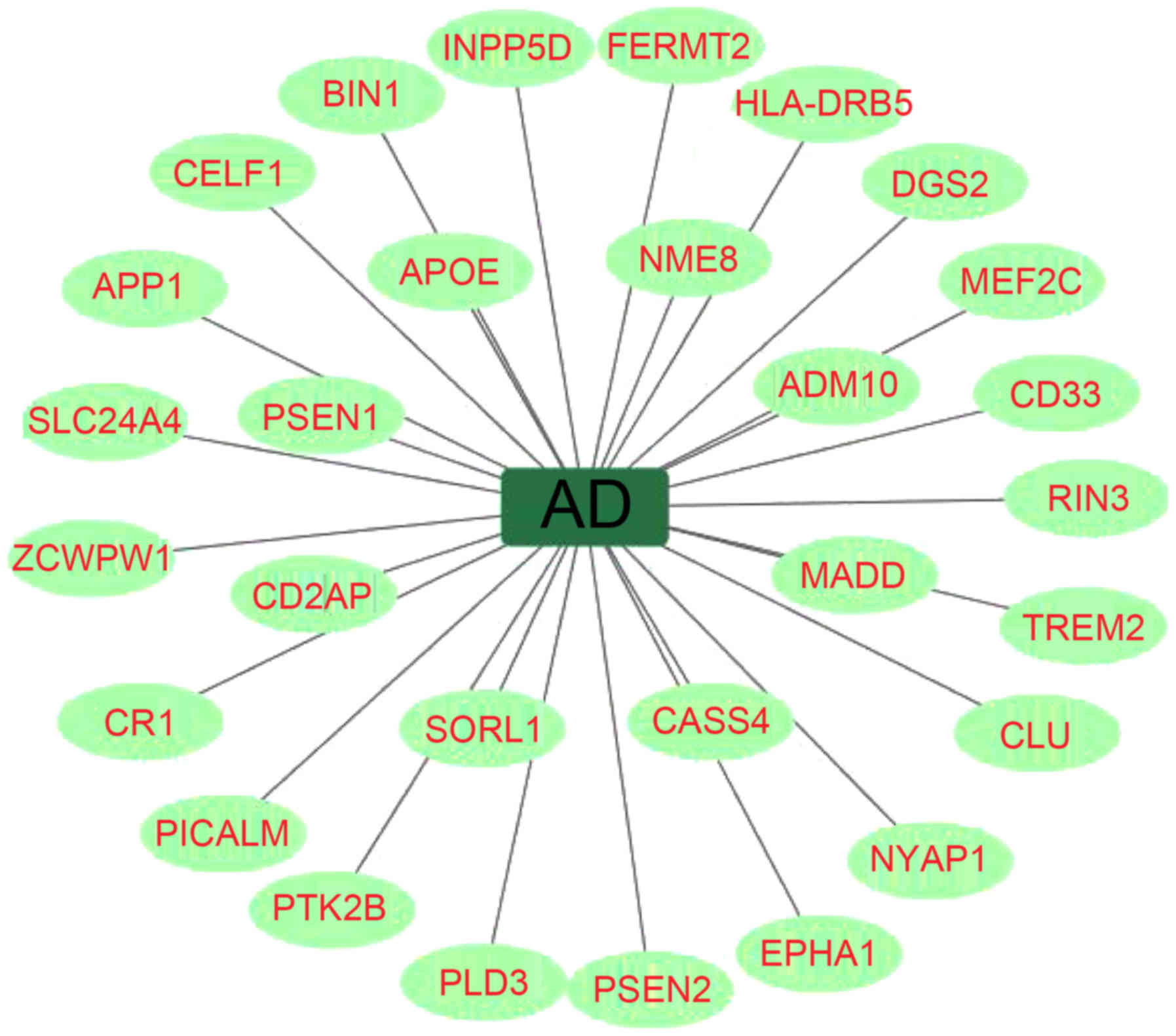
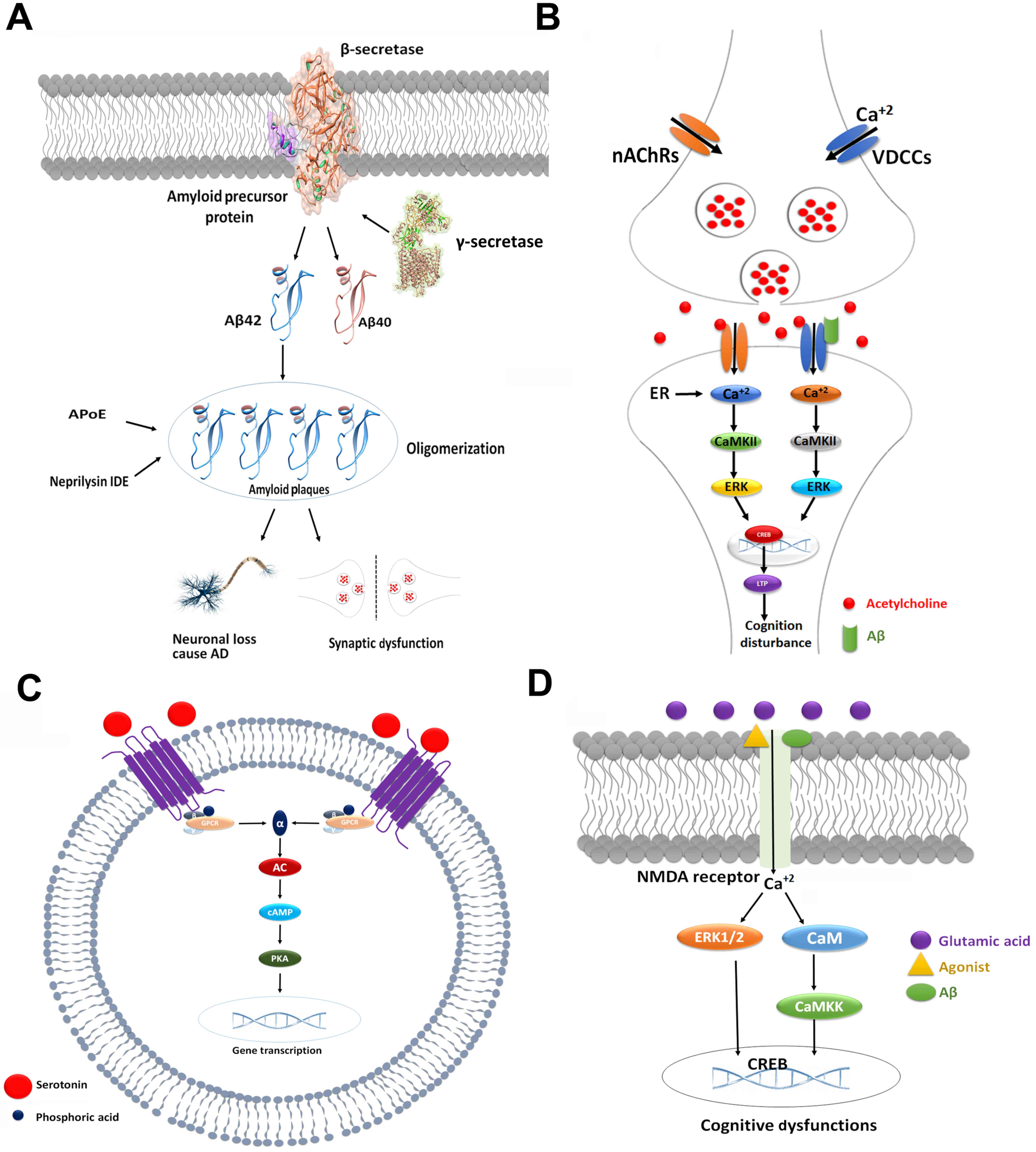
Neurobiological data have demonstrated that AD is
characterized by the degeneration of neurons and disturbances in
neuronal synapsis within cortical and subcortical areas (16). Amyloid plaques and neurofibrillary
tangle (NFT) accumulations have been reported to be governing
mechanisms of AD in humans (17).
Plaques are characterized by dense deposition of Aβ, while NFTs are
clumps of microtubules associated with tau protein. Aβ consists of
39–43 amino acids, which are also found in APPs. Proteomic studies
have demonstrated that APP is a transmembrane protein that aids
neuron growth and post-injury repair (18,19).
In AD, β- and γ-secretase are proteolytic enzymes that cleave APP
into smaller fragments, which accumulate outside the neurons to
form senile plaques (20,21). The basic mechanistic pathway of AD
is presented in Fig 2A.
Glycogen synthase kinase 3 (GSK-3) is also
associated with neuronal loss and is potentially implicated in AD,
as GSK-3 forms associations with Aβ and NFTs, which is considered
to be a major hallmark of AD (22). GSK-3 controls various metabolic
processes, including phosphorylation, protein complex formation and
subcellular distribution (23).
Additionally, GSK-3 is considered to increase the production of Aβ
and NFTs by hyperphosphorylation of tau proteins (24). Furthermore, disturbance in
hippocampal volume, inflammation and oxidative stress may also be
implicated in the pathology of AD.
ACh receptors are the most important target proteins
that specifically bind to ACh neurotransmitters. Based on the
affinities and specificities with neurotransmitters, ACh receptors
are divided into nicotinic Ach receptors (nAChRs) and muscarinic
receptors (MRs). nAChRs are localized to skeletal neuromuscular
junctions and autonomic ganglia, whilst MRs are present in the
brain and parasympathetic effector organs (25), and are associated with cognition in
AD (26,27). Tsang et al (28) identified that M1/G-protein coupling
significantly decreased with the progression of AD, whereas the
density of M1 receptors was not reduced. Furthermore, another in
vitro study reported that an M1 receptor agonist, TBPB, reduced
Aβ production, which indicates that the M1 receptor may be used as
a novel therapeutic target for the treatment of AD (29). Furthermore, in a knockout mouse
study, the M3 receptor was reported to be associated with fear
learning and memory conditions, which is relevant to AD symptoms
(30).
Another type of receptor that has been extensively
investigated is the nicotinic Ach receptor (nAChR), which consists
of two subtypes, α7 and α4β2 (31). Previous studies have reported that
the expression of these receptors is reduced with the progression
of AD (25,32). Young et al (33) also investigated the role of
α7-nAChR in knockout mice and demonstrated an impairment in the
attention of knockout mice compared with wild type mice (33). However, another study reported
conflicting results in α7-nAChR knockout mice by demonstrating
neuroprotective effects compared with normal groups (34). However, additional studies have
reported that α7-nAChR agonists have led to improvements in
cognitive deficits (35–37).
It has been observed that Ach receptors are
associated with improvements in cognitive deficits in patients with
AD (38). For example, ACh
receptors govern calcium signaling, which has been demonstrated to
improve learning and memory in aging (38,39).
Upon activation, ACh receptors trigger increases in calcium levels,
which induces various intracellular processes that mediate learning
and memory (40). Calcium
signaling mediates three different types of effects, which include
rapid, short and long-term effects. Short and long-term effects are
the result of signaling cascades and changes in gene expression,
respectively (41).
Specifically, following activation of calcium
influx, long-term effects involve the activation of
calcium/calmodulin-dependent protein kinase II/IV (CaMKII/IV),
extracellular signal-regulated kinase/mitogen-activated protein
kinase (ERK/MAPK) and cAMP response element-binding protein (CREB).
As a result, the activated enzymatic cascades alter the gene
expression and may govern cognition symptoms via long-term
potentiation (LTP) (42–45). An antagonistic association was
observed between Aβ peptides and cholinergic systems. The binding
of Aβ to nAChRs is also a factor in the activation of calcium, and
may induce certain downstream signaling pathways that lead to a
decline in cognition (Fig. 2B)
(46,47).
An increased serotonin (5-hydroxytryptamine)
concentration in the synaptic cleft has been reported to be a
potential therapeutic strategy to slow the progression of AD
(48,49). Serotonin targets specific receptors
at postsynaptic neurons and mediates downstream signaling pathways
that control cognition. It has been reported that ≥16 different
types of serotonin receptors exist, which are categorized into 7
subfamilies (5-HT1-5-HT7) (50).
All serotonin receptors are G-protein-coupled receptors (GPCRs),
excluding the 5-HT3 receptor (50). The activation of these receptors
stimulates downstream signal transduction pathways that govern
certain intracellular responses. The protein kinase A (PKA)
signaling cascade is responsible for the inhibition and stimulation
of phospholipase C/protein kinase C, which regulates the ERK/MAPK
pathways (51,52). Subsequently, these activations
affect cognitive impairment in neurodegenerative diseases.
Results from animal and clinical experiments have
also demonstrated the importance of 5-HT in cognitive dysfunction
and memory deficits (53).
Increases in the expression of 5-HT1A receptors were reported to be
associated with cognitive impairment, and these receptors are
therefore considered to be potential targets for the treatment of
AD (54). Furthermore,
Garcia-Alloza et al (55)
demonstrated that 5-HT1B/1D receptors were associated with
cognitive dysfunction in AD. It was also observed that the density
of the 5-HT2A receptor was significantly reduced in the frontal and
temporal cortical neurons in patients with AD compared with healthy
participants. Furthermore, various studies have reported an
important association between 5-HT2 receptors and cognitive decline
in AD (56–58).
The serotonin receptor 5-HT6 has an important role
in various mechanistic pathways within the brain (59). It is primarily expressed in the
striatal, hippocampal and cortical areas (60). Notably, it was previously reported
that inhibition of the 5-HT6 receptor improved learning and memory
(61,62). Another animal study also
demonstrated the importance of 5-HT6, as the agonist SB-271046
improved age-associated deficits and spatial recognition memory in
aged mice (63). It was also
reported that, another agonist, WAY-181187, may also be used to
modulate synaptic plasticity via attenuation of LTP (64).
5-HT receptor-mediated signaling pathways are
associated with improvements incognitive defects (65). The 5-HT6 receptor stimulates
G-proteins, which results in cAMP production via adenylyl cyclase
activation (66,67). cAMP subsequently triggers PKA,
which, via phosphorylation, activates CREB (67). A number of studies have indicated
that the 5-HT6 receptor modulates various neurotransmitters,
including glutamate and Ach, to aid memory processes (Fig. 2C) (68,69).
Adrenergic receptors are metabotropic GPCRs, which
are divided into two major groups, α and β. Adrenergic receptors
are typically sensitized for norepinephrine and epinephrine
neurotransmitters. A number of studies have reported that
adrenergic receptors (α and β) are closely associated with
cognitive decline in AD (70,71).
An expression study by Kalaria and Harik (72) demonstrated that β2 levels were
increased in the cortex and hippocampus of patients with AD. In
addition, a behavioral study reported that certain structural
changes in adrenergic receptors were associated with the presence
or absence of aggressive behavior in AD patients (73).
Dopamine receptors exhibit important roles in
various human functions, including cognition and learning (50). Dopamine receptors are divided into
two different classes, D1- and D2-like receptors, which consist of
five subtypes. D1-type receptors include D1 and D5 receptors,
whereas D2-type receptors include D2, D3 and D4 receptors (74). Functionally, D1- and D2-type
receptors function in synaptic plasticity and cognition by
stimulating the protein signaling cascade of cAMP/PKA and CREB
modulation (43,44). However, another study demonstrated
that dopamine receptors were directly associated with AD and
Parkinson's disease (50).
NMDA/glutamate receptors are have been extensively
studied, and are abundantly expressed in the cerebral cortex,
hippocampus, nucleus accumbens and striatum (75,76).
Variations in glutamatergic receptors are implicated in the
pathogenesis of neurodegenerative diseases, such as AD, as they are
associated with neuronal death (77). A reduced expression study on NMDA,
NMDA receptor subunit 1 and subunit 2B proteins in rat models
reported that there is a close association between NMDA receptors
and cognitive deficits (78).
Neuronal loss induced by amyloid plaques are a consequence of NMDA
receptor modulation. Amyloid plaques activate NMDA receptors, which
results in higher calcium influx into neurons, ERK1/2 activation
and mediation of respective downstream enzymes (79–82).
Therefore, NMDA signaling pathways have a potential role in the
pathogenesis of cognitive dysfunctions (Fig. 2D).
AChE is a type of hydrolase, and exhibits key
functions in cholinergic neurotransmission in the autonomic and
somatic nervous systems (83).
AChE interacts with Ach, converting it into choline and acetic acid
(82). Expression studies have
demonstrated that AChE is frequently present in motor neurons and
certain other types of conducting tissue, including nerve and
muscle, motor and sensory fibers, and cholinergic and
non-cholinergic fibers (84,85).
In AD, cholinergic neurons mediate memory deficits and cognitive
decline by reducing the level of ACh (86,87).
Therefore, AChE may be considered as a novel target to reverse AD
symptoms. Furthermore, butyrylcholinesterase (BChE) is also
considered to be a minor player in the regulation of synaptic ACh
levels (88). Therefore,
inhibition of BChE may also be considered a valid approach to
restore cholinergic function in AD (89,90).
The majority of neuromuscular problems are treated
by AChE inhibitors, which are also considered to be
first-generation drugs for the treatment of AD. There are four
established inhibitors (donepezil, galantamine, rivastigmine and
tacrine) that are commonly used to improve cognition (91). However, tacrine is not as reputable
due to poor tolerability (92,93).
Donepezil demonstrated its neuroprotective effects by diminishing
the excitotoxicity of glutamate by reducing Aβ load and cell
toxicity, as well as increasing cell life span (94,95).
Rivastigmine is a cholinergic agent that targets AChE and BChE.
Clinical trials have indicated that rivastigmine improves
cognition, with few side effects in patients with AD (96). Tacrine is another inhibitor that
increases ACh levels from cholinergic nerve endings. Tacrine
inhibits the activity of certain enzymes, including monoamine
oxidase, and suppresses γ-aminobutyric acid (GABA) signaling, which
results in the release of dopamine, noradrenaline and serotonin
from nerve endings, and improves memory in patients with AD
(97).
Recently, novel inhibitors have been synthesized
from natural and synthetic sources for patients with AD. Huperzine
A (Hup A) is an AChE inhibitor that is primarily used in the
treatment of memory disorders. Hup A is highly potent and has a
higher bioavailability compared with donepezil and tacrine, but is
less effective compared with BChE inhibitors for treating AD
symptoms (98). Recent attempts
have demonstrated that derivatives of Hup A, with aromatic rings,
exhibit potential therapeutic effects for AD symptoms (99). However, further studies are
required to assess the potential benefits of Hup A for treating AD
(100). Camps et al
(101) synthesized hybrids of
innovative tacrine and Hup A as a cholinesterase agonists to treat
AD. This designed agonist exhibits different functional moieties at
basic nuclei of chemical compounds, and provided good results at
various positions. The halogen moiety had a higher activity and
increased therapeutic effectiveness of treating AD compared with
tacrine. However, it also exhibits limited inhibition of BChE.
Furthermore, the agonist designed by Camps et al (101) also has the propensity to cross
the blood-brain barrier.
Huperzine B (Hup B) is also considered to be an AChE
inhibitor with reversible and effective properties. Hup B is less
potent compared with Hup A, and is also used as a template
structure to synthesize novel compounds that inhibit AChE (102). Another potent derivative is
bis-Hup B, which consists of two Hup B molecules connected to a
carbon-nitrogen chain by an amine group. The bis-Hup B compound has
exhibited higher inhibitory potential against AChE compared with
against BChE (103).
Berberine is another candidate compound with
multiple biological activities, including the potential to cross
the blood brain barrier and target the central nervous system
(CNS). Berberine acts as an inhibitor of AChE (104,105) and also performs a neuroprotective
function by reducing NMDA-induced excitotoxicity (105).
The designing of novel γ-secretase agonists remains
a challenging approach due to its non-amyloid behavior and
interaction with metabolic processes. Various undesired effects are
also generated, including gastrointestinal lethality, hematological
toxicity and skin reactions (114).
In addition, a number of candidate molecules have
been synthesized by considering α-secretase as a target molecule
(123). Of these, etazolate
(EHT-0202) activates neuronal α-secretase and, as a result,
enhances soluble APP production (124). Etazolate was investigated in a
phase II study in patients with mild-to-moderate AD. Results
revealed that etazolate exhibited good clinical efficacy in
patients with AD (124).
Bryostatin-1 and exebryl-1 are potent inhibitors of α-secretase,
which significantly affect Aβ production and improve memory
(125).
It is difficult to fully understand all of the
receptor-based mechanistic signaling pathways and the interactions
of neurotransmitters with drugs by experimentation. Therefore,
computational modeling and simulation approaches are considered to
be important for targeting and investigating the neurodegeneration
disorders. The present review will highlight a number of
computational modeling attempts and biomarker interpretations to
improve the understanding of the pathogenesis and symptoms of
AD.
To interpret the basic mechanism of AD,
computational models have been designed on the basis of amyloid
plaques, NFTs and hippocampus functions. Furthermore, additional
models are based on neuronal functionality and the synaptic
transmission of neurotransmitters.
Amyloid plaque formation is also considered a key
biochemical concept to design models (142,143). In addition, the kinetics of APP
processing and downstream intracellular interactions of calcium and
Aβ were observed in the AD brain (144–146). The secretases (α, β and γ)
function as cleaving agents of APP. It has been observed that
secretase agonists target APP and minimize the Aβ production, and
may slow the progression of AD (147). Based on intracellular calcium and
Aβ interactions, a computational model was built to account for
established characteristics of AD, which include its
irreversibility, acute to chronic pathology and inherent random
characteristics of sporadic AD.
In intracellular signaling, multiple proteins are
interconnected through specific receptor-mediated pathways. GSK-3b,
p53, Aβ and tau proteins are observed in computational modeling to
investigate the mechanistic pathways that mediate AD (152). A multi-compartment model for
GSK-3b, p53, Aβ and tau proteins was designed to determine the
associations among these proteins (152). The predicted results demonstrated
that abrupt changes in DNA damage the p53 and Mdm2 complex. As a
result, GSK-3b/p53 complexes are formed, which enhance the
transcriptional activity of p53 and GSK-3b. Consequently, there are
increases in the production of Aβ, Mdm2, mRNA and tau
phosphorylation. Computational model results indicate that, in
normal states, Aβ is degraded in cells and, upon dephosphorylation,
degradation become optimized within cells. However, under
conditions of stress, Aβ production and tau phosphorylation
increase. Therefore, adjusting the DNA damage parameter may clear
Aβ and stop the phosphorylation of the tau protein. Additionally,
plaque and tangle formation were independent, even with GSK-3b
overactivity (152).
In another computational model based on Aβ
functionality, which was developed by Diem et al (153), the results indicated that the
deposition of Aβ in human artery walls reflect the lymphatic
drainage pathway with the progression of AD. Initially, the
diffusion of Aβ occurs from the brain to basement membranes in
capillaries and arteries via extracellular spaces of gray matter in
the brain. However, the exact mechanism of perivascular elimination
of Aβ remains under consideration. Based on this mechanistic
approach, a computational model was designed to explain the process
of periarterial drainage with regards to diffusion in the brain,
and demonstrated that periarterial drainage along basement
membranes is rapid compared with diffusion. The predicted results
indicated that failure of periarterial drainage is a mechanism
underlying the pathogenesis of AD, in addition to complications
associated with its immunotherapy (153).
An additional network interaction model of Aβ,
neuroinflammation, mitochondrial dysfunction and lipid metabolism
dysregulation was designed by Kyrtsos and Baras (155). The basic purpose of this
computational model was to investigate the short and moderate level
effects of inflammation, and mutational effects on the ApoE allele.
Their model was based on cellular and molecular levels. In cellular
levels, four different types of cells, which included neurons,
astrocytes, microglia and brain endothelial cells, were used to
interact with each other. While at the molecular level, each
cellular downstream metabolic network was addressed to mediate the
metabolic responses of particular cell types. Modeling of chemical
species for each cell type was performed by average distribution.
The simulation results indicated that the ApoE4 allele ultimately
led to an increase in Aβ. This increase causes ATP to collapse and
an elevation of glutamate levels, which is the major cause of
neuronal loss in a local region. The computational model results
demonstrated that inflammation may be considered as a key component
in the pathogenesis of AD. Furthermore, inflammation strength and
duration are also important factors in AD progression (155).
To interpret the Aβ functionality more adequately
against AD, single-cell-based models were employed. The different
cell-based-models act as a single framework, which investigates the
different properties of individual cells. Chen (156) revealed that a higher expression
of Aβ in cells leads to intrinsic disruption of electrical
properties in the dendrites of the hippocampus. In the dendrites of
pyramidal cells, Aβ bocks the A-type potassium channels, which
results in enhanced membrane excitability and calcium influx
(156). Hyperexcitability of
dendrites gradually leads to degenerative changes or neuronal cell
death (157). The effect of Aβ
was modeled by decreasing the maximal conductance in transient
A-type potassium channels. The simulation results for this
experimental study demonstrated that, when Aβ affected the
potassium current, there was increased invasion of backpropagated
action potentials (bAPs) from the cell body into the apical
dendritic trunk of CA1 pyramidal neurons. In another study by
Hoffman et al (158),
similar results were observed following the administration of
pharmacological agents that blocked the A-type potassium current
(158). The simulation results
indicated that the disturbance of normal dendritic electrical
activity caused by an intra-articular blockade produces significant
differences between the depolarizations of Aβ and normal cases in
the distal oblique branches compared with the dendritic trunk.
Furthermore, a number of studies have reported that modified
synaptic membrane properties disturb the firing properties of CA1
pyramidal neurons under current and voltage clamp conditions by the
amalgamation of Aβ (159,160).
A neural network model of corticohippocampus
formation was designed to investigate the effects of scopolamine, a
drug that blocks cellular effects of ACh, on the encoding and
retrieval of memories in paired associate tasks (163). Four modules were present in this
model by Hasselmo and Wyble, which included the entorhinal cortex
(EC), dentate gyrus (DG), region CA3 and region CA1 (164). In each module, ‘memory’ was
represented as a pattern of neural activation. The information
following the patterns among the four modules were represented as
EC to DG to CA3 to CA1. The represented items, such as individual
words, in CA3 neurons exhibited weaker recurrent connections
compared with contextual information. Their detailed model
simulation demonstrated that scopolamine impaired the encoding of
new input patterns, but had no effect on previously learnt recalled
patterns. Results indicated that impairment is selective in free
recall, upon the recognition of items that are not already encoded.
This model was the first attempt to simulate the effects of a drug
on human memory. The experiment investigated and quantified the
physiological effects at a cellular level. To design novel drugs
against neurodegenerative diseases, modeling attempts to interlink
the behavior, physiology and molecular biological aspects in a
single constrained model for human memory functions.
An additional computational modeling approach was
established to investigate the modulation and control storage, and
AD dynamics within the hippocampal CA3 network on the basis of
subcortical cholinergic and GABAergic inputs (165). To build upon Meschnik and
Finkel's model (165), Buzsaki
developed a ‘two-stage’ memory model and highlighted the importance
of interneurons, basket cells and chandelier cells in memory
(166–168). Furthermore, Lisman et al
(168) designed a computational
model on the basis of embedded γ cycles within the θ cycles. Their
results demonstrated that attractor-based auto-associative memory
may be implemented by the synchronization of γ-frequency ranges.
Each newly arrived input pattern at the commencement of θ cycle
with embedded 5–10 γ-cycles generated a network activity to
congregate various γ-cycles as a steady attractor, which
characterizes the stored memory. Their predicted results support
the hypothesis that CA3 pyramidal cells generate distinct
behavioral functions by bursting and spiking patterns. In addition,
the change between behavioral states associated with the online
processing and recall of information is regulated by cholinergic
input in the hippocampus. A deficiency of cholinergic neurons is
associated with a reduction of γ frequency. The reduction of
γ-cycles within the θ rhythm results in memory loss and cognition,
which is associated with AD (169).
The initiation of synaptic deletion and
compensation model was initially proposed by Horn et al
(173) and further developed by
Ruppin and Reggia (174). The
artificial neural progression model for AD (173) deviates from the excitotoxicity,
which does not account for cognitive impairment. It has been
observed that a 50% loss of synaptic connections is considered a
primary factor for cognitive deficits. In earlier stages of AD, the
loss of connections is compensated by strengthening the remaining
connections. Horn's model demonstrated that synaptic connections
are associated with memory loss and disturbances in learning
patterns. The rate of memory deterioration may be minimized by
enhancing the remaining connection weight of a constant
multiplication factor (173).
The synaptic runway model was based on associative
memory and memory storage as a pattern of neuronal spatiotemporal
activation (175,176). Memory storage activates different
analog patterns that interact with previous associations. For
example, if there is an overlap between patterns or if the memory
capacity is exhausted. The results demonstrated that the
significant increase in the number of associations stored by the
network may govern pathological increases in the strength of
synaptic connections. As a result, such synaptic connections give
rise to an increase in neuronal activity, high metabolic demand and
may eventually cause excitotoxicity. The synaptic runway model
investigated two basic mechanistic approaches to memory, encoding
and retrieval, to attempt to reduce neuronal cell death (176). In normal conditions,
neuromodulation is satisfactory to preclude the variations in
runaway synaptic modification (RSM). Whereas, in manifestations of
disease conditions such as AD, the RSM neuromodulation is
inevitable. However, the threshold levels for RSM in AD are lower
compared with controls (177).
Computational exploration of AD has reported
changes in hippocampal functionality and behavioral performance
(178,179). For example, the ability to learn
and adapt learning to novel situations is impaired in AD. Moustafa
et al (180) also
identified that simulated learning occurred through an interaction
between the hippocampal region and basal ganglia (180). Hebbian learning and temporal
difference algorithms were used to train the model, and the results
indicated that hippocampal damage leads to impaired learning
performance.
Generally, a biomarker is a parameter of
physiological, biochemical or anatomical domains that indicates
normal biological and pathological processes or reactions to a
therapeutic intervention (182).
Biomarkers are currently considered to be important factors in the
diagnosis of neurodegeneration (183). AD biomarkers, which include Aβ
plaques, and tau-associated and fluid biomarkers, have been
validated in clinical trials (183), and are currently being used
within therapeutic trials (184).
There are two major categories of AD markers, which are Aβ plaques
and tau-associated neurodegeneration. Furthermore, certain types of
AD models based on imaging measurements and CSF analytes exist
(185–189). Various targeted proteins and
receptors may also be used as markers by inhibiting their
downstream signaling pathways by using antagonists. Aβ and tau
proteins are employed as early markers in the treatment of certain
cognitive disorders, including LOAD, lewy body dementia, mild
cognitive impairment, vascular dementia and frontotemporal lobar
degeneration (190,191).
Studies concerning AD have demonstrated that damage
may occur at various regions of the brain, including the neocortex,
EC, hippocampus, amygdala, nucleus basalis, anterior thalamus and
the corpus callosum within these lobes (201–204). Neuronal damage results in atrophy
of structures in the frontal, temporal and parietal lobes.
Consistent with the heterogeneous symptomatology of AD, damage may
be localized to numerous sites within these lobes. An abnormal
paleness of the ceruleus locus, which contains neuromelanin
neurons, is also considered to be a key feature of AD (205). The neuropathological structures,
NFTs and senile plaques, within affected brain regions of AD are
also considered to be markers. The accumulation of NFTs in the
affected regions following neuronal death causes abnormalities in
structure of the cytoskeleton, which is important for preserving
the cell structure as well as for transportation (206). In addition, the
hyperphosphorylation of tau interrupts axonal transport, which
leads to disturbances in various molecular movements and results in
neuron death (202,207).
The secretases (β- and γ-secretase) cleave APP into
various types of Aβ protein. It has been reported that an elevated
level of BACE1 activity may contribute to the amyloidgenic process
in AD (207,208). Therefore, BACE1 is considered to
be a biomarker for monitoring amyloidogenic APP metabolism in the
CNS (209).
Aβ is the fundamental element of senile plaques,
which is considered to be a common biomarker for AD (210). Depending on the structure of
senile plaques, they are classified as either neuritic or diffuse
plaques (201). Neuritic plaques
have spherical morphology with a periphery of neurites, which may
include axons, astrocytes and microglia, with neighboring dense
amyloid proteins (206). Diffuse
plaques have an amorphous morphological appearance without
neurites. Diffuse plaques may be present in normal aging brain
tissue (211). However, a number
of studies have also reported that diffuse plaques may or may not
be ancestors of neuritic plaques (201,212). Amyloid angiopathy is a generic
term for blood vessel (arteries, veins and capillaries) disease.
Amyloid angiopathy is also considered to be a marker for AD, as it
involves the accumulation of amyloid protein in the cerebral blood
vessels of patients with AD (213–215).
Additional reported biomarkers for the pathogenesis
of neurodegenerative diseases include glucose metabolism, oxidative
free radical damage to mitochondrial DNA, neuroreceptors and
neurotransmitter functional activity (216,217). It has been reported that
decreases in glucose metabolism (218,219) and augmented oxidative free
radical damage (220–222) are responsible for neuronal death
in the temporal and temporoparietal regions of the brain.
Furthermore, changes in the neurotransmitter activity may govern
abnormal types of neuroreceptor responses. Data mining revealed
that a reduced density of nAChR, and serotonin and α2-epinephrine
receptors (217), reduces the
binding of neurotransmitters and may disturb synaptic efficiency.
Any modulator of neurotransmitters, including ACh, serotonin and
GABA, may be considered beneficial in the improvement of cognition
in AD. In addition, other neurochemical markers, including
N-acetylaspartate and myoinositol, have also been reported as
potential treatments for AD (223).
Modeling AD biomarkers becomes more important in
the elderly state, due to the neurodegenerative nature of AD. A
previous study using autopsy demonstrated that the medial temporal
tauopathy may be decreased by two-thirds after the age of 50, and
is present in the majority of individuals >70 years of age
(224). Furthermore, a number of
studies have reported that tauopathy precedes LOAD (224–226). In Aβ deposition, CSF levels of
Aβ42 and amyloid PET scans are highly effective parameters for
biomarkers to accurately identify EOAD (227).
It has been observed that magnetic resonance
imaging (MRI) studies may be considered as quantitative biomarker
measures for AD, on the basis of calculating the values of
AD-signature regions (228). The
summation calculation is primarily performed by an anatomic atlas
that is spatially registered to the subject's imaging study
(228). Additional potential AD
biomarkers that may be employed to investigate the pathology of AD
include visinin-like protein 1, a CSF analyte (229), diffusion and perfusion MRI
(230) and agonist of tau PET
imaging (231). These biomarkers
are reported as novel suggestions and limited experimental data
exists currently.
AD is a slow neurodegenerative disorder in which
pathophysiological irregularities lead to obvious symptoms such as
severe memory loss. This review has demonstrated that mechanistic
gene/receptor-mediated signaling pathways may be used as novel
therapeutic targets to treat cognitive symptoms. To interpret such
receptors and their effects on Aβ, various computational modeling
and simulation approaches have been employed to identify novel
targets for AD. Furthermore, identification of potential biomarkers
may also be considered an important approach prior to the
implementation of in vitro and in vivo experiments.
Therefore, the design of interventional approaches (modeling and
simulations) that target the appropriate molecular pathways in
developmental stages of AD depends upon specific AD biomarkers.
This may improve treatment by allowing individual patients to
receive the most appropriate drug for them in the shortest amount
of time (236–240). However, current AD models have
limitations, which include not explaining the effects of
mechanistic pathways and cytotoxicity. Furthermore, there is no
comprehensive explanation of the ACh neuronal transmission that
leads to AD and other neurodegenerative diseases. Future models
should aim to investigate and explain the molecular mechanisms
underlying the implication of ACh in the development of AD in the
human hippocampus. In addition, drug simulations should also be
addressed to determine their effects on other brain compartments.
Notably a model has already been suggested to explain the
dysfunction of ACh in AD (164).
Finally, drug models may be more helpful if they considered key
knowledge regarding dosage form, targeted receptors and their
associated downstream signaling pathways. Detailed computational
modeling and simulation approaches are essential to understanding
what chemical compounds may be synthesized in order treat or cure
AD.
HA and NZ received financial support from the
United Arab Emirates University (grant no. CIT 31T085).
|
1
|
Hedden T and Gabrieli JD: Insights into
the ageing mind: A view from cognitive neuroscience. Nat Rev
Neurosci. 5:87–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ganguli M: Depression, cognitive
impairment and dementia: Why should clinicians care about the web
of causation? Indian J Psychiatry. 51 Suppl 1:S29–S34.
2009.PubMed/NCBI
|
|
3
|
Tarawneh R and Holtzman DM: The clinical
problem of symptomatic Alzheimer disease and mild cognitive
impairment. Cold Spring Harb Perspect Med. 2:a0061482012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burns A and Iliffe S: Alzheimer's disease.
BMJ. 338:b1582009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mendez MF: Early-onset alzheimer's
disease: Nonamnestic subtypes and type 2 AD. Arch Med Res.
43:677–685. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amaducci LA, Fratiglioni L, Rocca WA,
Fieschi C, Livrea P, Pedone D, Bracco L, Lippi A, Gandolfo C, Bino
G, et al: Risk factors for clinically diagnosed Alzheimer's
disease: A case-control study of an Italian population. Neurology.
36:922–931. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mayeux R: Understanding Alzheimer's
disease: Expect more genes and other things. Ann Neurol.
39:689–690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer's disease. Lancet. 368:387–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Waring SC and Osenberg RN: Genome-wide
association studies in Alzheimer disease. Arch Neurol. 65:329–334.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Selkoe DJ: Translating cell biology into
therapeutic advances in Alzheimer's disease. Nature. 399 6738
Suppl:S23–S31. 2008. View Article : Google Scholar
|
|
11
|
Mahley RW, Weisgraber KH and Huang Y:
Apolipoprotein E4: A causative factor and therapeutic target in
neuropathology, including Alzheimer's disease. Proc Natl Acad Sci
USA. 103:5644–5651. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strittmatter WJ, Saunders AM, Schmechel D,
Pericak-Vance M, Enghild J, Salvesen GS and Roses AD:
Apolipoprotein E: High-avidity binding to beta-amyloid and
increased frequency of type 4 allele in late-onset familial
Alzheimer disease. Proc Natl Acad Sci USA. 90:1977–1981. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertram L and Tanzi ER: Genome-wide
association studies in Alzheimer's disease. Hum Mol Genet.
18:R137–R145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Killin LO, Starr JM, Shiue IJ and Russ TC:
Environmental risk factors for dementia: A systematic review. BMC
Geriatr. 16:1752016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dosunmu R, Wu J, Basha MR and Zawia NH:
Environmental and dietary risk factors in Alzheimer's disease.
Expert Rev Neurother. 7:887–900. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wenk GL: Neuropathologic changes in
Alzheimer's disease. J Clin Psychiatry. 64 Suppl 9:S7–S10.
2003.
|
|
17
|
Tiraboschi P, Sabbagh MN, Hansen LA,
Salmon DP, Merdes A, Gamst A, Masliah E, Alford M, Thal LJ and
Corey-Bloom J: Alzheimer disease without neocortical
neurofibrillary tangles: ‘A second look’. Neurology. 62:1141–1147.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Priller C, Bauer T, Mitteregger G, Krebs
B, Kretzschmar HA and Herms J: Synapse formation and function is
modulated by the amyloid precursor protein. J Neurosci.
26:7212–7221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Turner PR, O'Connor K, Tate WP and Abraham
WC: Roles of amyloid precursor protein and its fragments in
regulating neural activity, plasticity and memory. Prog Neurobiol.
70:1–32. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hooper NM: Roles of proteolysis and lipid
rafts in the processing of the amyloid precursor protein and prion
protein. Biochem Soc Trans. 33:335–338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ohnishi S and Takano K: Amyloid fibrils
from the viewpoint of protein folding. Cell Mol Life Sci.
61:511–524. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hooper C, Killick R and Lovestone S: The
GSK3 hypothesis of Alzheimer's disease. J Neurochem. 104:1433–1439.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jope RS, Yuskaitis CJ and Beurel E:
Glycogen synthase kinase-3 (GSK3): Inflammation, diseases and
therapeutics. Neurochem Res. 32:577–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paterson D and Nordberg A: Neuronal
nicotinic receptors in the human brain. Prog Neurobiol. 61:75–111.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clader JW and Wang Y: Muscarinic receptor
agonists and antagonists in the treatment of Alzheimer's disease.
Curr Pharm Des. 11:3353–3361. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang S, Li Y, Zhang C, Zhao Y, Bu G, Xu H
and Zhang YW: M1 muscarinic acetylcholine receptor in Alzheimer's
disease. Neurosci Bull. 30:295–307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsang SW, Lai MK, Kirvell S, Francis PT,
Esiri MM, Hope T, Chen CP and Wong PT: Impaired coupling of
muscarinic M1 receptors to G-proteins in the neocortex is
associated with severity of dementia in Alzheimer's disease.
Neurobiol Aging. 27:1216–1223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones CK, Brady AE, Davis AA, Xiang Z,
Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR,
et al: Novel selective allosteric activator of the M1 muscarinic
acetylcholine receptor regulates amyloid processing and produces
antipsychotic-like activity in rats. J Neurosci. 28:10422–10433.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Poulin B, Butcher A, McWilliams P,
Bourgognon JM, Pawlak R, Kong KC, Bottrill A, Mistry S, Wess J,
Rosethorne EM, et al: The M3-muscarinic receptor regulates learning
and memory in a receptor phosphorylation/arrestin-dependent manner.
Proc Natl Acad Sci USA. 107:9440–9445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wevers A and Schröder H: Nicotinic
acetylcholine receptors in Alzheimer's disease. J Alzheimers Dis.
1:207–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rinne JO, Myllykylä T, Lönnberg P and
Marjamäki P: A Postmortem study of brain nicotinic receptors in
Parkinson's and Alzheimer's disease. Brain Res. 547:167–170. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Young JW, Meves JM, Tarantino IS, Caldwell
S and Geyer MA: Delayed procedural learning in α7-nicotinic
acetylcholine receptor knockout mice. Genes Brain Behav.
10:720–733. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dziewczapolski G, Glogowski CM, Masliah E
and Heinemann SF: Deletion of the alpha 7 nicotinic acetylcholine
receptor gene improves cognitive deficits and synaptic pathology in
a mouse model of Alzheimer's disease. J Neurosci. 29:8805–8815.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Wang H, Zhang Z, Li Z, He D,
Sokabe M and Chen L: DMXB (GTS-21) ameliorates the cognitive
deficits in beta amyloid (25–35(−)) injected mice through
preventing the dysfunction of alpha7 nicotinic receptor. J Neurosci
Res. 88:1784–1794. 2010.PubMed/NCBI
|
|
36
|
Faghih R, Gfesser GA and Gopalakrishnan M:
Advances in the discovery of novel positive allosteric modulators
of the alpha7 nicotinic acetylcholine receptor. Recent Pat CNS Drug
Discov. 2:99–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roncarati R, Scali C, Comery TA, Grauer
SM, Aschmi S, Bothmann H, Jow B, Kowal D, Gianfriddo M, Kelley C,
et al: Procognitive and neuroprotective activity of a novel alpha7
nicotinic acetylcholine receptor agonist for treatment of
neurodegenerative and cognitive disorders. J Pharmacol Exp Ther.
329:459–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gubbins EJ, Gopalakrishnan M and Li J:
Alpha7 nAChR-mediated activation of MAP kinase pathways in PC12
cells. Brain Res. 1328:1–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miwa JM, Stevens TR, King SL, Caldarone
BJ, Ibanez-Tallon I, Xiao C, Fitzsimonds RM, Pavlides C, Lester HA,
Picciotto MR and Heintz N: The prototoxin lynx1 acts on nicotinic
acetylcholine receptors to balance neuronal activity and survival
in vivo. Neuron. 51:587–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Turner TJ: Nicotine enhancement of
dopamine release by a calcium-dependent increase in the size of the
readily releasable pool of synaptic vesicles. J Neurosci.
24:11328–11336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen JX and Yakel JL: Nicotinic
acetylcholine receptor-mediated calcium signaling in the nervous
system. Acta Pharmacol Sin. 30:673–680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bitner RS, Bunnelle WH, Anderson DJ,
Briggs CA, Buccafusco J, Curzon P, Decker MW, Frost JM, Gronlien
JH, Gubbins E, et al: Broad-spectrum efficacy across cognitive
domains by alpha7 nicotinic acetylcholine receptor agonism
correlates with activation of ERK1/2 and CREB phosphorylation
pathways. J Neurosci. 27:10578–10587. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang KT and Berg DK: Voltage-gated
channels block nicotinic regulation of CREB phosphorylation and
gene expression in neurons. Neuron. 32:855–865. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hu M, Liu QS, Chang KT and Berg DK:
Nicotinic regulation of CREB activation in hippocampal neurons by
glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci.
21:616–625. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ji D, Lape R and Dani JA: Timing and
location of nicotinic activity enhances or depresses hippocampal
synaptic plasticity. Neuron. 31:131–141. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Auld DS, Kornecook TJ, Bastianetto S and
Quirion R: Alzheimer's disease and the basal forebrain cholinergic
system: Relations to beta-amyloid peptides, cognition, and
treatment strategies. Prog Neurobiol. 68:209–245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lilja AM, Porras O, Storelli E, Nordberg A
and Marutle A: Functional interactions of fibrillar and oligomeric
amyloid-β with alpha7 nicotinic receptors in Alzheimer's disease. J
Alzheimers Dis. 23:335–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Claeysen S, Bockaert J and Giannoni P:
Serotonin: A new hope in alzheimer's disease? ACS Chem Neurosci.
6:940–943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Geldenhuys WJ and Van der Schyf CJ: Role
of serotonin in Alzheimer's disease: A new therapeutic target? CNS
Drugs. 25:765–781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu Y, Yan J, Zhou P, Li J, Gao H, Xia Y
and Wang Q: Neurotransmitter receptors and cognitive dysfunction in
Alzheimer's disease and Parkinson's disease. Prog Neurobiol.
97:1–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li Y, Huang XF, Deng C, Meyer B, Wu A, Yu
Y, Ying W, Yang GY, Yenari MA and Wang Q: Alterations in 5-HT2A
receptor binding in various brain regions among
6-hydroxydopamine-induced Parkinsonian rats. Synapse. 3:224–230.
2010. View Article : Google Scholar
|
|
52
|
Polter AM and Li X: 5-HT1A
receptor-regulated signal transduction pathways in brain. Cell
Signal. 22:1406–1412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sumiyoshi T, Park S, Jayathilake K, Roy A,
Ertugrul A and Meltzer HY: Effect of buspirone, a serotonin1A
partial agonist, on cognitive function in schizophrenia: A
randomized, double-blind, placebo-controlled study. Schizophr Res.
95:158–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lai MK, Tsang SW, Francis PT, Keene J,
Hope T, Esiri MM, Spence I and Chen CP: Postmortem serotoninergic
correlates of cognitive decline in Alzheimer's disease.
Neuroreport. 13:1175–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Garcia-Alloza M, Hirst WD, Chen CP,
Lasheras B, Francis PT and Ramírez MJ: Differential involvement of
5-HT(1B/1D) and 5-HT6 receptors in cognitive and non-cognitive
symptoms in Alzheimer's disease. Neuropsychopharmacology.
29:410–416. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Blin J, Baron JC, Dubois B, Crouzel C,
Fiorelli M, Attar-Lévy D, Pillon B, Fournier D, Vidailhet M and
Agid Y: Loss of brain 5-HT2 receptors in Alzheimer's disease. In
vivo assessment with positron emission tomography and
[18F]setoperone. Brain. 116:497–510. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hasselbalch SG, Madsen K, Svarer C,
Pinborg LH, Holm S, Paulson OB, Waldemar G and Knudsen GM: Reduced
5-HT2A receptor binding in patients with mild cognitive impairment.
Neurobiol Aging. 29:1830–1838. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lai MK, Tsang SW, Alder JT, Keene J, Hope
T, Esiri MM, Francis PT and Chen CP: Loss of serotonin 5HT2A
receptors in the postmortem temporal cortex correlates with rate of
cognitive decline in Alzheimer's disease. Psychopharmacology
(Berl). 179:673–677. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ramírez MJ: 5-HT6 receptors and
Alzheimer's disease. Alzheimers Res Ther. 5:152013.PubMed/NCBI
|
|
60
|
Ruat M, Traiffort E, Arrang JM,
Tardivel-Lacombe J, Diaz J, Leurs R and Schwartz JC: A novel rat
serotonin (5-HT6) receptor: Molecular cloning, localization and
stimulation of cAMP accumulation. Biochem Biophys Res Commun.
193:268–276. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mitchell ES and Neumaier JF: 5-HT6
receptors: A novel target for cognitive enhancement. Pharmacol
Ther. 108:320–333. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Perez-García G and Meneses A: Oral
administration of the 5-HT6 receptor antagonists SB-357134 and
SB-399885 improves memory formation in an autoshaping learning
task. Phar Biochem Behav. 81:673–682. 2005. View Article : Google Scholar
|
|
63
|
Da Silva Costa V, Duchatelle P, Boulouard
M and Dauphin F: Selective 5-HT6 receptor blockade improves spatial
recognition memory and reverses age-related deficits in spatial
recognition memory in the mouse. Neuropsychopharmacology.
34:488–500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
West PJ, Marcy VR, Marino MJ and
Schaffhauser H: Activation of the 5-HT(6) receptor attenuates
longterm potentiation and facilitates GABAergic neurotransmission
in rat hippocampus. Neuroscience. 164:692–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang G and Stackman RW Jr: The role of
serotonin 5-HT2A receptors in memory and cognition. Front
Pharmacol. 6:2252015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yun HM and Rhim H: The serotonin-6
receptor as a novel therapeutic target. Exp Neurobiol. 20:159–168.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nichols DE and Nichols CD: Serotonin
receptors. Chem Rev. 108:1614–1641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hirst WD, Stean TO, Rogers DC, Sunter D,
Pugh P, Moss SF, Bromidge SM, Riley G, Smith DR, Bartlett S, et al:
SB-399885 is a potent, selective 5-HT6 receptor antagonist with
cognitive enhancing properties in aged rat water maze and novel
object recognition models. Eur J Pharmacol. 553:109–119. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Schechter LE, Lin Q, Smith DL, Zhang G,
Shan Q, Platt B, Brandt MR, Dawson LA, Cole D, Bernotas R, et al:
Neuropharmacological profile of novel and selective 5-HT6 receptor
agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology.
33:1323–1335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Laureys G, Clinckers R, Gerlo S, Spooren
A, Wilczak N, Kooijman R, Smolders I, Michotte Y and De Keyser J:
Astrocytic beta(2)-adrenergic receptors: From physiology to
pathology. Prog Neurobiol. 91:189–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shimohama S, Taniguchi T, Fujiwara M and
Kameyama M: Biochemical characterization of alphaadrenergic
receptors in human brain and changes in Alzheimer-type dementia. J
Neurochem. 47:1295–1301. 1986.PubMed/NCBI
|
|
72
|
Kalaria RN and Harik SI: Increased alpha
2- and beta 2-adrenergic receptors in cerebral microvessels in
Alzheimer disease. Neurosci Lett. 106:233–238. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Russo-Neustadt A and Cotman CW: Adrenergic
receptors in Alzheimer's disease brain: Selective increases in the
cerebella of aggressive patients. J Neurosci. 17:5573–5580. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Contreras F, Fouillioux C, Bolívar A,
Simonovis N, Hernández-Hernández R, Armas-Hernandez MJ and Velasco
M: Dopamine, hypertension and obesity. J Hum Hypertens. 16 Suppl
1:S13–S17. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nilsson A, Eriksson M, Muly EC, Akesson E,
Samuelsson EB, Bogdanovic N, Benedikz E and Sundström E: Analysis
of NR3A receptor subunits in human native NMDA receptors. Brain
Res. 1186:102–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Janssen WG, Vissavajjhala P, Andrews G,
Moran T, Hof PR and Morrison JH: Cellular and synaptic distribution
of NR2A and NR2B in macaque monkey and rat hippocampus as
visualized with subunit-specific monoclonal antibodies. Exp Neurol.
191 Suppl 1:S28–S44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Proctor DT, Coulson EJ and Dodd PR:
Post-synaptic scaffolding protein interactions with glutamate
receptors in synaptic dysfunction and Alzheimer's disease. Prog
Neurobiol. 93:509–521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sun H, Zhang J, Zhang L, Liu H, Zhu H and
Yang Y: Environmental enrichment influences BDNF and NR1 levels in
the hippocampus and restores cognitive impairment in chronic
cerebral hypoperfused rats. Curr Neurovasc Res. 7:268–280. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Amadoro G, Ciotti MT, Costanzi M, Cestari
V, Calissano P and Canu N: NMDA receptor mediates tau-induced
neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad
Sci USA. 103:2892–2897. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fortin DA, Davare MA, Srivastava T, Brady
JD, Nygaard S, Derkach VA and Soderling TR: Long-term
potentiation-dependent spine enlargement requires synaptic
Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J
Neurosci. 30:11565–11575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guetg N, Aziz Abdel S, Holbro N, Turecek
R, Rose T, Seddik R, Gassmann M, Moes S, Jenoe P, Oertner TG, et
al: NMDA receptor-dependent GABAB receptor internalization via
CaMKII phosphorylation of serine 867 in GABAB1. Proc Natl Acad Sci
USA. 107:13924–13929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Silva T, Reis J, Teixeira J and Borges F:
Alzheimer's disease, enzyme targets and drug discovery struggles:
From natural products to drug prototypes. Ageing Res Rev.
15:116–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Colović MB, Krstić DZ, Lazarević-Pašti TD,
Bondžić AM and Vasić VM: Acetylcholinesterase Inhibitors:
Pharmacology and Toxicology. Curr Neuropharmacol. 11:315–335. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
de Almeida JP and Saldanha C: Nonneuronal
cholinergic system in human erythrocytes: Biological role and
clinical relevance. J Membr Biol. 234:227–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Massoulié J, Pezzementi L, Bon S, Krejci E
and Vallette FM: Molecular and cellular biology of cholinesterases.
Prog Neurobiol. 41:31–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Schliebs R and Arendt T: The significance
of the cholinergic system in the brain during aging and in
Alzheimer's disease. J Neural Transm (Vienna). 113:1625–1644. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Schliebs R and Arendt T: The cholinergic
system in aging and neuronal degeneration. Behav Brain Res.
221:555–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Greig NH, Lahiri DK and Sambamurti K:
Butyrylcholinesterase: An important new target in Alzheimer's
disease therapy. Int Psychogeriatr. 14 Suppl 1:S77–S91. 2002.
View Article : Google Scholar
|
|
89
|
Lane RM, Kivipelto M and Greig NH:
Acetylcholinesterase and its inhibition in Alzheimer disease. Clin
Neuropharmacol. 27:141–149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lane RM, Potkin SG and Enz A: Targeting
acetylcholinesterase and butyrylcholinesterase in dementia. Int J
Neuropsychopharmacol. 9:101–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Grossberg GT: Cholinesterase Inhibitors
for the treatment of alzheimer's disease: Getting on and staying
on. Curr Ther Res Clin Exp. 64:216–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Francis PT, Palmer AM, Snape M and Wilcock
GK: The cholinergic hypothesis of Alzheimer's disease: A review of
progress. J Neurol Neurosurg Psychiatry. 66:137–147. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Prakash A, Kalra J, Mani V, Ramasamy K and
Majeed AB: Pharmacological approaches for Alzheimer's disease:
Neurotransmitter as drug targets. Expert Rev Neurother. 15:53–71.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Akasofu S, Kimura M, Kosasa T, Sawada K
and Ogura H: Study of neuroprotection of donepezil, a therapy for
Alzheimer's disease. Chem Biol Interact. 175:222–226. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Takada Y, Yonezawa A, Kume T, Katsuki H,
Kaneko S, Sugimoto H and Akaike A: Nicotinic acetycholine
receptor-mediated neuroprotection by donepezil against glutamate
neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther.
306:772–777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Farlow M, Veloso F, Moline M, Yardley J,
Brand-Schieber E, Bibbiani F, Zou H, Hsu T and Satlin A: Safety and
tolerability of donepezil 23 mg in moderate to severe Alzheimer's
disease. BMC Neurol. 11:572011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Barar FSK: Essentials of
Pharmacotherapeutics, Antiparkinsonian drugs. 4th edition. S. Chand
and Company Ltd.; New Delhi: pp. 1692007
|
|
98
|
Bai DL, Tang XC and He XC: Huperzine A, a
potential therapeutic agent for treatment of Alzheimer's disease.
Curr Med Chem. 7:355–374. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yan J, Sun L, Wu G, Yi P, Yang F, Zhou L,
Zhang X, Li Z, Yang X, Luo H and Qiu M: Rational design and
synthesis of highly potent antiacetylcholinesterase activity
huperzine A derivatives. Bioorg Med Chem. 17:6937–6941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li J, Wu HM, Zhou RL, Liu GJ and Dong BR:
Huperzine A for Alzheimer's disease. Cochrane Database Syst Rev.
16:CD0055922008.
|
|
101
|
Camps PE, El Achab R, Morral J,
Muñoz-Torrero D, Badia A, Baños JE, Vivas NM, Barril X, Orozco M
and Luque FJ: New tacrine-huperzine A hybrids (huprines): Highly
potent tight-binding acetylcholinesterase inhibitors of interest
for the treatment of Alzheimer's disease. J Med Chem. 43:4657–4666.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mehta M, Adem A and Sabbagh M: New
Acetylcholinesterase Inhibitors for Alzheimer's Disease. Int J
Alzheimers Dis. 2012:7289832012.PubMed/NCBI
|
|
103
|
Feng S, Wang Z, He X, Zheng S, Xia Y,
Jiang H, Tang X and Bai D: Bis-huperzine B: Highly potent and
selective acetylcholinesterase inhibitors. J Med Chem. 48:655–657.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Jung HA, Min BS, Yokozawa T, Lee JH, Kim
YS and Choi JS: Anti-Alzheimer and antioxidant activities of
Coptidis Rhizoma alkaloids. Biol Pharm Bull. 32:1433–1438. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kulkarni SK and Dhir A: Berberine: A plant
alkaloid with therapeutic potential for central nervous system
disorders. Phytother Res. 24:317–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Turner AJ, Fisk L and Nalivaeva NN:
Targeting amyloid-degrading enzymes as therapeutic strategies in
neurodegeneration. Ann N Y Acad Sci. 1035:1–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ghosh AK, Gemma S and Tang J:
Beta-Secretase as a therapeutic target for Alzheimer's disease.
Neurotherapeutics. 5:399–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sathya M, Premkumar P, Karthick C, Moorthi
P, Jayachandran KS and Anusuyadevi M: BACE1 in Alzheimer's disease.
Clin Chim Acta. 414:171–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Asai M, Hattori C, Iwata N, Saido TC,
Sasagawa N, Szabó B, Hashimoto Y, Maruyama K, Tanuma S, Kiso Y and
Ishiura S: The novel beta-secretase inhibitor KMI-429 reduces
amyloid beta peptide production in amyloid precursor protein
transgenic and wild-type mice. J Neurochem. 96:533–540. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Luo X and Yan R: Inhibition of BACE1 for
therapeutic use in Alzheimer's disease. Int J Clin Exp Pathol.
3:618–628. 2010.PubMed/NCBI
|
|
111
|
Hussain I, Hawkins J, Harrison D, Hille C,
Wayne G, Cutler L, Buck T, Walter D, Demont E, Howes C, et al: Oral
administration of a potent and selective non peptidic BACE-1
inhibitor decreases beta-cleavage of amyloid precursor protein and
amyloid-beta production in vivo. J Neurochem. 100:802–809. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Iserloh U, Pan J, Stamford AW, Kennedy ME,
Zhang Q, Zhang L, Parker EM, McHugh NA, Favreau L, Strickland C and
Voigt J: Discovery of an orally efficaceous
4-phenoxypyrrolidine-based BACE-1 inhibitor. Bioorg Med Chem Lett.
18:418–422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chang WP, Huang X, Downs D, Cirrito JR,
Koelsch G, Holtzman DM, Ghosh AK and Tang J: Beta-secretase
inhibitor GRL-8234 rescues age-related cognitive decline in APP
transgenic mice. FASEB J. 25:775–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mangialasche F, Solomon A, Winblad B,
Mecocci P and Kivipelto M: Alzheimer's disease: Clinical trials and
drug development. Lancet Neurol. 9:702–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dovey HF, John V, Anderson JP, Chen LZ, de
Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E,
Holsztynska EJ, et al: Functional gamma-secretase inhibitors reduce
beta-amyloid peptide levels in brain. J Neurochem. 76:173–181.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wolfe MS: Inhibition and modulation of
gamma-secretase for Alzheimer's disease. Neurotherapeutics.
5:391–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lanz TA, Himes CS, Pallante G, Adams L,
Yamazaki S, Amore B and Merchant KM: The gamma-secretase inhibitor
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl
ester reduces A beta levels in vivo in plasma and cerebrospinal
fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice.
J Pharmacol Exp Ther. 305:864–871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Imbimbo BP: Alzheimer's disease:
γ-secretase inhibitors. Drug Discov Today. 5:169–175. 2008.
|
|
119
|
De Strooper B, Vassar R and Golde T: The
secretases: Enzymes with therapeutic potential in Alzheimer
disease. Nat Rev Neurol. 6:99–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Panza F, Frisardi V, Imbimbo BP, Capurso
C, Logroscino G, Sancarlo D, Seripa D, Vendemiale G, Pilotto A and
Solfrizzi V: REVIEW: γ -Secretase inhibitors for the treatment of
alzheimer's disease: The current state. CNS Neurosci Ther.
16:272–284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Martone RL, Zho H, Atchison K, Comery T,
Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, et al:
Begacestat (GSI-953): A novel, selective thiophene sulphonamide
inhibitor of amyloid precursor protein gamma-secretase for the
treatment of Alzheimer's disease. J Pharmacol Exp Ther.
331:598–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Hopkins CR: ACS chemical neuroscience
molecule spotlight on begacestat (GSI-953). ACS Chem Neurosci.
3:3–4. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Han SH and Mook-Jung I: Diverse molecular
targets for terapeutic strategies in alzheimer's disease. J Korean
Med Sci. 29:893–902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Desire L, Marcade M, Peillon H, Drouin D,
Sol O and Pando M: Clinical trials of EHT 0202, a neuroprotective
and procognitive alpha-secretase stimulator for Alzheimer's
disease. Alzheimers Dement. 5:P255–P256. 2009. View Article : Google Scholar
|
|
125
|
Snow AD, Cummings J, Lake T, Hu Q,
Esposito L, Cam J, Hudson M, Smith E and Runnels S: Exebryl-1: A
novel small molecule currently in human clinical trials as a
disease-modifying drug for the treatment of Alzheimer's disease.
Alzheimer's Dement. 5:P4182009. View Article : Google Scholar
|
|
126
|
Hu S, Begum AN, Jones MR, Oh MS, Beech WK,
Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, et al: GSK3 inhibitors
show benefits in an Alzheimer's disease (AD) model of
neurodegeneration but adverse effects in control animals. Neurobiol
Dis. 33:193–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Serrano-Pozo A, Frosch MP, Masliah E and
Hyman BT: Neuropathological alterations in alzheimer disease. Cold
Spring Harb Perspect Med. 1:a0061892011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Harper JD and Lansbury PT Jr: Models of
amyloid seeding in Alzheimer's disease and scrapie: Mechanistic
truths and physiological consequences of the time-dependent
solubility of amyloid proteins. Annu Rev Biochem. 66:385–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Inouye H and Kirschner DA: A beta
fibrillogenesis: Kinetic parameters for fibril formation from congo
red binding. J Struct Biol. 130:123–129. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Jarrett JT, Berger EP and Lansbury PT: The
carboxy terminus of the beta amyloid protein is critical for the
seeding of amyloid formation: Implications for the pathogenesis of
Alzheimer's disease. Biochemistry. 32:4693–4697. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kim JR, Lee Muresan KY and Murphy RM: Urea
modulation of beta-amyloid fibril growth: Experimental studies and
kinetic models. Protein Sci. 13:2888–2898. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Lomakin A, Chung DS, Benedek GB, Kirschner
DA and Teplow DB: On the nucleation and growth of amyloid
beta-protein fibrils: Detection of nuclei and quantitation of rate
constants. Proc Natl Acad Sci USA. 93:1125–1129. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lomakin A, Teplow DB, Kirschner DA and
Benedek GB: Kinetic theory of fibrillogenesis of amyloid
beta-protein. Proc Natl Acad Sci USA. 94:7942–7947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
McLaurin J, Franklin T, Zhang X, Deng J
and Fraser PE: Interactions of Alzheimer amyloid-beta peptides with
glycosaminoglycans effects on fibril nucleation and growth. Eur J
Biochem. 266:1101–1110. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Murphy RM and Pallitto MM: Probing the
kinetics of beta-amyloid self-association. J Struct Biol.
130:109–122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Naiki H and Nakakuki K: First-order
kinetic model of Alzheimer's beta-amyloid fibril extension in
vitro. Lab Invest. 74:374–383. 1996.PubMed/NCBI
|
|
137
|
Tomski SJ and Murphy RM: Kinetics of
aggregation of synthetic beta-amyloid peptide. Arch Biochem
Biophys. 294:630–638. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Walsh DM, Lomakin A, Benedek GB, Condron
MM and Teplow DB: Amyloid beta-protein fibrillogenesis: Detection
of a protofibrillar intermediate. J Biol Chem. 272:22364–22372.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Harper JD, Wong SS, Lieber CM and Lansbury
PT Jr: Assembly of A beta amyloid protofibrils: An in vitro model
for a possible early event in Alzheimer's disease. Biochemistry.
38:8972–8980. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Pallitto MM and Murphy RM: A mathematical
model of the kinetics of beta-amyloid fibril growth from the
denaturated state. Biophys J. 81:1805–1822. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Barrow CJ, Yasuda A, Kenny PT and Zagorski
MG: Solution conformations and aggregational properties of
synthetic amyloid beta peptides of Alzheimer's disease. analysis of
circular dichroism spectra. J Mol Biol. 225:1075–1093. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Cruz L, Urbanc B, Buldyrev SV, Christie R,
Gómez-Isla T, Havlin S, McNamara M, Stanley HE and Hyman BT:
Aggregation and disaggregation of senile plaques in Alzheimer
disease. Proc Natl Acad Sci USA. 94:7612–7616. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Urbanc B, Cruz L, Buldyrev SV, Havlin S,
Hyman BT and Stanley HE: Dynamic feedback in an
aggregation-disaggregation model. Phys Rev E Stat Phys Plasmas
Fluids Relat Interdiscip Topics. 60:2120–2126. 1999.PubMed/NCBI
|
|
144
|
De Caluwé J and Dupont G: The progression
towards Alzheimer's disease described as a bistable switch arising
from the positive loop between amyloids and Ca(2+). J Theor Biol.
331:12–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ortega F, Stott J, Visser S and Bendtsen
C: Interplay between α-, β-, and γ-secretases determines biphasic
amyloid-β protein level in the presence of γ-secretases inhibitor.
J Biol Chem. 288:785–792. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Schmidt V, Baum K, Lao A, Rateitschak K,
Schmitz Y, Teichmann A, Wiesner B, Petersen CM, Nykjaer A, Wolf J,
et al: Quantative modelling of amyloidogenic processing and its
influence by SORLA in Alzheimer's disease. EMBO J. 31:187–200.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Guardia-Laguarta C, Pera M and Lleó A:
Gamma-Secretase as a therapeutic target in Alzheimer's disease.
Curr Drug Targets. 11:506–517. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Anastasio TJ: Data driven modelling of
Alzheimer's disease pathogenesis. J Theor Biol. 290:60–72. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Anastasio TJ: Exploring the contribution
of estrogen to amyloid-beta regulation: A novel multifactorial
computational modelling approach. Front Pharmacol. 4:162013.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Anastasio TJ: Computational identification
of potential multitarget treatments for ameliorating the adverse
effects of amyloid-β on synaptic plasticity. Front Pharmacol.
5:852014. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Craft DL, Wein LM and Selkoe DJ: A
mathematical model of the impact of novel treatments on the A beta
burden in the Alzheimer's brain, CSF and plasma. Bull Math Biol.
64:1011–1031. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Proctor CJ and Gray DA: GSK3 and p53-is
there a link in Alzheimer's disease? Mol Neurodegener. 5:72010.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Diem AK, Tan M, Bressloff NW, Hawkes C,
Morris AW, Weller RO and Carare RO: A simulation model of
periarterial clearance of amyloid-β from the brain. Front Aging
Neurosci. 8:182016. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Proctor CJ, Boche D, Gray DA and Nicoll
JA: Investigating interventions in alzheimer's disease with
computer simulation models. PLoS ONE. 8:e736312013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Kyrtsos CR and Baras JS: Studying the role
of ApoE in Alzheimer's disease pathogenesis using a systems biology
model. J Bioinform Comput Biol. 11:13420032013. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Chen C: beta-Amyloid increases dendritic
Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1
pyramidal neurons. Biochem Biophys Res Commun. 338:1913–1919. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Good TA and Murphy RM: Effect of
beta-amyloid block of the fast-inactivating K+ channel on
intracellular Ca2+ and excitability in a modeled neuron. Proc Natl
Acad Sci USA. 93:15130–15135. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hoffman DA, Magee JC, Colbert CM and
Johnston D: K+ channel regulation of signal propagation in
dendrites of hippocampal pyramidal neurons. Nature. 387:869–875.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Culmone V and Migliore M: Progressive
effect of beta amyloid peptides accumulation on CA1 pyramidal
neurons: A model study suggesting possible treatments. Front Comp
Neurosci. 6:522012.
|
|
160
|
Wilson RS, Boyle PA, Yu L, Barnes LL,
Schneider JA and Bennett DA: Life-span cognitive activity,
neuropathologic burden, and cognitive aging. Neurology. 81:314–321.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Abramov E, Dolev I, Fogel H, Ciccotosto
GD, Ruff E and Slutsky I: Amyloid-beta as a positive endogenous
regulator of release probability at hippocampal synapses. Nat
Neurosci. 12:1567–1576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Parodi J, Sepulveda FJ, Roa J, Opazo C,
Inestrosa NC and Aguayo LG: Beta-amyloid causes depletion of
synaptic vesicles leading to neurotransmission failure. J Biol
Chem. 285:2506–2514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Romani A, Marchetti C, Bianchi D,
Leinekugel X, Poirazi P, Migliore M and Marie H: Computational
modeling of the effects of amyloid-beta on release probability at
hippocampal synapses. Front Comp Neurosci. 7:12013.
|
|
164
|
Hasselmo ME and Wyble BP: Free recall and
recognition in a network model of the hippocampus: Simulating
effects of scopolamine on human memory function. Behav Brain Res.
89:1–34. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Menschik ED and Finkel LH: Neuromodulatory
control hippocampal function: Towards a model of Alzheimer's
disease. Artif Intell Med. 13:99–121. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Buzsáki G: Two-stage model of memory trace
formation: A role for noisy brain states. Neuroscience. 31:551–570.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Buzsáki G and Chrobak JJ: Temporal
structure in spatially organized neuronal ensembles: A role for
interneuronal networks. Curr Opin Neurobiol. 5:504–510. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Lisman JE and Idiart MA: Storage of 7 +/-
2 short-term memories in oscillatory subcycles. Science.
267:1512–1515. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Lisman J: The theta/gamma discrete phase
code occurring during the hippocampal phase precession may be a
more general brain coding scheme. Hippocampus. 15:913–922. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Roberts PD, Spiros A and Geerts H:
Simulations of symptomatic treatments for Alzheimer's disease:
Computational analysis of pathology and mechanisms of drug action.
Alzheimers Res Ther. 4:502012. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Bianchi D, De Michele P, Marchetti C,
Tirozzi B, Cuomo S, Marie H and Migliore M: Effects of increasing
CREB-dependent transcription on the storage and recall processes in
a hippocampal CA1 microcircuit. Hippocampus. 24:165–177. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Cutsuridis V, Cobb S and Graham BP:
Encoding and retrieval in the hippocampal CA1 microcircuit model.
Hippocampus. 20:423–446. 2010.PubMed/NCBI
|
|
173
|
Horn D, Ruppin E, Usher M and Hermann M:
Neural network modeling of memory deterioration in Alzheimer's
disease. Neural Comput. 5:736–749. 1993. View Article : Google Scholar
|
|
174
|
Ruppin E and Reggia JA: A neural model of
memory impairment in diffuse cerebral atrophy. Br J Psychiatry.
166:19–28. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hasselmo ME: Runaway synaptic modification
in models of cortex: Implications for Alzheimer's disease. Neural
Netw. 7:13–40. 1994. View Article : Google Scholar
|
|
176
|
Hasselmo ME: A computational model of the
progression of Alzheimer's disease. MD Comput. 14:181–191.
1997.PubMed/NCBI
|
|
177
|
Siegle GJ and Hasselmo ME: Using
connectionist models to guide assessment of psychological disorder.
Psychol Assess. 14:263–278. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Bhattacharya BS, Coyle D and Maguire LP: A
thalamo-cortico-thalamic neural mass model to study alpha rhythms
in Alzheimer's Disease. Neural Netw. 24:631–645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Gluck MA, Myers CE, Nicolle MM and Johnson
S: Computational models of the hippocampal region: Implications for
prediction of risk for Alzheimer's disease in non-demented elderly.
Curr Alzheimer Res. 3:247–257. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Moustafa AA, Keri S, Herzallah MM, Myers
CE and Gluck MA: A neural model of hippocampal-striatal
interactions in associative learning and transfer generalization in
various neurological and psychiatric patients. Brain Cogn.
74:132–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
McAuley MT, Kenny RA, Kirkwood TB,
Wilkinson DJ, Jones JJ and Miller VM: A mathematical model of
aging-related and cortisol induced hippocampal dysfunction. BMC
Neurosci. 10:262009. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Jack CR Jr and Holtzman DM: Biomarker
modeling of Alzheimer's disease. Neuron. 80:1347–1358. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Mayeux R: Biomarkers: Potential uses and
limitations. NeuroRx. 1:182–188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Blennow K, Hampel H and Zetterberg H:
Biomarkers in amyloid-β immunotherapy trials in Alzheimer's
disease. Neuropsychopharmacology. 39:189–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Blennow K, Zetterberg H and Fagan AM:
Fluid biomarkers in alzheimer disease. Cold Spring Harb Perspect
Med. 2:a0062212012. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Albert MS, DeKosky ST, Dickson D, Dubois
B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen
RC, et al: The diagnosis of mild cognitive impairment due to
Alzheimer's disease: Recommendations from the national institute on
aging-alzheimer's association workgroups on diagnostic guidelines
for alzheimer's disease. Alzheimers Dement. 7:270–279. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Dubois B, Feldman HH, Jacova C, Cummings
JL, Dekosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox
NC, Galasko D, et al: Revising the definition of Alzheimer's
disease: A new lexicon. Lancet Neurol. 9:1118–1127. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Jack CR Jr, Albert MS, Knopman DS, McKhann
GM, Sperling RA, Carrillo MC, Thies B and Phelps CH: Introduction
to the recommendations from the national institute on
aging-alzheimer's association workgroups on diagnostic guidelines
for alzheimer's disease. Alzheimers Dement. 7:257–262. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
189
|
McKhann GM, Knopman DS, Chertkow H, Hyman
BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux
R, et al: The diagnosis of dementia due to Alzheimer's disease:
Recommendations from the national institute on aging-alzheimer's
association workgroups on diagnostic guidelines for alzheimer's
disease. Alzheimers Dement. 7:263–269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Castro-Chavira SA, Fernandez T, Nicolini
H, Diaz-Cintra S and Prado-Alcala RA: Genetic markers in biological
fluids for aging-related major neurocognitive disorder. Curr
Alzheimer Res. 12:200–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Sonnen JA, Montine KS, Quinn JF, Kaye JA,
Breitner JCS and Montine TJ: Biomarkers for cognitive impairment
and dementia in elderly people. Lancet Neurol. 7:704–714. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Dekkers MP, Nikoletopoulou V and Barde YA:
Cell biology in neuroscience: Death of developing neurons: New
insights and implications for connectivity. J Cell Biol.
203:385–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Terry RD: Basis of structural Alzheimer
disease and some pathogenic conceptsAlzheimer's disease: From
molecular biology to therapy. Becker P and Giacobini F: Birkhäuser
Publishing Ltd.; Cambridge, MA: pp. 19–23. 1996
|
|
194
|
Burggren A and Brown J: Imaging markers of
structural and functional brain changes that precede cognitive
symptoms in risk for Alzheimer's disease. Brain Imaging Behav.
8:251–261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Zlokovic BV: Neurovascular pathways to
neurodegeneration in Alzheimer's disease and other disorders. Nat
Rev Neurosci. 12:723–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Desikan RS, Cabral HJ, Hess CP, Dillon WP,
Glastonbury CM, Weiner MW, Schmansky NJ, Greve DN, Salat DH,
Buckner RL, et al: Automated MRI measures identify individuals with
mild cognitive impairment and Alzheimer's disease. Brain.
132:2048–2057. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Dickerson BC and Wolk DA: Alzheimer's
Disease Neuroimaging Initiative: MRI cortical thickness biomarker
predicts AD-like CSF and cognitive decline in normal adults.
Neurology. 78:84–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Hua X, Leow AD, Lee S, Klunder AD, Toga
AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, et al: 3D
characterization of brain atrophy in Alzheimer's disease and mild
cognitive impairment using tensor-based morphometry. Neuroimage.
41:19–34. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Morra JH, Tu Z, Apostolova LG, Green AE,
Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR Jr,
et al: Validation of a fully automated 3D hippocampal segmentation
method using subjects with Alzheimer's disease mild cognitive
impairment, and elderly controls. Neuroimage. 43:59–68. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Morra JH, Tu Z, Apostolova LG, Green AE,
Avedissian C, Madsen SK, Parikshak N, Toga AW, Jack CR Jr, Schuff
N, et al: Automated mapping of hippocampal atrophy in 1-year repeat
MRI data from 490 subjects with Alzheimer's disease, mild cognitive
impairment and elderly controls. Neuroimage. 45 1 Suppl:S3–S15.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Mirra SS and Markesbery WR: The
Neuropathology of Alzheimer's DiseaseAlzheimer's Disease: Cause
(s), Diagnosis, Treatment and Care. Khachaturian ZS and Radebaugh
TS: CRC press; New York, NY: pp. 111–123. 1996
|
|
202
|
Price DL: Aging of the brain and dementia
of the Alzheimer typePrinciples of Neural Sciences. Kandel ER and
Jessel TM: McGraw-Hill; New York, NY: pp. 1149–1168. 2000
|
|
203
|
Teipel SJ, Bayer W, Alexander GE, Bokde
AL, Zebuhr Y, Teichberg D, Müller-Spahn F, Schapiro MB, Möller HJ,
Rapoport SI and Hampel H: Regional pattern of hippocampus and
corpus callosum atrophy in Alzheimer's disease in relation to
dementia severity: Evidence for early neocortical degeneration.
Neurobiol Aging. 24:85–94. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Xanthakos S, Krishnan KR, Kim DM and
Charles HC: Magnetic resonance imaging of Alzheimer's disease. Prog
Neuropsychopharmacol Biol Psychiatry. 20:597–626. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Moon WJ, Park JY, Yun WS, Jeon JY, Moon
YS, Kim H, Kwak KC, Lee JM and Han SH: A comparison of substantia
nigra T1 hyperintensity in parkinson's disease dementia,
alzheimer's disease and age-matched controls: Volumetric analysis
of neuromelanin imaging. Korean J Radiol. 17:633–640. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Serrano-Pozo A, Frosch MP, Masliah E and
Hyman BT: Neuropathological alterations in alzheimer disease. Cold
Spring Harb Perspect Med. 1:a0061892011. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Avila J, Lim F, Moreno F, Belmonte C and
Cuello AC: Tau function and dysfunction in neurons: Its role in
neurodegenerative disorders. Mol Neurobiol. 25:213–231. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Cole SL and Vassar R: The Alzheimer's
disease beta-secretase enzyme, BACE1. Mol Neurodegener. 2:222007.
View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Zetterberg H, Andreasson U, Hansson O, Wu
G, Sankaranarayanan S, Andersson ME, Buchhave P, Londos E, Umek RM,
Minthon L, et al: Elevated cerebrospinal fluid BACE1 activity in
incipient Alzheimer disease. Arch Neurol. 65:1102–1107. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Selkoe DJ: Cell biology of protein
misfolding: The examples of Alzheimer's and Parkinson's diseases.
Nat Cell Biol. 6:1054–1061. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Cairns NJ, Ikonomovic MD, Benzinger T,
Storandt M, Fagan AM, Shah AR, Reinwald LT, Carter D, Felton A,
Holtzman DM, et al: Absence of Pittsburgh compound B detection of
cerebral amyloid beta in a patient with clinical, cognitive, and
cerebrospinal fluid markers of Alzheimer disease: A case report.
Arch Neurol. 66:1557–1562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Selkoe DJ: Alzheimer's disease: A central
role for amyloid. J Neuropathol Exp Neurol. 53:438–447. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
de Courten-Myers GM: Cerebral amyloid
angiopathy and Alzheimer's disease. Neurobiol Ageing. 25:603–604.
2004. View Article : Google Scholar
|
|
214
|
Haglund M, Sjöbeck M and Englund E: Severe
cerebral amyloid angiopathy characterizes an underestimated variant
of vascular dementia. Dement Geriatr Cogn Disord. 18:132–137. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Tian J, Shi J, Bailey K and Mann DM:
Negative association between amyloid plaques and cerebral amyloid
angiopathy in Alzheimer's disease. Neurosci Lett. 352:137–140.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Hassan M, Sehgel SA and Sajid R:
Regulatory cascade of neuronal loss and glucose metabolism. CNS
Neurol Disord Drug Targets. 13:1232–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Budinger TF: Neuroimaging Applications for
the Study of Alzheimer's DiseaseAlzheimer's Disease: Cause (s),
Diagnosis, Treatment and Care. Khachaturian ZS and Radebaugh TS:
CRC press; New York, NY: pp. 146–169. 1996
|
|
218
|
Hoyer S: Oxidative metabolism deficiencies
in brains of patients with Alzheimer's disease. Acta Neurol Scand.
94:18–24. 1996. View Article : Google Scholar
|
|
219
|
Planel E, Miyasaka T, Launey T, Chui DH,
Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y and
Takashima A: Alterations in glucose metabolism induce hypothermia
leading to tau hyperphosphorylation through differential inhibition
of kinase and phosphatase activities: Implications for alzheimer's
disease. J Neurosci. 24:2401–2411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Evans PH: Free radicals in brain
metabolism and pathology. Br Med Bull. 49:577–587. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Eckert A, Keil U, Kressmann S, Schindowski
K, Leutner S, Leutz S and Müller WE: Effects of EGb 761 Ginkgo
biloba extract on mitochondrial function and oxidative stress.
Pharmacopsychiatry. 1 Suppl 1:S15–S23. 2003.
|
|
222
|
Baloyannis SJ, Costa V and Michmizos D:
Mitochondrial alterations in Alzheimer's disease. Am J Alzheimers
Dis Other Demen. 19:89–93. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Choi JK, Carreras I, Aytan N,
Jenkins-Sahlin E, Dedeoglu A and Jenkins BG: The effects of aging,
housing and ibuprofen treatment on brain neurochemistry in a triple
transgene Alzheimer's disease mouse model using magnetic resonance
spectroscopy and imaging. Brain Res. 1590:85–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Braak H and Braak E: Frequency of stages
of Alzheimer-related lesions in different age categories. Neurobiol
Aging. 18:351–357. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Haroutunian V, Purohit DP, Perl DP, Marin
D, Khan K, Lantz M, Davis KL and Mohs RC: Neurofibrillary tangles
in nondemented elderly subjects and mild Alzheimer disease. Arch
Neurol. 56:713–718. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Price JL and Morris JC: Tangles and
plaques in nondemented aging and ‘preclinical’ Alzheimer's disease.
Ann Neurol. 45:358–368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Jack CR Jr, Dickson DW, Parisi JE, Xu YC,
Cha RH, O'Brien PC, Edland SD, Smith GE, Boeve BF, Tangalos EG, et
al: Antemortem MRI findings correlate with hippocampal
neuropathology in typical aging and dementia. Neurology.
58:750–757. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Senjem ML, Gunter JL, Shiung MM, Petersen
RC and Jack CR Jr: Comparison of different methodological
implementations of voxel-based morphometry in neurodegenerative
disease. Neuroimage. 26:600–608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Tarawneh R, D'Angelo G, Macy E, Xiong C,
Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, et
al: Visinin like protein-1: Diagnostic and prognostic biomarker in
Alzheimer disease. Ann Neurol. 70:274–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Struyfs H, Van Hecke W, Veraart J, Sijbers
J, Slaets S, De Belder M, Wuyts L, Peters B, Sleegers K, Robberecht
C, et al: Diffusion kurtosis imaging: A possible MRI biomarker for
AD diagnosis? J Alzheimers Dis. 48:937–948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
James OG, Doraiswamy PM and Borges-Neto S:
PET Imaging of tau pathology in alzheimer's disease and
tauopathies. Front Neurol. 6:382015. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Baird AL, Westwood S and Lovestone S:
Blood-based proteomic biomarkers of alzheimer's disease pathology.
Front Neurol. 6:2362015. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Anderso NL and Anderson NG: The human
plasma proteome: History, character, and diagnostic prospects. Mol
Cell Proteomics. 1:845–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Montagne A, Barnes SR, Sweeney MD,
Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua
L, et al: Blood-brain barrier breakdown in the aging human
hippocampus. Neuron. 85:296–302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Lewczuk P, Esselmann H, Bibl M, Paul S,
Svitek J, Miertschischk J, Meyrer R, Smirnov A, Maler JM, Klein C,
et al: Electrophoretic separation of amyloid beta peptides in
plasma. Electrophoresis. 25:3336–3343. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Fox NC, Black RS, Gilman S, Rossor MN,
Griffith SG, Jenkins L and Koller M: AN1792(QS-21)-201 Study:
Effects of Abeta immunization (AN1792) on MRI measures of cerebral
volume in Alzheimer disease. Neurology. 64:1563–1572. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Frisoni GB and Delacourte A: Neuroimaging
outcomes in clinical trials in Alzheimer's disease. J Nutr Health
Aging. 13:209–212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Rinne JO, Brooks DJ, Rossor MN, Fox NC,
Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, et
al: 11C-PiB PET assessment of change in fibrillar amyloid-beta load
in patients with Alzheimer's disease treated with bapineuzumab: A
phase 2, double-blind, placebo-controlled, ascending-dose study.
Lancet Neurol. 9:363–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Sperling RA, Jack CR Jr and Aisen PS:
Testing the right target and right drug at the right stage. Sci
Transl Med. 3:111cm332011. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Geerts H, Dacks PA, Devanarayan V, Haas M,
Khachaturian ZS, Gordon MF, Maudsley S, Romero K and Stephenson D:
Brain Health Modeling Initiative (BHMI): Big data to smart data in
Alzheimer's disease: The brain health modeling initiative to foster
actionable knowledge. Alzheimers Dement. 12:1014–1021. 2016.
View Article : Google Scholar : PubMed/NCBI
|