. Biochemistry. 30:4480–4486. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smiley ST, Reers M, Mottola-Hartshorn C,
Lin M, Chen A, Smith TW, Steele GD Jr and Chen LB: Intracellular
heterogeneity in mitochondrial membrane potentials revealed by a
J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA.
88:pp. 3671–3675. 1991; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Novgorodov SA, Gudz TI, Kushnareva YE,
Zorov DB and Kudrjashov YB: Effect of ADP/ATP antiporter
conformational state on the suppression of the nonspecific
permeability of the inner mitochondrial membrane by cyclosporine A.
FEBS Lett. 277:123–126. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu W, Zhang X, Liu J, Wang X, Li S, Liu R,
Liao N, Zhang T and Hai C: Cyclosporine a suppressed glucose
oxidase induced P53 mitochondrial translocation and hepatic cell
apoptosis through blocking mitochondrial permeability transition.
Int J Biol Sci. 12:198–209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han L, Song S, Niu Y, Meng M and Wang C:
Eicosapentaenoic acid (EPA) induced macrophages activation through
GPR120-mediated Raf-ERK1/2-IKKβ-NF-κB p65 signaling pathways.
Nutrients. 9:pii: E9372017. View Article : Google Scholar
|
|
15
|
Leist M, Single B, Castoldi AF, Kühnle S
and Nicotera P: Intracellular adenosine triphosphate (ATP)
concentration: A switch in the decision between apoptosis and
necrosis. J Exp Med. 185:1481–1486. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nicotera P and Leist M: Energy supply and
the shape of death in neurons and lymphoid cells. Cell Death
Differ. 4:435–442. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Skulachev VP: Bioenergetic aspects of
apoptosis, necrosis and mitoptosis. Apoptosis. 11:473–485. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nicotera P, Leist M and Ferrando-May E:
Intracellular ATP, a switch in the decision between apoptosis and
necrosis. Toxicol Lett 102–103. 1–142. 1998.
|
|
19
|
Eguchi Y, Srinivasan A, Tomaselli KJ,
Shimizu S and Tsujimoto Y: ATP-dependent steps in apoptotic signal
transduction. Cancer Res. 59:2174–2181. 1999.PubMed/NCBI
|
|
20
|
Ernster L and Schatz G: Mitochondria: A
historical review. J Cell Biol. 91:227s–255s. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kadenbach B, Ramzan R, Moosdorf R and Vogt
S: The role of mitochondrial membrane potential in ischemic heart
failure. Mitochondrion. 11:700–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lemasters JJ, Qian T, He L, Kim JS, Elmore
SP, Cascio WE and Brenner DA: Role of mitochondrial inner membrane
permeabilization in necrotic cell death, apoptosis, and autophagy.
Antioxid Redox Signal. 4:769–781. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakagawa Y, Suzuki T, Kamimura H and Nagai
F: Role of mitochondrial membrane permeability transition in
N-nitrosofenfluramine-induced cell injury in rat hepatocytes. Eur J
Pharmacol. 529:33–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Plin C, Haddad PS, Tillement JP, Elimadi A
and Morin D: Protection by cyclosporin A of mitochondrial and
cellular functions during a cold preservation-warm reperfusion of
rat liver. Eur J Pharmacol. 495:111–118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Onishi A, Miyamae M, Kaneda K, Kotani J
and Figueredo VM: Direct evidence for inhibition of mitochondrial
permeability transition pore opening by sevoflurane preconditioning
in cardiomyocytes: Comparison with cyclosporine A. Eur J Pharmacol.
675:40–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andreeva L, Tanveer A and Crompton M:
Evidence for the involvement of a membrane-associated
cyclosporin-A-binding protein in the Ca(2+)-activated inner
membrane pore of heart mitochondria. Eur J Biochem. 230:1125–1132.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Halestrap AP, Connern CP, Griffiths EJ and
Kerr PM: Cyclosporin A binding to mitochondrial cyclophilin
inhibits the permeability transition pore and protects hearts from
ischaemia/reperfusion injury. Mol Cell Biochem. 174:167–172. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oh DY, Walenta E, Akiyama TE, Lagakos WS,
Lackey D, Pessentheiner AR, Sasik R, Hah N, Chi TJ, Cox JM, et al:
A Gpr120-selective agonist improves insulin resistance and chronic
inflammation in obese mice. Nat Med. 20:942–947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Im DS: Functions of omega-3 fatty acids
and FFA4 (GPR120) in macrophages. Eur J Pharmacol. 785:36–43. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Janssen S, Laermans J, Iwakura H, Tack J
and Depoortere I: Sensing of fatty acids for octanoylation of
ghrelin involves a gustatory G-protein. PLoS One. 7:e401682012.
View Article : Google Scholar : PubMed/NCBI
|