Introduction
Chronic kidney disease (CKD) is a global healthcare
concern that causes significant morbidity and mortality in the
human population (1). CKD
patients, even with similar etiologies, frequently exhibit
different susceptibility and severity of renal fibrosis, leading to
different clinical outcomes (2).
Epithelial-mesenchymal transition (EMT) contributes to the
pathogenesis of renal fibrosis, which is characterized by the loss
of intercellular contacts caused by downregulation of E-cadherin,
de novo expression of α-smooth muscle actin (αSMA) and
accumulation of collagen (3).
Pro-fibrotic growth factors, especially transforming growth factor
β1 (TGF-β1) is considered to be the most important factor
contributing to EMT during the fibrogenic phase of fibrosis
(4,5). Regulation of EMT may be a promising
target for the prevention of the progression of renal fibrosis.
MicroRNAs (miRNAs or miRs) are non-coding,
single-stranded RNA molecules that can regulate target mRNAs
predominantly by binding to the 3′ untranslated region (UTR) at the
post-transcriptional level (6).
Aberrant expression of miRNAs is associated with the initiation and
progression of several pathological processes, including autoimmune
diseases, cancer and cardiovascular disease (7–9). In
addition, previous studies have revealed a role for miRNAs in
kidney injury and repair, providing novel insights into the
mechanism underlying EMT in renal fibrosis (10). It has been demonstrated that
miR-205 and other members of the miR-200 family can inhibit the
TGF-β-induced EMT by downregulating zinc finger E-box-binding
homeobox 1 and 2, two major transcriptional repressors of
E-cadherin (11,12). Chen et al (13) suggested that miR-328-mediated
transient upregulation of CD44 triggers pressure-induced EMT in
renal fibrosis. Expression of miR-192 was upregulated by
stimulation of mouse mesangial cells with TGF-β1, and miR-192 is
responsible for increased collagen II in diabetic kidney glomeruli
(14). The above results suggest
important roles of miRNAs in renal fibrosis and EMT of tubular
epithelial cells. A recent study demonstrated that TGF-β1 inhibits
the expression of miR-152/30a, therfore enhancing DNA
methyltransferase 1/3a and contributing to the promotion of
pro-fibrotic protein expression and renal fibrosis (15). However, the association between
miR-152 and TGF-β1-induced tubular epithelial cell EMT remains to
be elucidated.
Based on the results of the previous studies, the
present study investigated the expression of miR-152 in the
TGF-β1-treated tubular epithelial HK-2 cell line and examined
whether miR-152 modification could ameliorate TGF-β1-induced EMT.
The results of the present study demonstrated that miR-152
expression is significantly reduced in HK-2 cells following
stimulation with TGF-β1 and that enhanced expression of miR-152
prevents EMT induced by TGF-β1, possibly via negative regulation of
hematopoietic pre-B-cell leukemia transcription factor
(PBX)-interacting protein (HPIP). The results of the present study
further support the role of miR-152 in TGF-β1-induced EMT,
suggesting that it may be an effective therapeutic target for the
treatment of renal fibrosis.
Materials and methods
Cell culture and treatment
The human kidney proximal tubule cell line (HK-2)
was obtained from the American Type Culture Collection (Manassas,
VA, USA) and maintained in keratinocyte serum-free medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cells were incubated at 37°C in a 5% CO2 atmosphere.
TGF-β1 was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany) and used at a working concentration of 10 ng/ml, as
previously described (16–18). miR-152 mimic and control RNA mimic
(miR-NC) were obtained from GeneCopoeia Inc. (Rockville, MD, USA).
Cells were plated on 6-well plates and grown to 60% confluency and
then transfected with 35 nM miR-152 mimic or miR-NC using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. A rescue experiment
was performed by HPIP overexpression using the HPIP ORF expression
clone (GeneCopoecia, Inc.) and pcDNA3.1 empty vector was used as a
negative control.
Plasmid construction and 3′ UTR target
assay
Potential targets of miR-152 were predicted using
miRbase (www.mirbase.org), miTarget
(cbit.snu.ac.kr/~miTarget), and TargetScanS (www.targetscan.org/vert_71), and luciferase assay was
performed to determine whether miR-152 targeted the 3′-UTR of HPIP.
Luciferase reporter vectors were constructed using the 3′ UTR of
the HPIP gene, which was amplified in a polymerase chain reaction
(PCR), using the following primers: Forward,
5′-CTGAGCACGTCGCAATCTCTACTCACCAGA-3′ and reverse,
5′-GATAACGTCTTGAGCGATCTCTGTATCCTT-3′; extracted from the HK-2 cell
genomic DNA and inserted into the luciferase coding region in the
psiCHECK™-2 vector (Promega Corporation, Madison, WI, USA). A
mutant vector with the HPIP 3′UTR was identical to the wild-type
sequences, apart from the seed region, which was generated using
the QuikChange™ Site-Directed Mutagenesis kit (Biocompare Inc.,
South San Francisco, CA, USA) and served as a negative control. The
primers used to amplify the mutant HPIP 3′-UTR were as follows:
Forward, 5′-GAGTTCCGCATGCACCCTATACTCAGACAC-3′ and reverse,
5′-GCAGTTAACTGTTCGTCAGACTCGTATTCT-3′. Renilla luciferase,
encoded by the vector, served as an internal control. HK-2 cells
were seeded in 6-well plates overnight prior to transfection. The
following day, each luciferase reporter construct, including the
miR-152 mimic or miR-NC, was co-transfected into HK-2 cells using
Lipofectamine 2000. Following 24 h incubation, cells were
collected, and firefly and Renilla luciferase activities
were determined using a dual-luciferase reporter assay system
(Promega Corporation). All experiments were performed in triplicate
and repeated three times.
RNA extraction and reverse
transcription quantitative PCR (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from cultured
cells, according to the manufacturer's protocol. For mRNA
detection, cDNA synthesis was performed using the PrimeScript
RT-PCR kit (Takara Bio, Inc., Otsu, Japan) in a reaction system of
20 µl, at 16°C (30 min), 45°C (30 min), and 85°C (5 min). RT-qPCR
was performed using GoTaq qPCR Master Mix (Promega Corporation)
using the ABI PRISM 7500 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. GAPDH served as an internal control. The following
thermocycling conditions were used for the PCR: Initial
denaturation at 94°C for 5 min; 40 cycles of 95°C for 15 sec, 65°C
for 30 sec and 72°C for 30 sec; and a final extension at 72°C for 5
min. Expression of mature miR-152 was determined with the
Bulge-Loop™ miRNA qRT-PCR Primer Set (Guanghzou RiboBio Co., Ltd.,
Guanghzou, China). U6 RNA was used as an internal control. All
sequences used are presented in Table
I. The fold-change in gene expression was analyzed using the
2−ΔΔCq method (19).
Each sample was detected in triplicate.
 | Table I.Primer sequences for reverse
transcription quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription quantitative polymerase chain reaction.
| Target | Forward primer
5′-3′ | Reverse primer
5′-3′ |
|---|
| Has-miR-152 |
GTCGTATCCAGTGCGTGTCGTGGA |
GTCGGCAATTGCACTGGATACGACAGTCGG |
| α-SMA |
GCGCAGGTTCTGTGATACACT |
TGGTGTCGTGGAGTCG |
| E-cadherin |
GTGTTGCCCCTGAAGAGCAT |
GGGTGTCGAGGGAAAAATAGG |
| Collagen I |
GCTCCTCTTAGGGGCCACT |
CCACGTCTCACCATTGGGG |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| GAPDH |
CTGGGCTACACTGAGCACC |
AAGTGGTCGTTGAGGGCAATG |
Western blotting
Cells were lysed using cold radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China) and the protein concentrations were determined using the
Bicinchoninic Acid protein assay kit (Beyotime Institute of
Biotechnology). Subsequently, 30 µg protein was separated by 10–12%
sodium dodecyl sulfate polyacrylamide gels, transferred onto
polyvinylidene fluoride membranes. After blocked with 5% non-fat
milk powder at room temperature for 1 h, membranes were probed with
primary antibodies: anti-αSMA (1:5,000, cat no. EPR5308),
anti-E-cadherin (1:1,000, cat no. BS1098) (both from Bioworld
Technology, Inc., St. Louis Park, MN, USA), anti-collagen I
(1:2,000, cat no. ab34710), anti-HPIP (1:500, cat no. ab197260) and
anti-GAPDH (1:1,000, ab8245) (all from Abcam, Cambridge, MA, USA),
overnight at 4°C. The membranes were then incubated with goat
anti-rabbit IgG H&L antibodies (1:5,000, cat no. ab6721; Abcam)
for 2 h at room temperature. Results were visualized using the
enhanced chemiluminescence detection reagent (Beyotime Institute of
Biotechnology) and quantified by ImageJ software (version 6.0; the
National Institute of Health, Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
SPSS (version 20.0; IBM Corp., Armonk, NY, USA) was used to perform
statistical analyses using a two-tailed Student's t-test or one-way
analysis of variance followed by least significant difference test,
where appropriate. GraphPad Prism (version 6.0; GraphPad Software
Inc., La Jolla, CA, USA) was used to generate all graphs. All
experiments were repeated at least three times. P<0.05 was
considered to indicate a statistically significant difference.
Results
TGF-β1 induces downregulation of
miR-152 and initiation of EMT in HK-2 cells
The expression profile of miR-152, as well as
EMT-associated genes in TGF-β1-treated HK-2 cells was investigated.
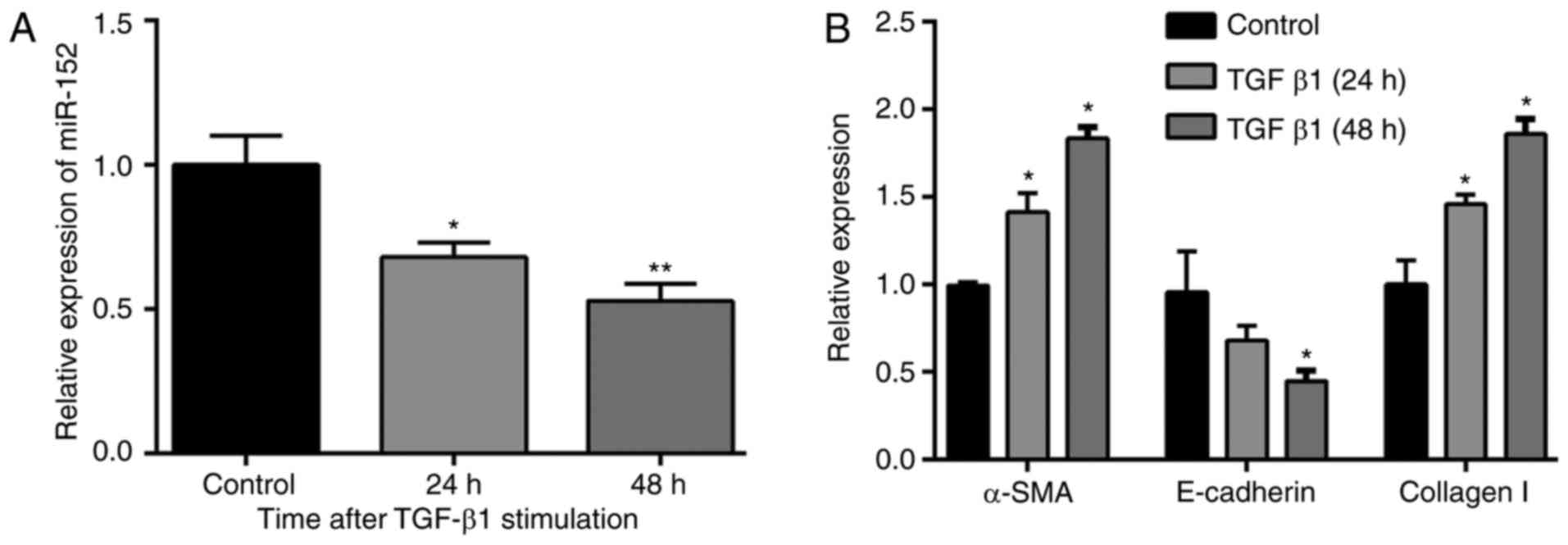
The results of RT-qPCR (Fig. 1A)
demonstrated that the expression of miR-152 was significantly
down-regulated in HK-2 cells following stimulation with TGF-β1 (10
ng/ml) for 24 and 48 h, compared with the control group (P<0.05
and P<0.01, respectively). As expected, the expression of the
epithelial marker E-cadherin was decreased, whereas mesenchymal
markers, including αSMA and collagen I were upregulated in HK-2
cells treated with TGF-β1 (10 ng/ml; Fig. 1B), especially for 48 h. Therefore,
HK-2 cells treated with 10 ng/ml TGF-β1 at 48 hwere used in the
following experiments.
miR-152 regulates the EMT in TGF-β1
treated HK-2 cells
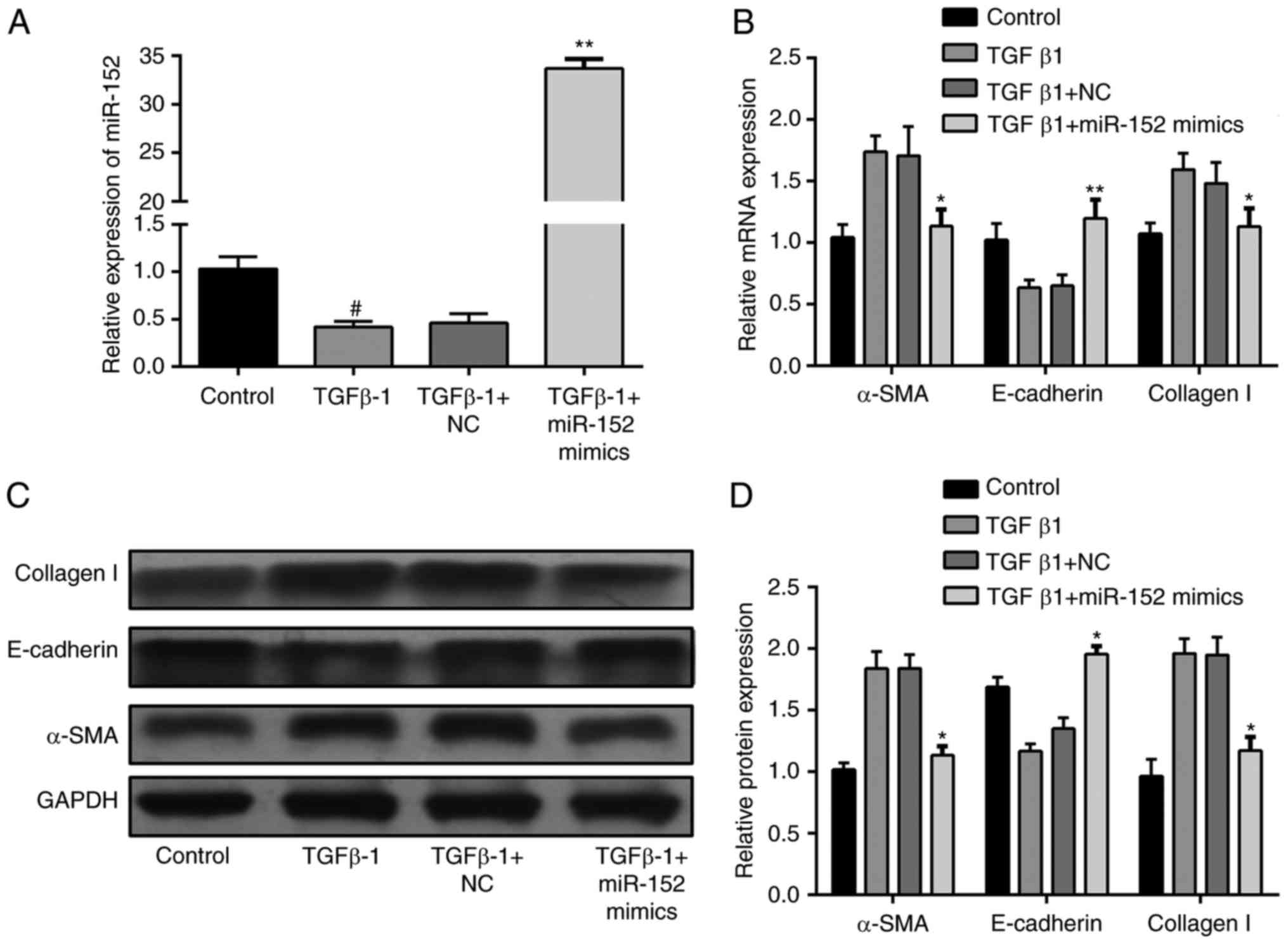
In order to elucidate the role of miR-152 in
TGF-β1-induced EMT in HK-2 cells, the expression of miR-152 was
altered by transfection with miR-152 mimic. As presented in
Fig. 2A, miR-152 expression was
significantly increased in the TGF-β1+miR-152 mimic group compared
with the TGF-β1+NC group (P<0.01). The effects of miR-152 on the
expression of EMT markers were also tested. mRNA expression of
E-cadherin was increased, while mRNA expression of αSMA and
collagen I were suppressed in miR-152-overexpressing HK-2 cells
compared with the control HK2 cells stimulated with TGF-β1
(Fig. 2B). Consistent with the
results of RT-qPCR, E-cadherin, αSMA and collagen I protein
expression demonstrated a similar response (Fig. 2C and D). Collectively, the results
of the present study indicate that miR-152 serves a role in the
regulation of EMT.
Overexpression of miR-152 suppresses
the expression of HPIP in HK-2 cells
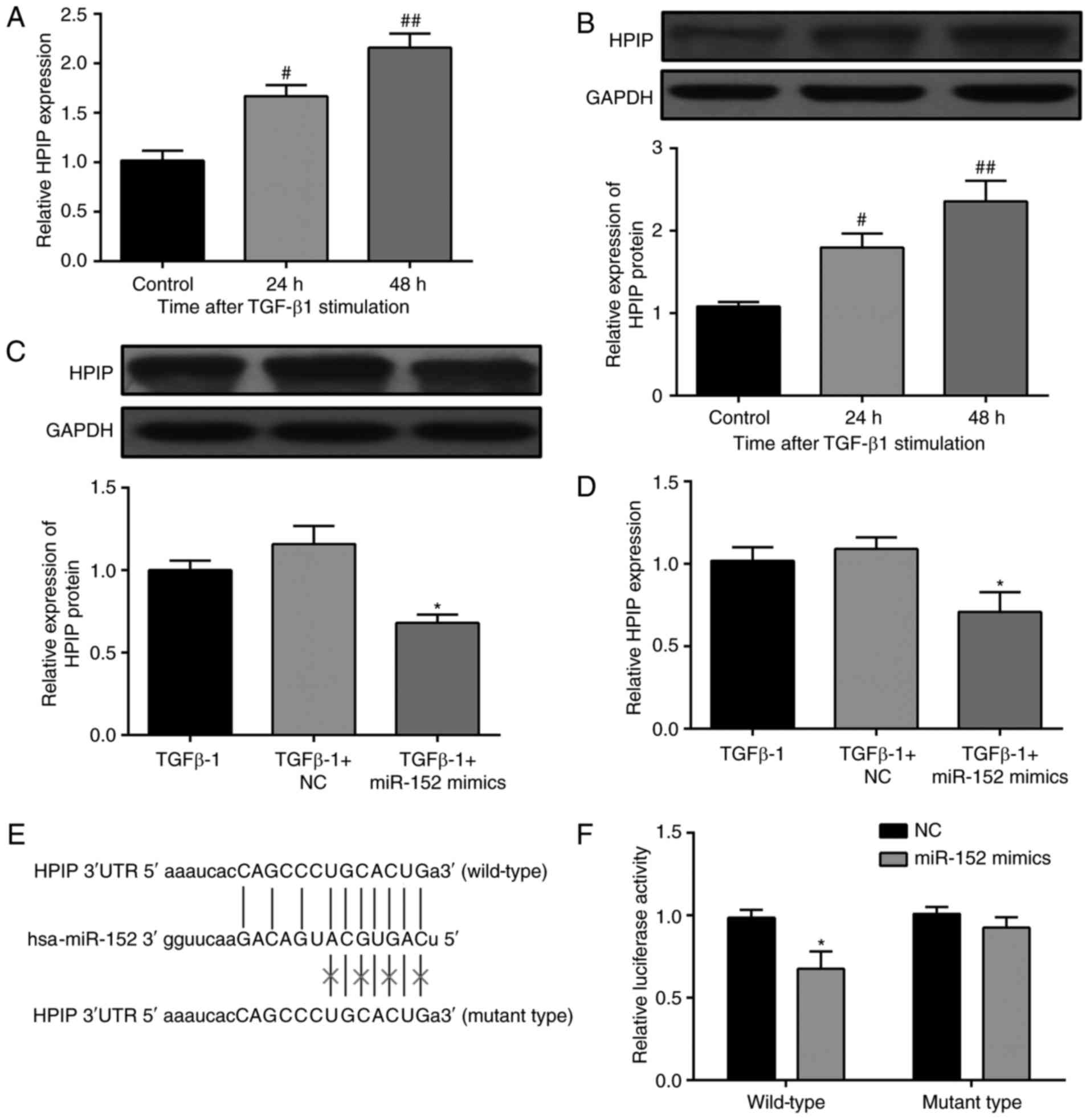
To determine the downstream target(s) of miR-152,
miRBase, miTarget and TargetScanS were employed, and the putative
complementary sequence to miR-152 was identified in the 3′-UTR of
HPIP mRNA. The encoded protein has been reported to be involved in
the TGF-β1-induced EMT in a variety of cancer cells (20). Therefore, HPIP expression was
investigated in HK-2 cells with or without TGF-β1 treatment. HPIP
mRNA expression levels were significantly increased in
TGF-β1-treated cells compared with the control group (Fig. 3A). Additionally, western blotting
analysis demonstrated that HPIP protein expression was upregulated
following stimulation with TGF-β1 (Fig. 3B). Subsequently, the effects of
miR-152 overexpression on HPIP in HK-2 cells were investigated.
HPIP was significantly downregulated at protein (Fig. 3C) and mRNA (Fig. 3D) levels following overexpression
of miR-152. A dual luciferase reporter assay was performed to
validate the results. Fig. 3E
demonstrates the putative position of the miR-152 target site in
the 3′ UTR of HPIP mRNA. Relative luciferase activity was
significantly reduced by co-transfection with miR-152 mimic and
luciferase reporters containing 3′ UTR mRNA of HPIP, while the
inhibition was abolished when the nucleotides were mutated in the
3′-UTR (Fig. 3F). The above
results demonstrate that miR-152 regulates HPIP expression by
directly targeting its 3′ UTR in HK-2 cells.
Overexpression of HPIP partly reverses
miR-152-mediated EMT induced by TGF-β
A rescue assay was designed to investigate whether
HPIP is involved in the miR-152-mediated regulation of EMT in HK-2
cells. Following transfection with the HPIP ORF clone, mRNA and
protein expression levels of HPIP in HK-2 cells were increased
compared with cells transfected with the pcDNA-3.1 vector (Fig. 4A-C). Overexpression of HPIP
reversed the effects of miR-152 on TGF-β1-induced EMT by decreasing
E-cadherin and upregulating αSMA and collagen I mRNA expression
(Fig. 4D). EMT-associated proteins
demonstrated the same pattern of alterations at the protein level
in HK-2 cells (Fig. 4E and F). The
above results demonstrate that miR-152 reverses TGF-β1-induced EMT
by negatively regulating HPIP expression in HK-2 cells.
Discussion
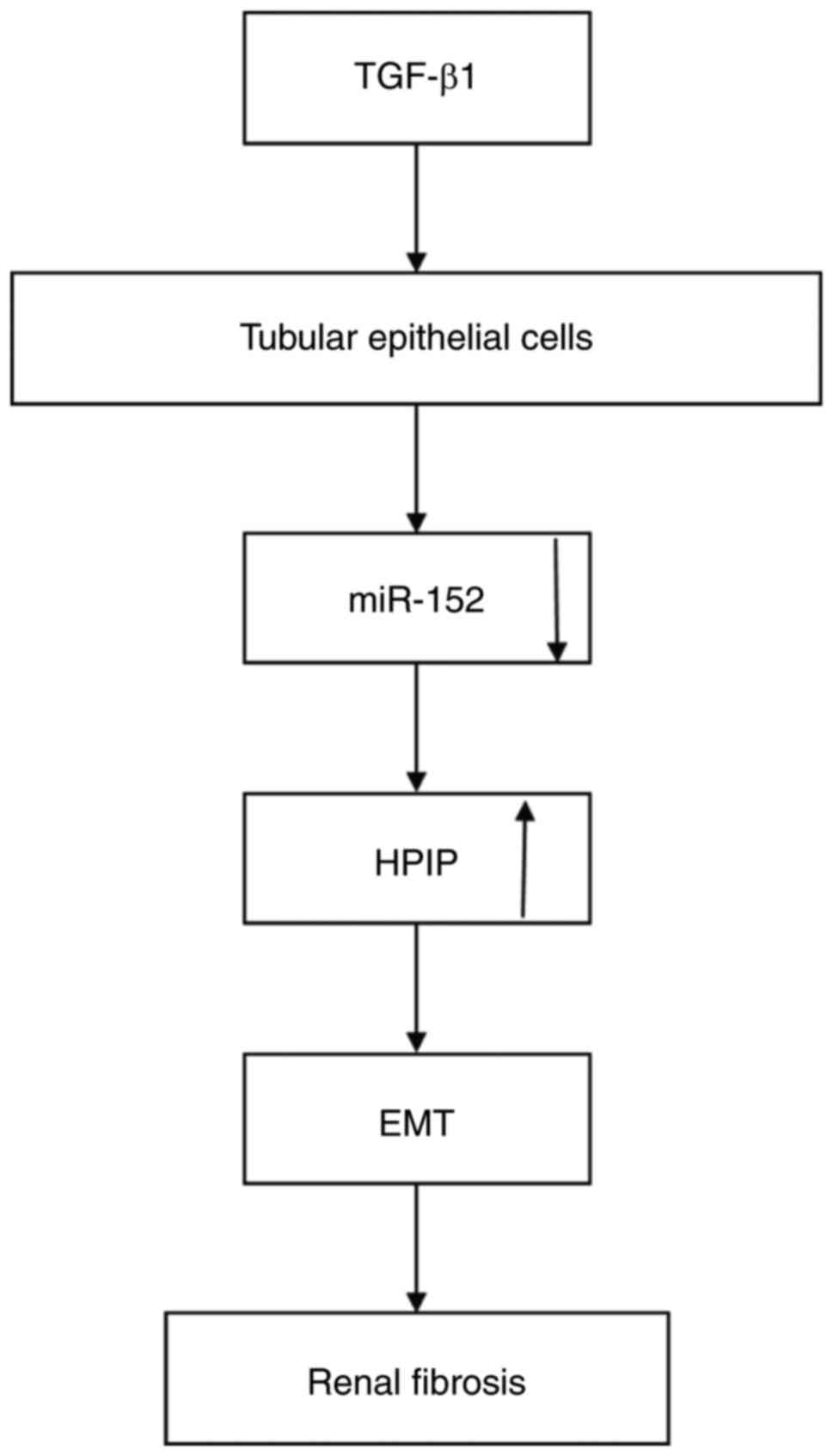
In the present study, miR-152-mediated HPIP
upregulation stimulated TGF-β1-mediated induction of EMT in renal
fibrosis (Fig. 5). miR-152
inhibited TGF-β1-induced EMT of human renal tubular epithelial
cells through the negative regulation of HPIP. A previous study
reported that tubular epithelial and epithelial parenchymal cells
of the kidney are involved in the progression of renal fibrosis
(21). Tubular epithelial cells
demonstrate unique plasticity that enables them to transform form
epithelial and mesenchymal phenotypes, and vice versa (22). An increasing number of publications
suggest that the pathological process of the EMT of tubular
epithelial cells could result in renal fibrosis and chronic renal
disease (23,24). Therefore, inhibition of specific
pathways involved in the EMT offers a novel therapeutic target to
inhibit renal fibrogenesis. Nevertheless, the molecular mechanisms
underlying the control of the onset of EMT of tubular epithelial
cells remains to be elucidated.
TGF-β1, which can be secreted by all types of renal
cells and infiltrated inflammatory cells, is a profibrotic agent in
renal cells (25). In the present
study, TGF-β1 was used as an inducer of EMT in tubular HK-2
epithelial cells in vitro, aiming to investigate its
underlying mechanisms of action. Stimulation with 10 ng/ml TGF-β1
resulted in the loss of E-cadherin expression and elevated
expression of αSMA and collagen I, signifying the induction of EMT
of HK-2 cells. Recently published data focused on the contribution
of specific miRNAs to the progression of EMT in renal fibrosis
(26,27). In the present study, downregulation
of miR-152 in HK-2 cells was observed following stimulation with
TGF-β1, which is consistent with the results of a previous study
(15). Investigation of miRNA
regulation in the kidney will improve the understanding of renal
pathology and may eventually lead to the development of novel
treatment strategies for reversing renal fibrosis and dysfunction.
Results of previous studies revealed a critical role for miR-152 in
human diseases, and miR-152 has been classified as an onco-miRNA in
a variety of cancers, including breast, gastric and bladder
cancers, and glioma (28–30). However, to date, a limited number
of studies investigated the role of miR-152 in the urinary system.
In a recent study, Lin et al (31) demonstrated that miR-152 expression
was significantly downregulated in a rat model of peritoneal
fibrosis, suggesting its involvement in the pathogenesis of
peritoneal fibrosis. Therefore, in the present study, it was
hypothesized that miR-152 may serve similar roles in EMT of tubular
epithelial cells and the progression of renal fibrosis. In the
present study, miR-152 was overexpressed to investigate its role in
the modification of the EMT, and it was identified that
overexpression of miR-152 prevents TGF-β1-induced EMT in HK-2
cells. These results provide novel insights into the role miR-152
in renal disease.
Subsequently, to determine the potential mechanisms
of miR-152 function, downstream targets were investigated and it
was demonstrated that the 3′ UTR of HPIP contained a sequence
complementary to miR-152. HPIP has emerged as an important
regulator of organogenesis and tumorigenesis. It has been
previously reported that HPIP is highly expressed in a variety of
cancers (32–34). Recently, Shi et al (35) demonstrated that HPIP silencing
suppresses TGF-β1-induced EMT in lung cancer cells by inhibiting
activation of mothers against decapentaplegic homolog 2. Similarly,
a study conducted by Zhang et al (36) demonstrated that HPIP silencing
prevents TGF-β1-induced EMT in ovarian cancer cells. This data
indicates the regulatory effect of HPIP during TGF-β1-induced EMT.
However, the expression profile of HPIP in TGF-β1-treated tubular
epithelial cells and the involvement of HPIP in the TGF-β1-induced
EMT remain to be elucidated. A recent study by Mai et al
(37) indicated that
overexpression of HPIP promoted EMT, whereas knockdown of HPIP
repressed EMT in renal carcinoma cells. Elevated HPIP mRNA and
protein levels were observed in TGF-β1-treated HK-2 cells in the
present study. The present study also indicated that transfection
with the miR-152 mimic resulted in a decrease in HPIP expression at
mRNA and protein level, suggesting that miR-152 serves a role in
HPIP mRNA degradation and post-transcriptional regulation. One
previous study has suggested the regulatory role of miRNA on HPIP
(38). Consistent with this study,
overexpression of HPIP partially abolished miR-152-mediated
suppression of the EMT in HK-2 cells, suggesting that HPIP is a
potential therapeutic target for EMT-associated renal fibrosis.
HPIP controls modulation of serine/threonine-protein
kinase mTOR phosphorylation and expression in liver cancer
(38). Knockdown of HPIP
significantly blocked the phosphatidylinositol 4,5-bisphosphate
3-kinase/RAC-alpha serine/threonine-protein kinase signaling
pathway in TGF-β1-stimulated ovarian cancer cells (36,39).
However, the effects of TGF-β1-induced EMT on the HPIP signaling
pathway were not identified in the present study. Further studies
are required to elucidate the role and mechanism of HPIP in renal
fibrosis. In addition, the present study only investigated the
expression profile of miR-152 and HPIP in HK-2 cells. Further
studies should validate their expression levels in human renal
fibrosis tissues and reveal their role in vivo.
In conclusion, the results of the present study
provide evidence that miR-152 controls TGF-β1-induced EMT in
tubular epithelial cells. Furthermore, it was demonstrated that
overexpression of miR-152 downregulates HPIP, contributing to the
inhibition of EMT progression. The results of the present study
contribute to better understanding of the mechanisms underlying
antifibrotic therapies. The results of the present study suggest
that upregulation of miR-152 or inhibition of HPIP may be useful
strategies for the treatment of renal fibrosis.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Gansu Province (grant no. 1308RJZA246).
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
miRNA
|
microRNA
|
|
TGF-β1
|
transforming growth factor β1
|
|
HPIP
|
hematopoietic pre-B-cell leukemia
transcription factor (PBX)-interacting protein
|
References
|
1
|
Webster AC, Nagler EV, Morton RL and
Masson P: Chronic kidney disease. Lancet. 389:1238–1252. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wing MR, Ramezani A, Gill HS, Devaney JM
and Raj DS: Epigenetics of progression of chronic kidney disease:
Fact or fantasy? Semin Nephrol. 33:363–374. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li Z, Liu X, Wang B, Nie Y, Wen J, Wang Q
and Gu C: Pirfenidone suppresses MAPK signaling pathway to reverse
epithelial-mesenchymal transition and renal fibrosis. Nephrology
(Carlton). 22:589–597. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bani-Hani AH, Campbell MT, Meldrum DR and
Meldrum KK: Cytokines in epithelial-mesenchymal transition: A new
insight into obstructive nephropathy. J Urol. 180:461–468. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y: Epithelial to mesenchymal
transition in renal fibrogenesis: Pathologic significance,
molecular mechanism, and therapeutic intervention. J Am Soc
Nephrol. 15:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: MicroRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Song YX and Wang ZN: The
microRNA-148/152 family: Multi-faceted players. Mol Cancer.
12:432013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miao CG, Yang YY, He X, Huang C, Huang Y,
Qin D, Du CL and Li J: MicroRNA-152 modulates the canonical Wnt
pathway activation by targeting DNA methyltransferase 1 in
arthritic rat model. Biochimie. 106:149–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu Y, Huang A, Li T, Su X, Ding H, Li H,
Qin X, Hou L, Zhao Q, Ge X, et al: miR-152 reduces human umbilical
vein endothelial cell proliferation and migration by targeting
ADAM17. FEBS Lett. 588:2063–2069. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chandrasekaran K, Karolina DS, Sepramaniam
S, Armugam A, Wintour EM, Bertram JF and Jeyaseelan K: Role of
microRNAs in kidney homeostasis and disease. Kidney Int.
81:617–627. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen CH, Cheng CY, Chen YC, Sue YM, Liu
CT, Cheng TH, Hsu YH and Chen TH: MicroRNA-328 inhibits renal
tubular cell epithelial-to-mesenchymal transition by targeting the
CD44 in pressure-induced renal fibrosis. PLoS One. 9:e998022014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kato M, Zhang J, Wang M, Lanting L, Yuan
H, Rossi JJ and Natarajan R: MicroRNA-192 in diabetic kidney
glomeruli and its function in TGF-beta-induced collagen expression
via inhibition of E-box repressors. Proc Natl Acad Sci USA.
104:3432–3437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin S, Zhang Q, Yang J, Lin W, Li Y, Chen
F and Cao W: TGFβ-incurred epigenetic aberrations of miRNA and DNA
methyltransferase suppress Klotho and potentiate renal fibrosis.
Biochim Biophys Acta. 1864:1207–1216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Y, Tong J, He F, Yu X, Fan L, Hu J,
Tan J and Chen Z: miR-141 regulates TGF-β1-induced
epithelial-mesenchymal transition through repression of HIPK2
expression in renal tubular epithelial cells. Int J Mol Med.
35:311–318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lan A, Qi Y and Du J: Akt2 mediates
TGF-β1-induced epithelial to mesenchymal transition by deactivating
GSK3β/snail signaling pathway in renal tubular epithelial cells.
Cell Physiol Biochem. 34:368–382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li SS, Liu QF, He AL and Wu FR: Tranilast
attenuates TGF-β1-induced epithelial-mesenchymal transition in the
NRK-52E cells. Pak J Pharm Sci. 27:51–55. 2014.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng Y, Li L, Zhang X, Zhang Y, Liang Y,
Lv J, Fan Z, Guo J, Hong T, Ji B, et al: Hematopoietic pre-B cell
leukemia transcription factor interacting protein is overexpressed
in gastric cancer and promotes gastric cancer cell proliferation,
migration, and invasion. Cancer Sci. 106:1313–1322. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hudson BG, Tryggvason K, Sundaramoorthy M
and Neilson EG: Alport's syndrome, Goodpasture's syndrome, and type
IV collagen. N Engl J Med. 348:2543–2556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeisberg M, Strutz F and Muller GA: Renal
fibrosis: An update. Curr Opin Nephrol Hypertens. 10:315–320. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ng YY, Huang TP, Yang WC, Chen ZP, Yang
AH, Mu W, Nikolic-Paterson DJ, Atkins RC and Lan HY: Tubular
epithelial-myofibroblast transdifferentiation in progressive
tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int.
54:864–876. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thuault S, Valcourt U, Petersen M,
Manfioletti G, Heldin CH and Moustakas A: Transforming growth
factor-beta employs HMGA2 to elicit epithelial-mesenchymal
transition. J Cell Biol. 174:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bijkerk R, de Bruin RG, van Solingen C,
van Gils JM, Duijs JM, van der Veer EP, Rabelink TJ, Humphreys BD
and van Zonneveld AJ: Silencing of microRNA-132 reduces renal
fibrosis by selectively inhibiting myofibroblast proliferation.
Kidney Int. 89:1268–1280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loboda A, Sobczak M, Jozkowicz A and Dulak
J: TGF-β1/Smads and miR-21 in renal fibrosis and inflammation.
Mediators Inflamm. 2016:83192832016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chhabra R, Dubey R and Saini N:
Cooperative and individualistic functions of the microRNAs in the
miR-23a~27a~24-2 cluster and its implication in human diseases. Mol
Cancer. 9:2322010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu G, Jia Z and Dou Z: miR-24-3p regulates
bladder cancer cell proliferation, migration, invasion and
autophagy by targeting DEDD. Oncol Rep. 37:1123–1131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roscigno G, Puoti I, Giordano I,
Donnarumma E, Russo V, Affinito A, Adamo A, Quintavalle C, Todaro
M, Vivanco MD and Condorelli G: MiR-24 induces chemotherapy
resistance and hypoxic advantage in breast cancer. Oncotarget.
8:19507–19521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin F, Wu X, Zhang H, You X, Zhang Z, Shao
R and Huang C: A microrna screen to identify regulators of
peritoneal fibrosis in a rat model of peritoneal dialysis. BMC
Nephrol. 16:482015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bugide S, David D, Nair A, Kannan N,
Samanthapudi VS, Prabhakar J and Manavathi B: Hematopoietic
PBX-interacting protein (HPIP) is over expressed in breast
infiltrative ductal carcinoma and regulates cell adhesion and
migration through modulation of focal adhesion dynamics. Oncogene.
34:4601–4612. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Vuurden DG, Aronica E, Hulleman E,
Wedekind LE, Biesmans D, Malekzadeh A, Bugiani M, Geerts D, Noske
DP, Vandertop WP, et al: Pre-B-cell leukemia homeobox interacting
protein 1 is overexpressed in astrocytoma and promotes tumor cell
growth and migration. Neuro Oncol. 16:946–959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng Y, Xu X, Zhang Y, Ding J, Wang Y,
Zhang X, Wu Z, Kang L, Liang Y, Zhou L, et al: HPIP is upregulated
in colorectal cancer and regulates colorectal cancer cell
proliferation, apoptosis and invasion. Sci Rep. 5:94292015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi S, Zhao J, Wang J, Mi D and Ma Z: HPIP
silencing inhibits TGF-β1-induced EMT in lung cancer cells. Int J
Mol Med. 39:479–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang GY, Liu AH, Li GM and Wang JR: HPIP
silencing prevents epithelial-mesenchymal transition induced by
TGF-β1 in human ovarian cancer cells. Oncol Res. 24:33–39. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mai H, Xu X, Mei G, Hong T, Huang J, Wang
T, Yan Z, Li Y, Liang Y, Li L, et al: The interplay between HPIP
and casein kinase 1alpha promotes renal cell carcinoma growth and
metastasis via activation of mTOR pathway. Oncogenesis. 5:e2602016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng
X, Zhu Z, Jiao H, Lin J, Jiang K, et al: Hepatitis B virus X
protein represses miRNA-148a to enhance tumorigenesis. J Clin
Invest. 123:630–645. 2013.PubMed/NCBI
|
|
39
|
Bugide S, Gonugunta VK, Penugurti V,
Malisetty VL, Vadlamudi RK and Manavathi B: HPIP promotes
epithelial-mesenchymal transition and cisplatin resistance in
ovarian cancer cells through PI3K/AKT pathway activation. Cell
Oncol (Dordr). 40:133–144. 2017. View Article : Google Scholar : PubMed/NCBI
|