Introduction
Hepatocellular carcinoma (HCC) is recognized as the
sixth most common cancer in the world and the third leading cause
of cancer-related deaths (1). HCC
occurs at a higher annual incidence in Asia and Africa than in
western countries, and finding effective treatments has therefore
been a top priority in these countries. Since HCC is caused by
chronic liver disease and is often diagnosed at an advanced stage,
drugs are one of the main treatment options for patients with
advanced HCC. Chemotherapeutics such as doxorubicin are currently
the primary agents used to treat HCC; however, these cytotoxic
molecules are non-selective and cause severe side effects (2). Therefore, researchers have begun to
investigate targeted therapeutics. Sorafenib is the only approved
targeted drug for HCC patients (2). It inhibits RAF serine/threonine
kinases (mutant and wild-type B-RAF and C-RAF/Raf-1), which play a
pivotal role in mitogenic and oncogenic signal transduction through
the RAF/mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase (ERK) kinase (MEK)/ERK (RAF/MEK/ERK)
cascade involved in tumor proliferation (3). In addition, sorafenib strongly
inhibits tyrosine kinase receptors that promote angiogenesis, such
as vascular endothelial growth factor receptor (VEGFR)2, VEGFR 3,
platelet-derived growth factor receptor-β (PDGFR-β), Flt3, and
c-Kit (4). In addition to blocking
the RAF/MEK/ERK pathway, sorafenib induces tumor cell apoptosis in
hepatocellular PLC/PRF/5 cells independent of caspase activation
(3). Though sorafenib is the only
available therapeutic agent that can prolong the overall survival
of patients with advanced HCC, sorafenib resistance in HCC often
prevents its long-term efficacy (5). Generally, resistance to sorafenib can
be attributed to two causes. First, HCC cells can harbor intrinsic
resistance prior to drug treatment. Ezzoukhry et al
(6) reported that the sensitivity
of different types of HCC cells to sorafenib is diverse and
suggested that the drug resistance of some HCC cell lines (e.g.,
Hep3B) may occur spontaneously when sorafenib is used at clinically
relevant concentrations (6).
Additionally, previous studies have demonstrated the paradoxical
ability of sorafenib to activate RAF kinases by transactivating RAF
dimers (7). The biochemical
rationale for sorafenib-induced activation of RAF kinases seems to
be in line with the mechanisms underlying intrinsic sorafenib
resistance in HCC. Second, acquired resistance to sorafenib during
the course of therapy is often encountered in HCC (5,8–10).
For example, somatic mutations in oncogenes and the activation of
numerous oncogenic signaling pathways, e.g., the MAPK/RAS/ERK,
TGFβ, and PI3K/PTEN/AKT pathways, contribute to the development of
sorafenib resistance (2). With the
emergence of sorafenib, clinical studies have been performed with a
range of drugs, primarily molecular targeted therapies; however,
these trials have terminated in failure (11). Despite this difficulty, there is
still an urgent need to identify novel pharmaceutical drugs beyond
sorafenib. CBI-5725 is a novel bi-aryl urea; the patent for this
compound is owned by Crown Bioscience, Inc. (China). Due to its
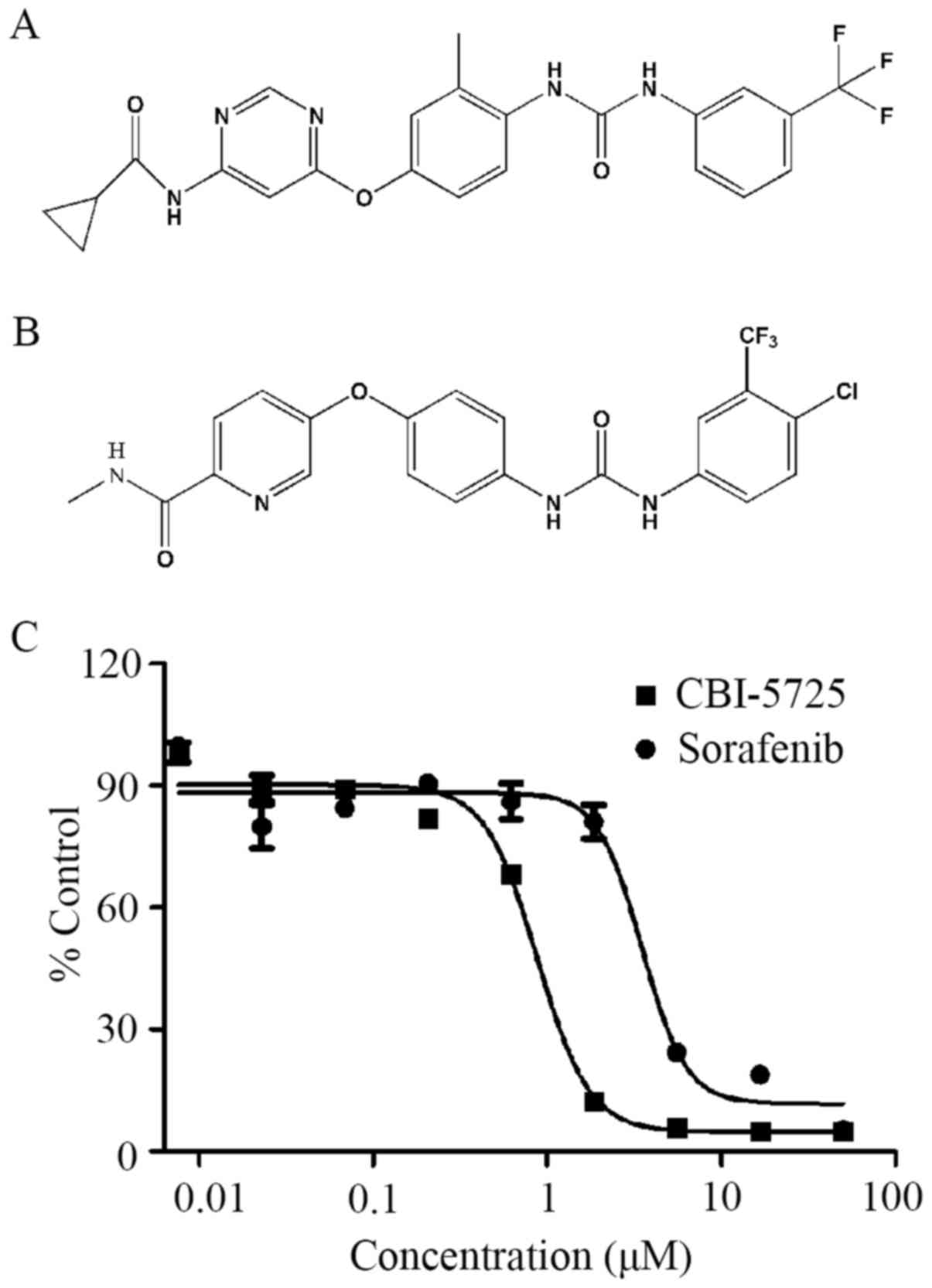
potent inhibitory effect on HCC cell proliferation (Fig. 1), CBI-5725 was selected for further
investigation. In this paper, the anti-cancer effects of this
compound on HCC tumor cells were assessed in vitro and
compared with those of sorafenib in the PLC/PRF/5 (mutant K-RAS and
wild-type B-RAF) HCC cell line. Furthermore, the mechanism
underlying CBI-5725 activity was explored. A comparison of the
anti-cancer effects of CBI-5725 and sorafenib is of great
importance for understanding the underlying mechanism. The aim of
this investigation was to identify a novel multikinase inhibitor as
a potential candidate for the treatment of liver cancer.
Materials and methods
Compounds
Sorafenib was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). CBI-5725 was synthesized by
Crown Bioscience, Inc. (San Diego, CA, USA). The chemical name of
CBI-5725 is
N-(3-trifluoromethylphenyl)-N′-(2-methyl-4-(6-cyclopropanecarboxamido-pyrimidin-4-yl)
oxyphenyl) urea. The structural formula is displayed in Fig. 1. The compounds were dissolved in
100% DMSO (Applichem, Darmstadt, Germany) and diluted in minimum
essential medium (MEM; MP Biomedicals, Solon, OH, USA) to a range
of concentrations with a final DMSO concentration of 0.1%. Cells
were treated with 0.1% (v/v) DMSO as a solvent control.
Cell lines
PLC/PRF/5 (mutant p53, mutant K-RAS, and wild-type
B-RAF) human HCC cells were purchased from American Type Culture
Collection (Manassas, VA, USA) and incubated in MEM with 10% fetal
bovine serum (FBS; PAA, Austria) in a 5% CO2 atmosphere
at 37°C.
Alamar blue assay
Tumor cells were trypsinized, plated at a density of
5,000 cells per well in 96-well plates and cultured overnight in a
humidified chamber with 5% CO2 at 37°C. The next day,
compounds were added to the wells at a final concentration ranging
from 7.6 nM to 50 µM. The cells were treated with test compounds
for 72 h at 37°C. Subsequently, alamar blue was added at a 10-fold
dilution in complete growth medium. This assay evaluates the number
of viable cells per well based on fluorescent signal
measurements.
Cellular B-RAF, C-RAF, MEK1/2, ERK1/2,
caspase-3, poly(ADP-ribose) polymerase (PARP), and Akt
activation
Cells were plated at a density of 600,000 cells per
dish in 60×15 mm tissue culture dishes (Nunclon; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The following day, cells were
washed once with serum-free medium and treated with compounds in
MEM with 10% FBS for 2 h. Afterwards, cells were washed with cold
PBS containing 0.1 mM vanadate (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) to inhibit dephosphorylation and then lysed in
RIPA lysis buffer (Applygen Technologies, Inc., Beijing, China)
containing protease inhibitor cocktail tablets (Roche Diagnostics,
Basel, Switzerland). The lysates were centrifuged, and 90 µg of
soluble protein was separated by SDS-PAGE, transferred to PVDF
membranes (Hybond-C extra; Amersham Pharmacia Biotech, Piscataway,
NJ, USA) and blocked in TBS-BSA (Amresco LLC, Solon, OH, USA) (for
phosphorylated proteins) or TBS-milk. The membranes were probed
with anti-pB-RAF, anti-pC-RAF, anti-pMEK1/2, anti-pERK1/2,
anti-caspase-3, and anti-PARP primary antibodies (Cell Signaling
Technology, Inc.) and horseradish peroxidase (HRP)-conjugated
secondary antibodies (Santa Cruz Biotechnology, Inc.). The
membranes were then treated with an enhanced chemiluminescence
reagent (EMD Millipore, Billerica, MA, USA), and the protein
signals were detected by an EC3 imaging system (UVP EC3 Imaging
System, UVP Inc., Upland, CA, USA).
Cell cycle analysis
Flow cytometry was utilized to determine the cell
cycle distribution. Cells (5×105) were plated into
6-well plates, incubated with test compounds for 24 h, collected,
fixed in cold 70% ethanol overnight at −20°C, washed once with PBS,
and finally stained with propidium iodide (PI) solution (50 µg/ml
PI and 50 µg/ml RNase A in PBS) for 30 min in the dark. The samples
were then examined by flow cytometry.
Annexin V-fluorescein isothiocyanate
(FITC)/PI apoptosis assay
FITC-conjugated Annexin V (Annexin V-FITC)/PI double
staining was used to quantitatively examine the proportion of
apoptotic cells. Annexin V detects externalized phosphatidylserine,
a marker of early apoptosis, while PI binds to nuclear DNA and is
indicative of the loss of plasma membrane integrity associated with
late apoptosis (12,13). Early apoptotic cells are Annexin V
positive and PI negative (Annexin
V-FITC+/PI−), whereas late apoptotic cells
are Annexin V/PI double-positive (Annexin
V-FITC+/PI+) (14,15).
The total number of apoptotic cells is the sum of early and late
apoptotic cells (16–18). Cells were treated with tested
compounds for 48 h, harvested, washed twice with PBS and then
resuspended in 500 µl of binding buffer. A total of 5 µl of Annexin
V-FITC was added to the cells, which were then incubated at room
temperature for 10 min. Subsequently, 5 µl of PI was added, and the
cells were incubated in the dark for another 10 min before analysis
by flow cytometry.
Tumor xenograft experiments
Male NCr-nu/nu mice, aged 5 weeks, were purchased
from Vital River (Beijing, China), for the in vivo study.
The mice were housed and received water and food ad libitum.
Experiments using these mice were performed in accordance with
protocols approved by the Animal Ethical Committee of Xuanwu
Hospital. Tumors were generated by harvesting PLC/PRF/5 cells from
mid-log phase cultures using trypsin-EDTA (Invitrogen; Thermo
Fisher Scientific, Inc.). PLC/PRF/5 cells (5×106)
suspended in 50% Matrigel (BD Biosciences, Franklin Lakes, NJ, USA)
in serum-free medium were injected s.c. into the dorsal flank of
each mouse. Sorafenib tosylate and CBI-5725 were dissolved in
Cremophor EL/95% ethanol (50:50; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). When PLC/PRF/5 tumors reached 200 to 220
mm3, sorafenib tosylate was administered p.o. once daily
for 14 days at 8 or 20 mg/kg body weight. CBI-5725 was administered
p.o. once daily for 14 days at 2, 6, or 15 mg/kg body weight. The
control group received vehicle treatment. Tumors were measured
using a Vernier caliper, and tumor volume was determined by
measuring the longest (a) and shortest (b) diameters and was
calculated using the equation axb2xπ/6.
Statistical analysis
All experiments were conducted at least three
independent times. The data were examined by one-way ANOVA, and
individual groups were compared by Tukey's post hoc test.
Statistical significance was determined at a confidence interval of
at least 95%.
Results
CBI-5725 inhibits PLC/PRF/5 cell
proliferation
Alamar blue was used to measure the inhibitory
effect of CBI-5725 on PLC/PRF/5 cell growth. Increased cell death
and debris, decreased cell density, and cell shrinkage were
observed after 72 h of CBI-5725 treatment. As shown in Fig. 1, the inhibitory effect of CBI-5725
on PLC/PRF/5 cell growth occurred in a dose-dependent manner. The
IC50 of CBI-5725 in PLC/PRF/5 cells was 0.83±0.09 µM,
whereas the IC50 of sorafenib was 4.99±0.13 µM,
indicating that CBI-5725 is more cytotoxic than sorafenib.
CBI-5725 inhibits the RAF/MEK/ERK
signaling cascade in PLC/PRF/5 cells
RAF kinases are essential regulators of the MEK/ERK
cascade, which regulates multiple physiological functions required
for cell growth and survival (19). However, overactivation of this
pathway induces abnormal cell growth and malignant behavior in
human tumors. Abnormal tumor growth is usually induced by the
activation of ERK signaling via mutations in RAS and RAF (20,21).
Up-regulated RAF/MEK/ERK signaling plays a prominent role in HCC
(22). To compare the effects of
CBI-5725 and sorafenib on RAF/MEK/ERK signal transduction in
PLC/PRF/5 cells, Western blot analyses were performed to examine
alterations in the phosphorylation level of key proteins in this
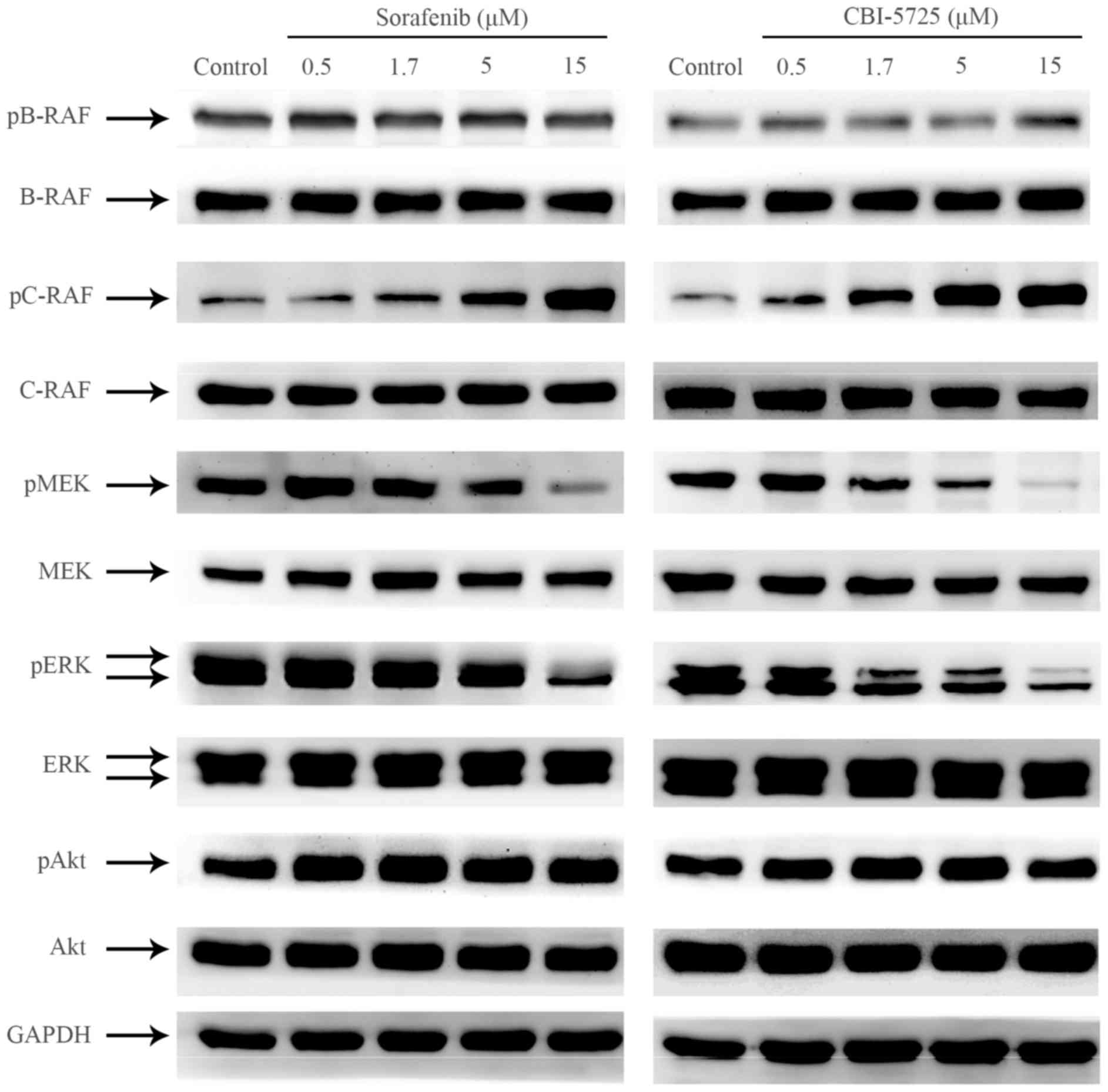
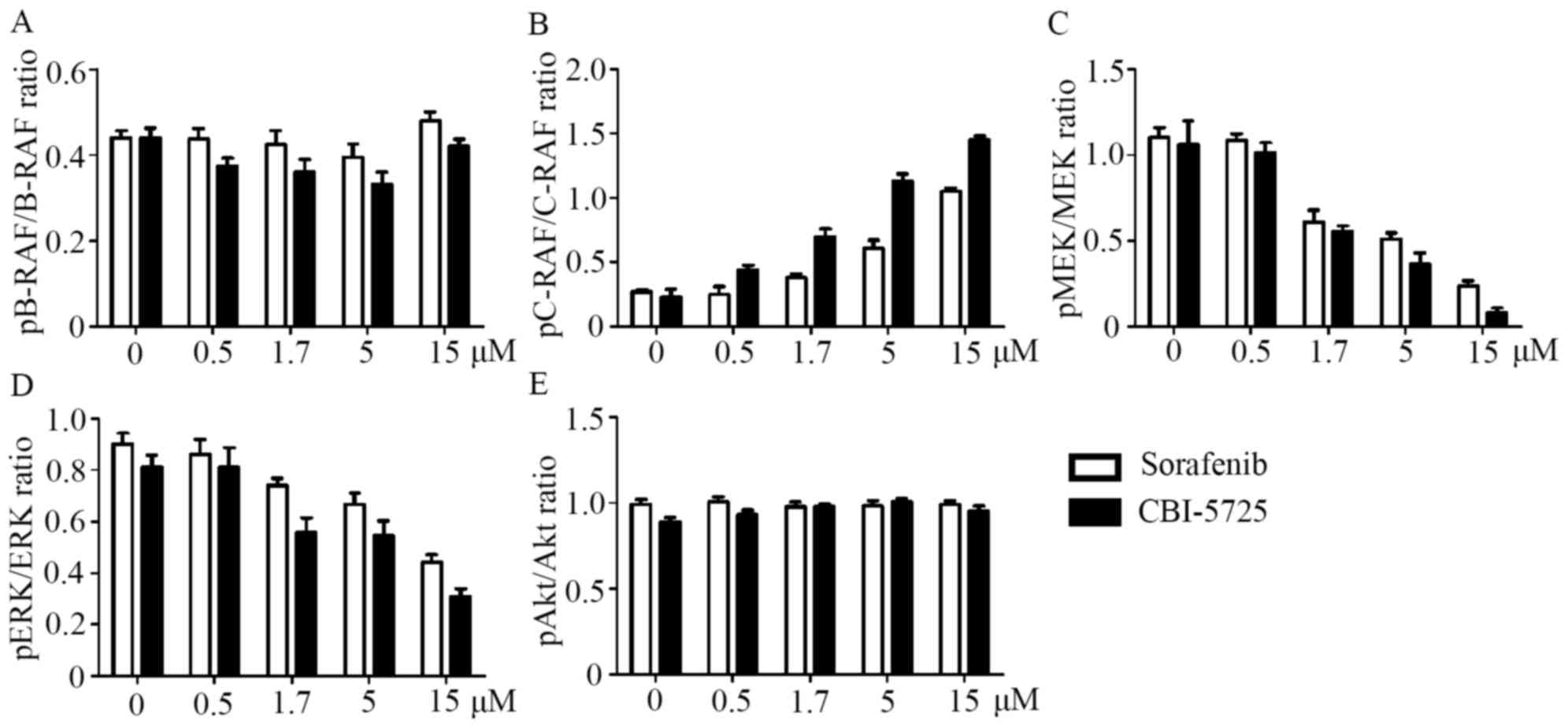
pathway. As shown in Figs. 2,
3A and 3B, neither CBI-5725 nor
sorafenib changed the level of phosphorylated B-RAF, but both drugs
markedly increased basal C-RAF phosphorylation in a dose-dependent
manner. Although both CBI-5725 and sorafenib paradoxically enhanced
C-RAF activation, they both suppressed the phosphorylation of MEK
and ERK in a dose-dependent manner (Figs. 2, 3C
and 3D), suggesting that both CBI-5725 and sorafenib are
pan-RAF inhibitors. The ratio of each phosphorylated protein to
total protein was examined by one-way ANOVA followed by Tukey's
post hoc test, and the data indicated that the effects of CBI-5725
and sorafenib on the RAF/MEK/ERK cascade were comparable (Fig. 3A-D). Total B-RAF, C-RAF, MEK, ERK
and Akt levels were unaltered, and no changes were detected in
phospho-Akt levels (Fig. 3E).
CBI-5725 modifies the cell cycle
In eukaryotic cells, the cell cycle is a highly
conserved mechanism by which replication occurs (23). Irreversible cell cycle arrest
serves as an alternative mechanism to prevent continued
proliferation in cancer (24). As
the above studies suggested that CBI-5725 prevents PLC/PRF/5 cell
proliferation, the effect of CBI-5725 on cell cycle progression was
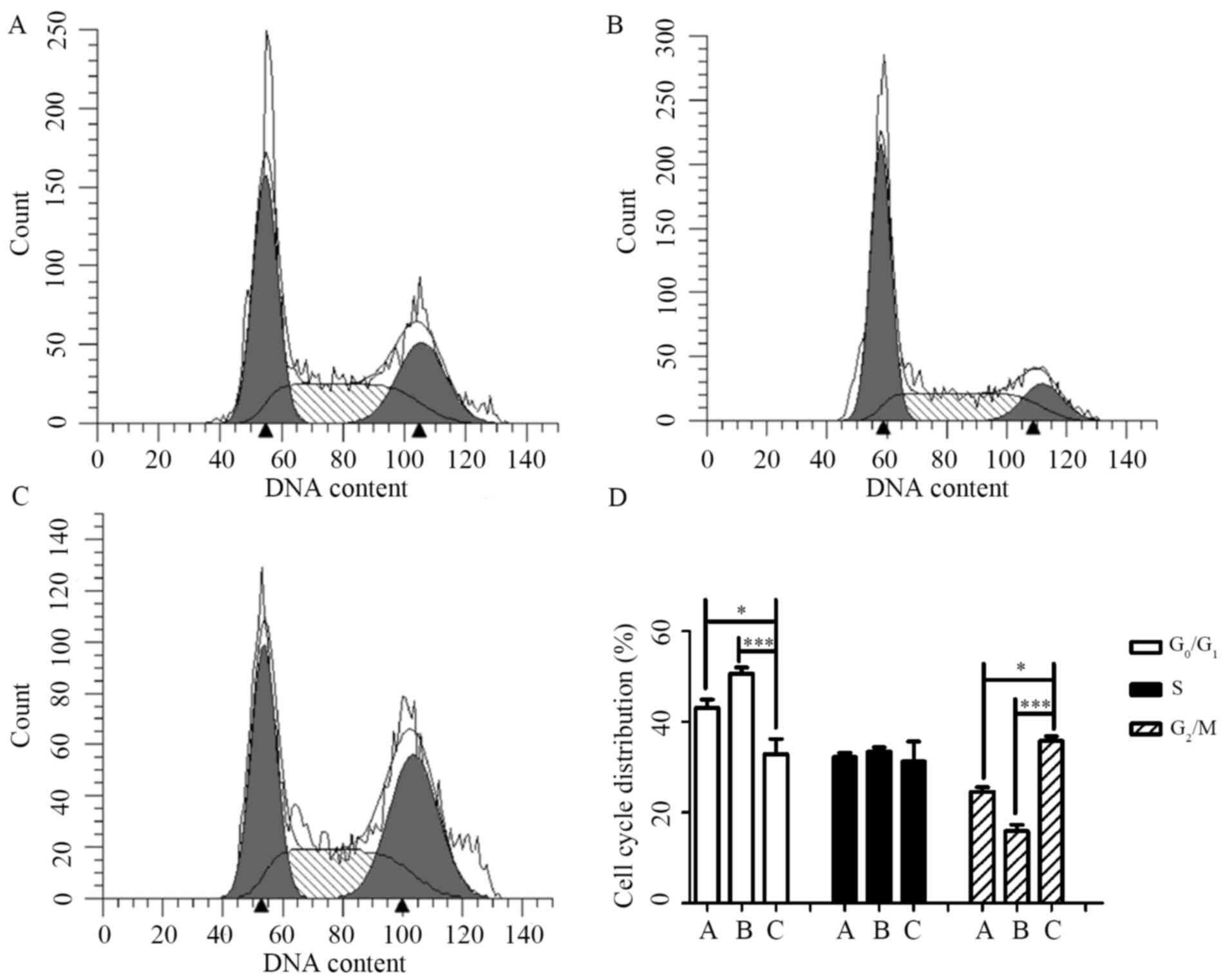
examined. As shown in Fig. 4,
CBI-5725 (15 µM; Fig. 4C)
significantly increased the proportion of cells in G2/M phase to
(35.77±1.08)%, whereas only (24.65±0.94)% and (15.92±1.39)% of
cells were in G2/M phase in the control (Fig. 4A) and sorafenib (15 µM; Fig. 4B) groups, respectively. The result
in Fig. 4D indicates that cells
treated for 24 h with CBI-5725 were arrested in G2/M phase. In
contrast, although sorafenib (15 µM) increased the percentage of
cells in G0/G1 phase, there was no significant difference in the
cell cycle distribution of sorafenib-treated cells and control
cells.
CBI-5725 induces the apoptosis of
PLC/PRF/5 cells
Because the cell cycle and apoptosis are intimately
linked, the effect of CBI-5725 and sorafenib on the apoptosis of
PLC/PRF/5 cells was examined using Annexin V-FITC and PI staining
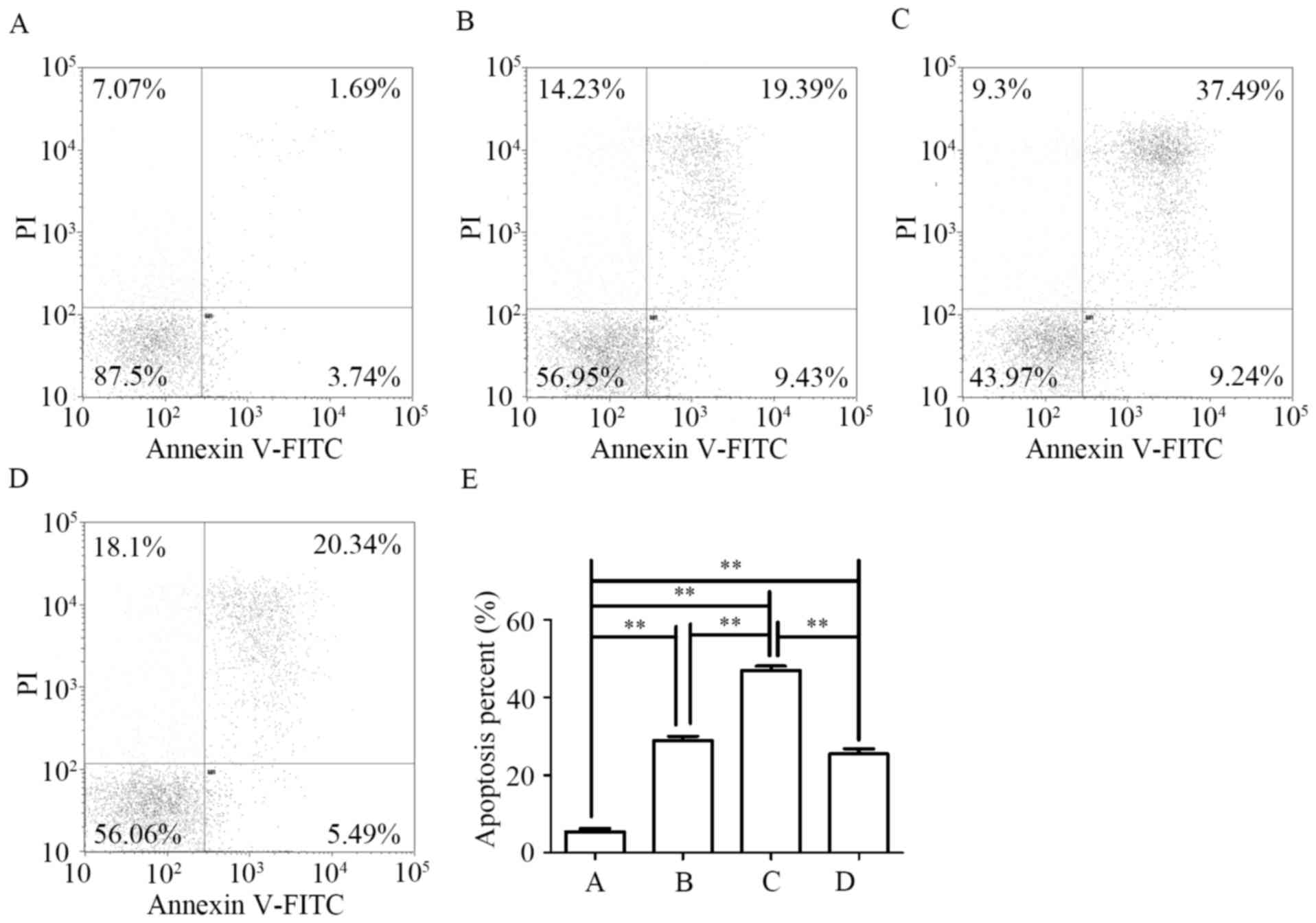
and flow cytometry. As shown in Fig.
5, each quadrant in the flow cytometry histogram represents a
specific cell population. The lower left quadrant exhibits live
cells (negative for Annexin V and PI), the lower right quadrant
shows early apoptotic cells, the upper right quadrant displays late
apoptotic cells, and the upper left quadrant exhibits dead
cells/debris (positive for PI only). When PLC/PRF/5 cells were
treated with CBI-5725 (5 or 15 µM) for 48 h, the proportion of
total apoptotic cells was 28.91±1.13 and 46.95±1.12%, respectively;
both percentages were higher than that for control treatment
(5.47±0.75%). These findings indicate that CBI-5725 induces the
apoptosis of PLC/PRF/5 cells in a dose-dependent manner. Fig. 5E shows that CBI-5725 (15 µM)
induced more apoptosis than sorafenib (15 µM) (46.95±1.12 vs.
25.50±1.34%, P<0.01).
Apoptosis induced by CBI-5725 in
PLC/PRF/5 cells relies on initiation of the caspase-3 signaling
pathway
Caspases, as critical effectors of apoptosis, are
divided into upstream initiator caspases and downstream executioner
caspases (25). There are two
common pathways by which caspases can be activated. Caspase-9 is
the upstream caspase for the intrinsic pathway, while caspase-8 has
this role in the extrinsic pathway (26). Both pathways converge on caspase-3,
a downstream executioner caspase. Once activated, pro-caspase-3 is
cleaved, giving rise to the active form of caspase-3. Active
caspase-3 cleaves PARP, ultimately leading to apoptosis (27). Cleaved PARP is among the most used
biomarkers of apoptosis (28).
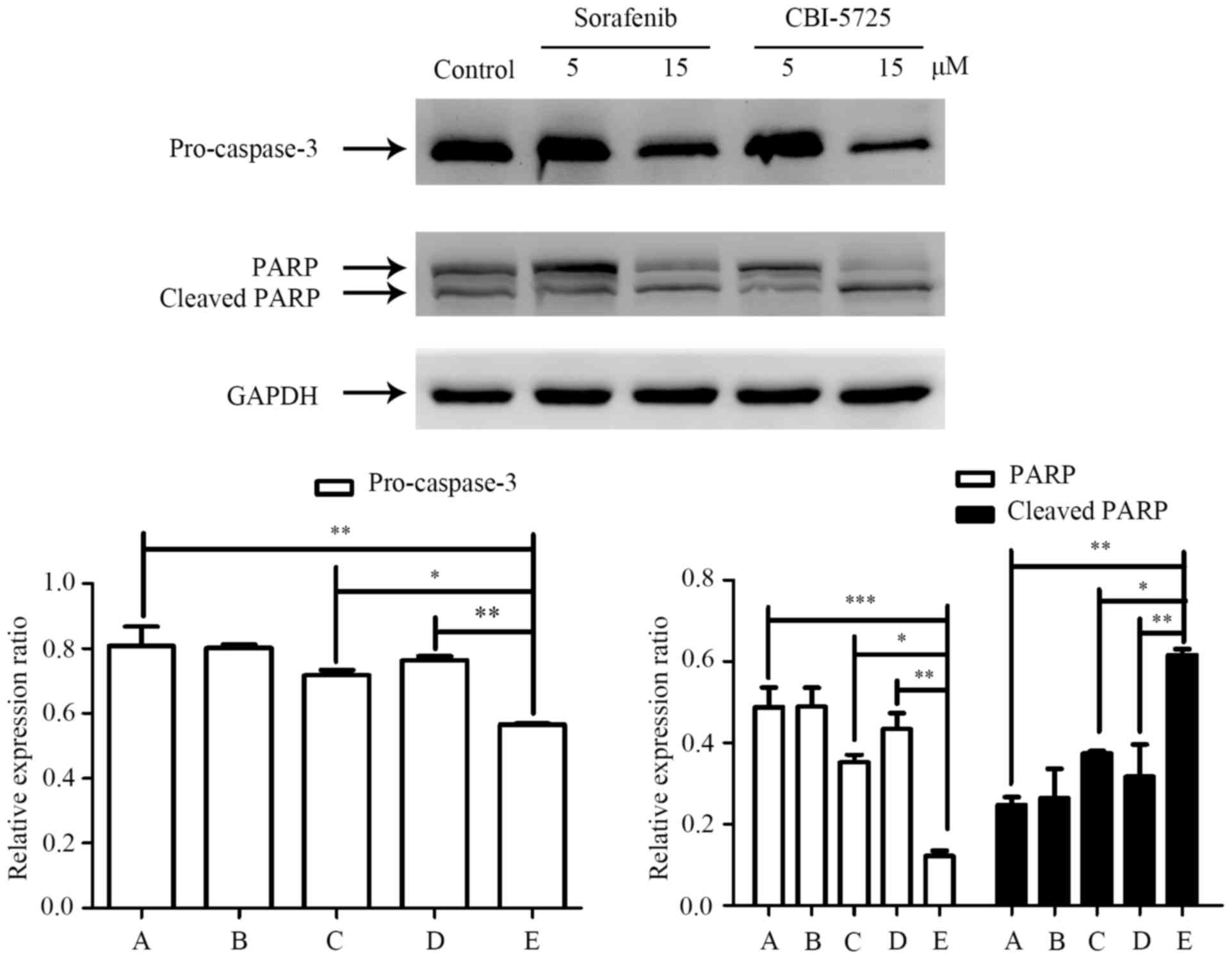
Changes in caspase-3 and PARP levels in PLC/PRF/5 cells after
treatment with CBI-5725 or sorafenib were measured by Western blot
analyses. As shown in Fig. 6, a
dose-dependent decrease in pro-caspase-3 and PARP levels and an
increase in cleaved PARP levels were observed in cells after 48 h
of CBI-5725 treatment. However, pro-caspase-3, PARP and cleaved
PARP levels were not distinctly altered in cells treated with
sorafenib. As shown in Fig. 6D and
E, pro-caspase-3 and PARP levels were much lower in cells
treated with 15 µM CBI-5725 than in control cells (0.566±0.004 vs.
0.807±0.061, P<0.01; 0.121±0.014 vs. 0.487±0.05, P<0.0001),
but cleaved PARP levels were much higher in CBI-5725-treated cells
than in control cells (0.615±0.015 vs. 0.247±0.019, P<0.01).
However, in cells treated with 15 µM sorafenib, there were no
significant changes in these levels (pro-caspase-3: 0.718±0.015 vs.
0.807±0.061; PARP: 0.352±0.018 vs. 0.487±0.05; cleaved PARP:
0.374±0.006 vs. 0.247±0.019). These data are consistent with the
results of the Annexin V-FITC/PI staining assay. The two main
apoptotic pathways both involve the activation of caspase-3. The
results indicate that the CBI-5725-induced apoptosis of PLC/PRF/5
cells occurs via the activation of caspase-3 and PARP.
CBI-5725 inhibits PLC/PRF/5 xenograft
tumor growth in vivo
To verify whether the effect of CBI-5725 on
PLC/PRF/5 cells has clinical relevance, we treated HCC xenografts
with CBI-5725 to assess its in vivo effect in comparison
with that of sorafenib. A single tumor was observed in each mouse
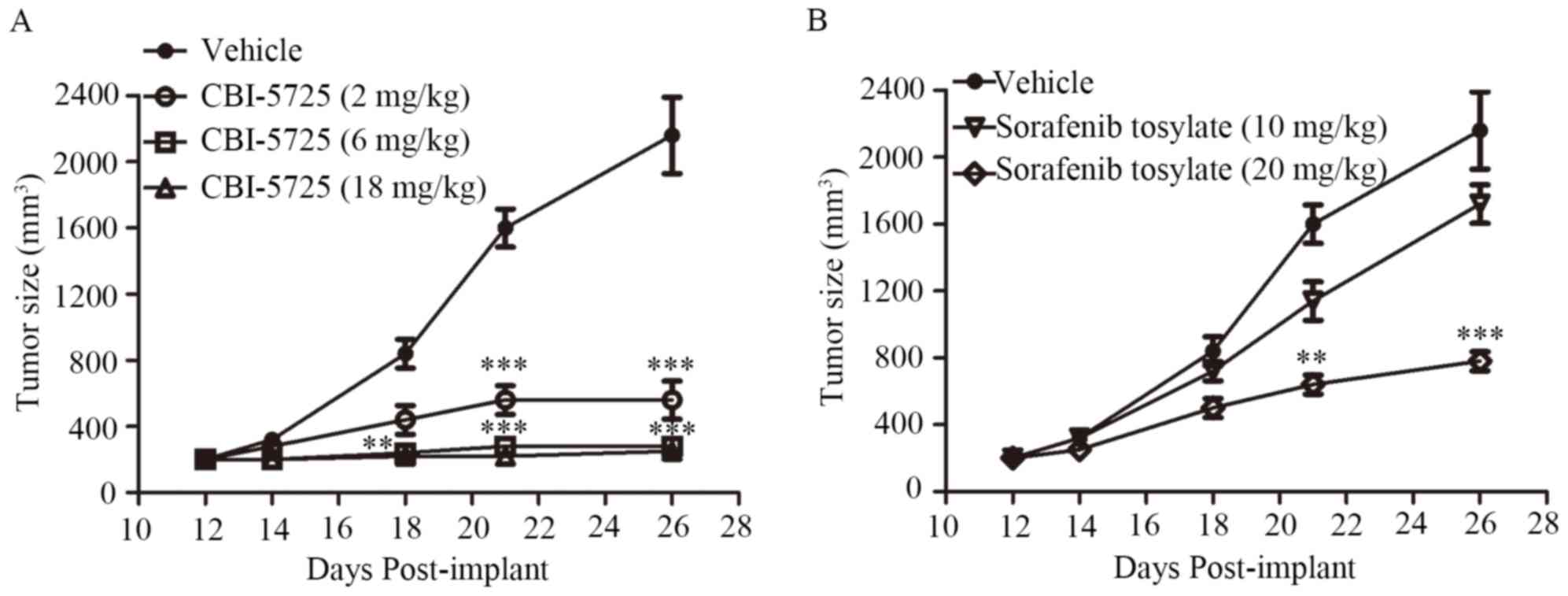
used in the experiments. As shown in Fig. 7A, CBI-5725 significantly inhibited
PLC/PRF/5 xenograft tumor growth. In comparison with the vehicle
group, in which the largest tumor diameter was 21 mm, 2 mg/kg
CBI-5725 inhibited tumor growth by approximately 73% at the end of
treatment (the largest tumor diameter was 13.4 mm), and 6 and 18
mg/kg CBI-5725 caused 89 and 92% tumor growth inhibition,
respectively (the largest tumor diameters were 10.7 and 10.3 mm,
respectively). Sorafenib tosylate inhibited PLC/PRF/5 tumor growth
in a dose-dependent manner. At 10 and 20 mg/kg, sorafenib inhibited
tumor growth by 19 and 64%, respectively (the largest tumor
diameters were 19.5 and 15 mm, respectively). There was no
increased weight loss in each treatment group relative to the
control group.
Discussion
Sorafenib is a multikinase inhibitor that acts in
opposition to the Ser/Thr kinase RAF and to a number of receptor
tyrosine kinases (RTKs), such as VEGFR2 and PDGFR, which are
essential for tumor cell proliferation and angiogenesis. Moreover,
sorafenib induces the apoptosis of PLC/PRF/5 cells (3). Although sorafenib is the only
approved targeted drug for HCC patients, increased sorafenib
resistance in HCC reduces its efficacy (29). Since the advent of sorafenib, many
trials for novel treatments for advanced HCC, primarily focused on
molecular targeted therapy, have been conducted; however, with the
exception of regorafenib, the results to date have been
disappointing (11). The
difficulty in advancing a new drug or treatment method can be
attributed to the diverse mechanisms of HCC carcinogenesis and
progression, as well as to the existence of background liver
diseases, such as chronic hepatitis and cirrhosis. Despite these
failures, novel pharmaceutical drugs are still urgently needed. In
this study, CBI-5725 was shown to more potently prevent the
proliferation and induce the apoptosis of a human HCC cell line
compared to sorafenib. CBI-5725 blocked the RAF/MEK/ERK pathway,
inhibited the cell cycle at G2/M phase and induced apoptosis
dependent on caspase 3/PARP activation. In addition, CBI-5725
exerted robust antitumor activity by inhibiting the growth of
PLC/PRF/5 xenografts. Therefore, CBI-5725 is a potential
therapeutic alternative to sorafenib for the treatment of HCC.
MAPKs are key signaling proteins that regulate
normal cell proliferation, survival and differentiation (30). Dysregulation of MAPK cascades plays
an important role in HCC occurrence and development (31). The MAPK pathway comprises a cascade
of phosphorylation events initiated from activated RAF proteins to
MEK and MEK to ERK (32). RAF
activity is strongly associated with cancer, and a diverse series
of ATP-competitive RAF inhibitors has been developed over the past
decade (33). Some of these
first-generation RAF inhibitors, such as the BRAF inhibitors
vemurafenib and dabrafenib, have markedly inhibited
BRAFV600E-dependent melanomas due to inhibition of the monomeric
form of this specific BRAF-mutant protein (34–36).
However, in wild-type BRAF tumors bearing activating RAS mutations
or increased RTK signaling, BRAF inhibitors were found to
paradoxically induce RAF activation by RAF dimerization, leading to
downstream ERK signaling and therefore enhancing tumor cell
proliferation (7,37–39).
To circumvent the limitation of first-generation RAF inhibitors, a
broad set of pan-RAF inhibitors were identified, including
sorafenib, LY3009120, TAK632, CCT196969 and CCT241161 (40–43).
Pan-RAF inhibitors such as sorafenib can inhibit BRAF or CRAF with
high affinity (44). Though
pan-RAF inhibitors induce BRAF-CRAF dimerization, they ultimately
inhibit the phosphorylation of downstream MEK and ERK, confirming
their efficacy in inhibiting the kinase activity of BRAF-CRAF
heterodimers (41). PLC/PRF/5
cells contain an activating mutation in the KRAS gene that
may drive their aberrant proliferation with a greater dependence on
signaling through the MAPK pathway for survival (4). The data in the present study
demonstrate that the response of the MAPK cascade to CBI-5725
resembles its response to sorafenib, suggesting that CBI-5725 may
also be a pan-RAF inhibitor. CBI-5725 can block MEK/ERK signaling
in PLC/PRF/5 cells harboring mutant K-RAS and wild-type BRAF.
Furthermore, cell cycle analysis by flow cytometry
revealed a significantly greater accumulation of cells in G2/M
phase after treatment with 15 µM CBI-5725 than after control
treatment, indicating that CBI-5725 induces cell cycle arrest in
G2/M phase. Since only approximately 35.77% of tumor cells
accumulated in G2/M phase, CBI-5725 did not elicit an irreversible
cell cycle arrest but rather slowed the progression through this
phase of the cell cycle, which contributed to the inhibition of
cell proliferation (45). By
contrast, the cell cycle distribution was nearly unaffected by 15
µM sorafenib, which complied with the result of a previous study
(3) that the cell cycle
distribution of PLC/PRF/5 cells was not affected by 24 h of
treatment with sorafenib at 3, 10, or 15 µM. G2/M phase arrest
relates to the down-regulation of cdc2, cdc25c and cyclin B levels,
as well as the up-regulation of p21 and p-cdc2 levels (46). Consequently, these regulators may
be associated with the inhibition of the G2/M transition by
CBI-5725.
Apoptosis, a tightly programmed cell death process,
plays a critical role not only in the growth and homeostasis of
normal tissues but also in the treatment of cancer, as it is a
target of many therapeutic approaches. In this study, the
CBI-5725-induced apoptosis of PLC/PRF/5 cells was examined by an
Annexin V-PI assay, and a remarkable dose-dependent effect was
detected. Moreover, CBI-5725 was shown to induce apoptosis more
potently than sorafenib. The mechanism of the action of CBI-5725 on
the apoptotic pathway was explored. Caspases are fundamental in
apoptosis pathways because they are both initiators and
executioners. Caspases can be activated through two common
pathways. The intrinsic and extrinsic pathways converge on
caspase-3, a downstream executioner caspase. Once activated,
pro-caspase-3 is cleaved to produce the active form of caspase-3,
which is primarily responsible for PARP cleavage during cell death
(47–49). PARP is an active participant in
pivotal biological processes such as transcription and cell cycle
modulation, the DNA damage response, apoptosis and genome integrity
maintenance (50). Cleavage of
PARP promotes cell destruction and functions as a marker of
apoptotic cells (51). After
treatment with CBI-5725, the levels of pro-caspase-3 and PARP
decreased, while cleaved PARP levels increased, indicating that
apoptosis induced by CBI-5725 in PLC/PRF/5 cells proceeded through
the caspase-dependent pathway. However, sorafenib did not
remarkably alter pro-caspase-3, PARP and cleaved PARP levels,
suggesting that sorafenib-induced apoptosis might not rely on
caspase activation in PLC/PRF/5 cells. This result is consistent
with previous findings (3).
In the PLC/PRF/5 tumor xenograft experiments,
CBI-5725 robustly prevented tumor growth. CBI-5725 at doses from 6
to 18 mg/kg produced nearly complete tumor growth inhibition after
14 days of oral administration. On the other hand, complete tumor
growth inhibition was not achieved with sorafenib tosylate at doses
up to 20 mg/kg. The above results obtained from the in vitro
experiments imply that blockage of the RAF/MEK/ERK pathway,
induction of cell cycle arrest and initiation of apoptosis may lead
to the tumor growth inhibition observed in PLC/PRF/5 tumor
xenografts treated with CBI-5725.
The findings described herein are promising and
should be verified in other HCC cell lines, such as HepG2 cells.
The mechanisms underlying the superior antitumor efficacy of
CBI-5725 remain to be determined.
In conclusion, this study demonstrates that CBI-5725
strongly inhibits HCC cell proliferation in vitro by
blocking the RAF/MEK/ERK pathway to the same extent as sorafenib,
elicits G2/M cell cycle arrest and induces tumor cell apoptosis
more potently than sorafenib. These findings may contribute to the
remarkable antitumor efficacy of CBI-5725 against a human HCC
xenograft model. Therefore, CBI-5725 may be an alternative to
sorafenib for the treatment of liver cancer patients.
Acknowledgements
We greatly appreciate the technical support and
discussions provided by previous members of the lab.
Funding
The present study was supported by the NSFC (grant
no. 81503158) and The Beijing Municipal Administration of
Hospitals' Youth Program (grant no. QML20160808).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
WW conceived, planned and carried out the
experiments, and analyzed and interpreted the data. BX performed
the flow cytometry to examine the cell cycle distribution and
apoptosis. WW wrote the manuscript with support from QXL, DCJ and
SYY. QXL contributed to study design, DCJ assisted with data
interpretation, and SYY provided critical revision and helped shape
the research, analysis and manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Experiments using mice were performed in accordance
with protocols approved by the Animal Ethical Committee of Xuanwu
Hospital (Beijing, China).
Consent for publication
Not applicable.
Competing interests
The present study included the use of the compound
CBI-5725; the patent for which is owned by Crown Bioscience, Inc.
(Beijing, China). The author, Dr Qixiang Li, is affiliated with
this company.
References
|
1
|
Lamarca A, Mendiola M and Barriuso J:
Hepatocellular carcinoma: Exploring the impact of ethnicity on
molecular biology. Crit Rev Oncol Hematol. 105:65–72. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swamy SG, Kameshwar VH, Shubha PB, Looi
CY, Shanmugam MK, Arfuso F, Dharmarajan A, Sethi G, Shivananju NS
and Bishayee A: Targeting multiple oncogenic pathways for the
treatment of hepatocellular carcinoma. Target Oncol. 12:1–10. 2016.
View Article : Google Scholar
|
|
3
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Azumi J, Tsubota T, Sakabe T and Shiota G:
miR-181a induces sorafenib resistance of hepatocellular carinoma
cells through downregulation of RASSF1 expression. Cancer Sci.
107:1256–1262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ezzoukhry Z, Louandre C, Trécherel E,
Godin C, Chauffert B, Dupont S, Diouf M, Barbare JC, Mazière JC and
Galmiche A: EGFR activation is a potential determinant of primary
resistance of hepatocellular carcinoma cells to sorafenib. Int J
Cancer. 131:2961–2969. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Poulikakos PI, Zhang C, Bollag G, Shokat
KM and Rosen N: RAF inhibitors transactivate RAF dimers and ERK
signalling in cells with wild-type BRAF. Nature. 464:427–430. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lackner MR, Wilson TR and Settleman J:
Mechanisms of acquired resistance to targeted cancer therapies.
Future Oncol. 8:999–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bagrodia S, Smeal T and Abraham RT:
Mechanisms of intrinsic and acquired resistance to kinase-targeted
therapies. Pigment Cell Melanoma Res. 25:819–831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bottsford-Miller JN, Coleman RL and Sood
AK: Resistance and escape from antiangiogenesis therapy: Clinical
implications and future strategies. J Clin Oncol. 30:4026–4034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moriguchi M, Umemura A and Itoh Y: Current
status and future prospects of chemotherapy for advanced
hepatocellular carcinoma. Clin J Gastroenterol. 9:184–190. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pillet S and von Messling V: Canine
distemper virus selectively inhibits apoptosis progression in
infected immune cells. J Virol. 83:6279–6287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thomas E, Gopalakrishnan V, Somasagara RR,
Choudhary B and Raghavan SC: Extract of Vernonia condensata,
inhibits tumor progression and improves survival of tumor-allograft
bearing mouse. Sci Rep. 6:232552016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brauchle E, Thude S, Brucker SY and
Schenke-Layland K: Cell death stages in single apoptotic and
necrotic cells monitored by Raman microspectroscopy. Sci Rep.
4:46982014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wlodkowic D, Telford W, Skommer J and
Darzynkiewicz Z: Apoptosis and beyond: Cytometry in studies of
programmed cell death. Methods Cell Biol. 103:55–98. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuo WT, Ho YJ, Kuo SM, Lin FH, Tsai FJ,
Chen YS, Dong GC and Yao CH: Induction of the mitochondria
apoptosis pathway by phytohemagglutinin erythroagglutinating in
human lung cancer cells. Ann Surg Oncol. 18:848–856. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lam CR, Tan MJ, Tan SH, Tang MB, Cheung PC
and Tan NS: TAK1 regulates SCF expression to modulate PKBα activity
that protects keratinocytes from ROS-induced apoptosis. Cell Death
Differ. 18:1120–1129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang LJ, Chen Y, He J, Yi S, Wen L, Zhao
J, Zhang BP and Cui GH: Betulinic acid inhibits autophagic flux and
induces apoptosis in human multiple myeloma cells in vitro. Acta
Pharmacol Sin. 33:1–1548. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buscà R, Pouysségur J and Lenormand P:
ERK1 and ERK2 map kinases: Specific roles or functional redundancy.
Front Cell Dev Biol. 4:532016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoshino R, Chatani Y, Yamori T, Tsuruo T,
Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J and Kohno
M: Constitutive activation of the 41-/43-kDa mitogen-activated
protein kinase signaling pathway in human tumors. Oncogene.
18:813–822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Q, Wei L, Yang H, Yang W, Yang Q,
Zhang Z, Wu K and Wu J: Bromodomain containing protein represses
the Ras/Raf/MEK/ERK pathway to attenuate human hepatoma cell
proliferation during HCV infection. Cancer Lett. 371:107–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pucci B, Kasten M and Giordano A: Cell
cycle and apoptosis. Neoplasia. 2:291–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A,
Wu Q, Zhang J and Hong Y: Caspases: A molecular switch node in the
crosstalk between autophagy and apoptosis. Int J Biol Sci.
10:1072–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duriez PJ and Shah GM: Cleavage of
poly(ADP-ribose) polymerase: A sensitive parameter to study cell
death. Biochem Cell Biol. 75:337–349. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prieto-Domínguez N, Ordóñez R, Fernández
A, García-Palomo A, Muntané J, González-Gallego J and Mauriz JL:
Modulation of autophagy by sorafenib: Effects on treatment
response. Front Pharmacol. 7:1512016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Zhao GD, Shi Z, Qi LL, Zhou LY and
Fu ZX: The Ras/Raf/MEK/ERK signaling pathway and its role in the
occurrence and development of HCC. Oncol Lett. 12:3045–3050. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jin T, Lavoie H, Sahmi M, David M, Hilt C,
Hammell A and Therrien M: RAF inhibitors promote RAS-RAF
interaction by allosterically disrupting RAF autoinhibition. Nat
Commun. 8:12112017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holderfield M, Deuker MM, McCormick F and
McMahon M: Targeting RAF kinases for cancer therapy: BRAF-mutated
melanoma and beyond. Nat Rev Cancer. 14:455–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li
Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI and Rosen
N: BRAF mutants evade ERK-dependent feedback by different
mechanisms that determine their sensitivity to pharmacologic
inhibition. Cancer Cell. 28:370–383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hatzivassiliou G, Song K, Yen I,
Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor
SL, Vigers G, et al: RAF inhibitors prime wild-type RAF to activate
the MAPK pathway and enhance growth. Nature. 464:431–435. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Heidorn SJ, Milagre C, Whittaker S, Nourry
A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer
CJ, Pritchard C and Marais R: Kinase-dead BRAF and oncogenic RAS
cooperate to drive tumor progression through CRAF. Cell.
140:209–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cox AD and Der CJ: The raf inhibitor
paradox: Unexpected consequences of targeted drugs. Cancer Cell.
17:221–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Peng SB, Henry JR, Kaufman MD, Lu WP,
Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al:
Inhibition of RAF isoforms and active dimers by LY3009120 leads to
anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell.
28:384–398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nakamura A, Arita T, Tsuchiya S, Donelan
J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T and
Yoshida S: Antitumor activity of the selective pan-RAF inhibitor
TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res.
73:7043–7055. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Girotti MR, Lopes F, Preece N,
Niculescu-Duvaz D, Zambon A, Davies L, Whittaker S, Saturno G,
Viros A, Pedersen M, et al: Paradox-breaking RAF inhibitors that
also target SRC are effective in drug-resistant braf mutant
melanoma. Cancer Cell. 31:4662017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Knievel J, Schulz WA, Greife A, Hader C,
Lübke T, Schmitz I, Albers P and Niegisch G: Multiple mechanisms
mediate resistance to sorafenib in urothelial cancer. Int J Mol
Sci. 15:20500–20517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Weber GF, Gaertner FC, Erl W, Janssen KP,
Blechert B, Holzmann B, Weighardt H and Essler M: IL-22-mediated
tumor growth reduction correlates with inhibition of ERK1/2 and AKT
phosphorylation and induction of cell cycle arrest in the G2-M
phase. J Immunol. 177:8266–8272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera
S and Ikejima T: Oridonin induces G2/M arrest and apoptosis via
activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK
survival pathway in murine fibrosarcoma L929 cells. Arch Biochem
Biophys. 490:70–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tewari M, Quan LT, O'Rourke K, Desnoyers
S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS and Dixit VM:
Yama/CPP32 beta, a mammalian homolog of CED-3, is a
CrmA-inhibitable protease that cleaves the death substrate
poly(ADP-ribose) polymerase. Cell. 81:801–809. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Le RY, Kirkland JB and Shah GM: Cellular
responses to DNA damage in the absence of Poly(ADP-ribose)
polymerase. Biochem Biophys Res Commun. 245:1–10. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vizetto-Duarte C, Custodio L, Gangadhar
KN, Lago JH, Dias C, Matos AM, Neng N, Nogueira JM, Barreira L,
Albericio F, et al: Isololiolide, a carotenoid metabolite isolated
from the brown alga Cystoseira tamariscifolia, is cytotoxic and
able to induce apoptosis in hepatocarcinoma cells through caspase-3
activation, decreased Bcl-2 levels, increased p53 expression and
PARP cleavage. Phytomedicine. 23:550–557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33955. 1998.
View Article : Google Scholar : PubMed/NCBI
|